Hyponatremia Associated with Prophylactic Low-Dose Trimethoprim during Systemic Corticosteroid Therapy for AQP4-Positive Optic Neuritis in a Diabetic Patient

 , ,
, ,
Abstract
:1. Introduction
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wrobel, A.; Arciszewska, K.; Maliszewski, D.; Drozdowska, D. Trimethoprim and other nonclassical antifolates an excellent template for searching modifications of dihydrofolate reductase enzyme inhibitors. J. Antibiot. (Tokyo) 2020, 73, 5–27. [Google Scholar] [CrossRef] [Green Version]
- Ho, J.M.W.; Juurlink, D.N. Considerations when prescribing trimethoprim-sulfamethoxazole. CMAJ Can. Med. Assoc. J. 2011, 183, 1851–1858. [Google Scholar] [CrossRef] [Green Version]
- Tsapepas, D.; Chiles, M.; Babayev, R.; Rao, M.K.; Jaitly, M.; Salerno, D.; Mohan, S. Incidence of Hyponatremia with High-Dose Trimethoprim-Sulfamethoxazole Exposure. Am. J. Med. 2016, 129, 1322–1328. [Google Scholar] [CrossRef]
- Perazella, M.A. Trimethoprim-induced hyperkalaemia: Clinical data, mechanism, prevention and management. Drug Saf. 2000, 22, 227–236. [Google Scholar] [CrossRef]
- Rodriguez Soriano, J. Renal tubular acidosis: The clinical entity. J. Am. Soc. Nephrol. 2002, 13, 2160–2170. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Kuroda, Y.; Imamura, S.; Toyoda, A.; Yoshida, I.; Kawakami, M.; Tabei, K. Hyponatremia and/or hyperkalemia in patients treated with the standard dose of trimethoprim-sulfamethoxazole. Intern. Med. (Tokyo, Japan) 2003, 42, 665–669. [Google Scholar] [CrossRef] [Green Version]
- Wald, R.; Jaber, B.L.; Price, L.L.; Upadhyay, A.; Madias, N.E. Impact of hospital-associated hyponatremia on selected outcomes. Arch. Intern. Med. 2010, 170, 294–302. [Google Scholar] [CrossRef]
- Liamis, G.; Milionis, H.; Elisaf, M. A review of drug-induced hyponatremia. Am. J. Kidney Dis. 2008, 52, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Velazquez, H.; Perazella, M.A.; Wright, F.S.; Ellison, D.H. Renal mechanism of trimethoprim-induced hyperkalemia. Ann. Intern. Med. 1993, 119, 296–301. [Google Scholar] [CrossRef]
- Choi, M.J.; Fernandez, P.C.; Patnaik, A.; Coupaye-Gerard, B.; D’Andrea, D.; Szerlip, H.; Kleyman, T.R. Brief report: Trimethoprim-induced hyperkalemia in a patient with AIDS. N. Engl. J. Med. 1993, 328, 703–706. [Google Scholar] [CrossRef]
- Alappan, R.; Perazella, M.A.; Buller, G.K. Hyperkalemia in hospitalized patients treated with trimethoprim-sulfamethoxazole. Ann. Intern. Med. 1996, 124, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Ahmet, A.; Ward, L.; Krishnamoorthy, P.; Mandelcorn, E.D.; Leigh, R.; Brown, J.P.; Cohen, A.; Kim, H. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin. Immunol. 2013, 9, 30. [Google Scholar] [CrossRef] [Green Version]
- Lanier, J.B.; Mote, M.B.; Clay, E.C. Evaluation and management of orthostatic hypotension. Am. Fam. Phys. 2011, 84, 527–536. [Google Scholar]
- Annane, D.; Renault, A.; Brun-Buisson, C.; Megarbane, B.; Quenot, J.P.; Siami, S.; Cariou, A.; Forceville, X.; Schwebel, C.; Martin, C.; et al. Hydrocortisone plus Fludrocortisone for Adults with Septic Shock. N. Engl. J. Med. 2018, 378, 809–818. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 2001, 345, 851–860. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, T.; Gomes, T.; Juurlink, D.N.; Loutfy, M.R.; Glazier, R.H.; Mamdani, M.M. Trimethoprim-sulfamethoxazole-induced hyperkalemia in patients receiving inhibitors of the renin-angiotensin system: A population-based study. Arch. Intern. Med. 2010, 170, 1045–1049. [Google Scholar] [CrossRef] [Green Version]
- Fralick, M.; Macdonald, E.M.; Gomes, T.; Antoniou, T.; Hollands, S.; Mamdani, M.M.; Juurlink, D.N. Co-trimoxazole and sudden death in patients receiving inhibitors of renin-angiotensin system: Population based study. BMJ Clin. Res. Ed. 2014, 349, g6196. [Google Scholar] [CrossRef] [Green Version]
- Antoniou, T.; Hollands, S.; Macdonald, E.M.; Gomes, T.; Mamdani, M.M.; Juurlink, D.N. Trimethoprim-sulfamethoxazole and risk of sudden death among patients taking spironolactone. CMAJ Can. Med. Assoc. J. 2015, 187, E138–E143. [Google Scholar] [CrossRef] [Green Version]
- Sousa, A.G.; Cabral, J.V.; El-Feghaly, W.B.; de Sousa, L.S.; Nunes, A.B. Hyporeninemic hypoaldosteronism and diabetes mellitus: Pathophysiology assumptions, clinical aspects and implications for management. World J. Diabetes 2016, 7, 101–111. [Google Scholar] [CrossRef]
- Hoorn, E.J.; Zietse, R. Diagnosis and Treatment of Hyponatremia: Compilation of the Guidelines. J. Am. Soc. Nephrol. 2017, 28, 1340–1349. [Google Scholar] [CrossRef]
- Babayev, R.; Terner, S.; Chandra, S.; Radhakrishnan, J.; Mohan, S. Trimethoprim-associated hyponatremia. Am. J. Kidney Dis. 2013, 62, 1188–1192. [Google Scholar] [CrossRef]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet (London, England) 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Amiry-Moghaddam, M.; Ottersen, O.P. The molecular basis of water transport in the brain. Nat. Rev. Neurosci. 2003, 4, 991–1001. [Google Scholar] [CrossRef]
- Pittock, S.J.; Weinshenker, B.G.; Lucchinetti, C.F.; Wingerchuk, D.M.; Corboy, J.R.; Lennon, V.A. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch. Neurol. 2006, 63, 964–968. [Google Scholar] [CrossRef] [Green Version]
- Pu, S.; Long, Y.; Yang, N.; He, Y.; Shan, F.; Fan, Y.; Yin, J.; Gao, Q.; Cong, G. Syndrome of inappropriate antidiuretic hormone secretion in patients with aquaporin-4 antibody. J. Neurol. 2015, 262, 101–107. [Google Scholar] [CrossRef]
- Inoue, K.; Nakayama, T.; Kamisawa, A.; Saito, J. Syndrome of inappropriate antidiuretic hormone accompanied by bilateral hypothalamic and anterior thalamic lesions with serum antiaquaporin 4 antibody. BMJ Case Rep. 2017, 2017. [Google Scholar] [CrossRef]
- Iorio, R.; Lucchinetti, C.F.; Lennon, V.A.; Costanzi, C.; Hinson, S.; Weinshenker, B.G.; Pittock, S.J. Syndrome of inappropriate antidiuresis may herald or accompany neuromyelitis optica. Neurology 2011, 77, 1644–1646. [Google Scholar] [CrossRef] [Green Version]
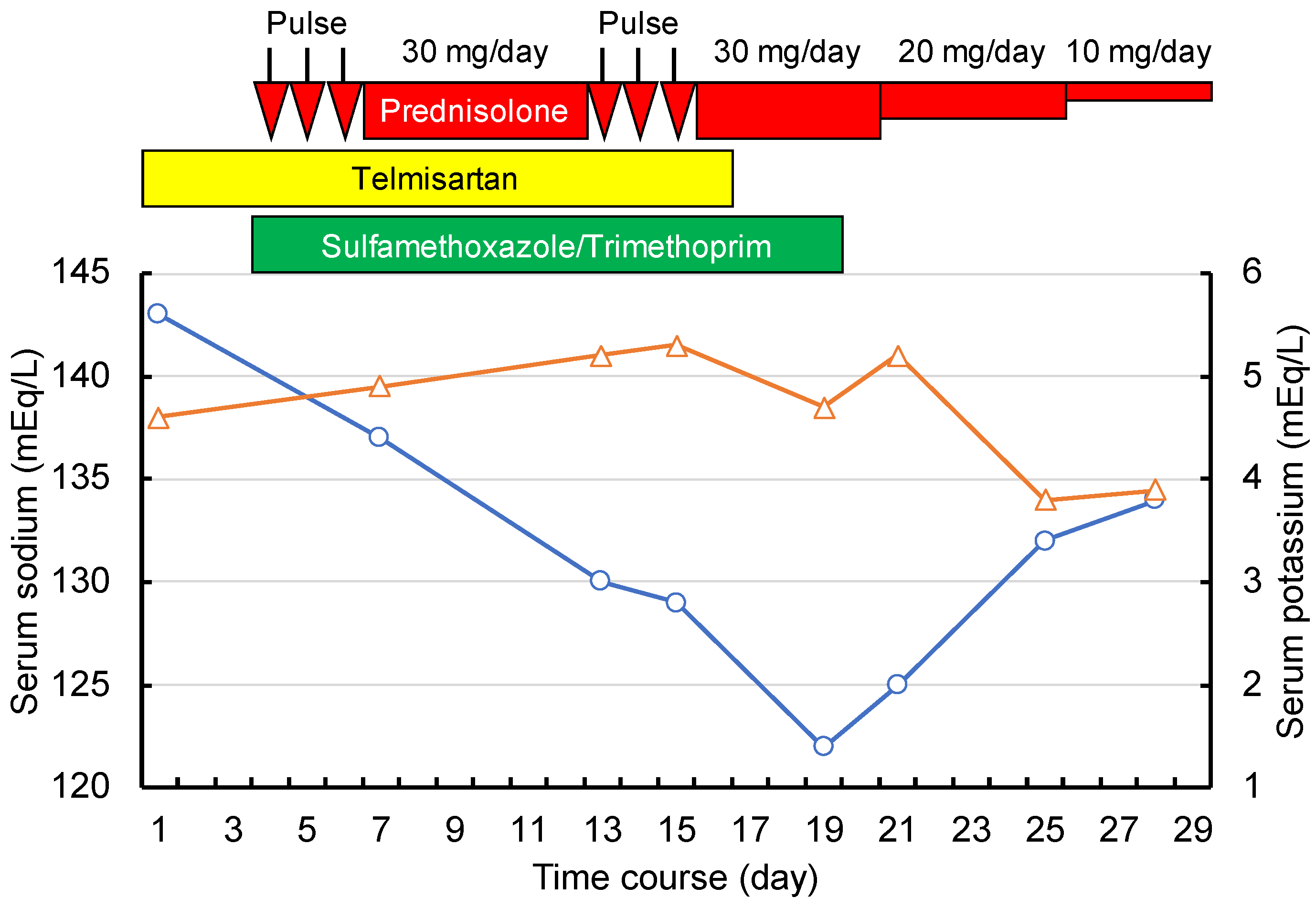

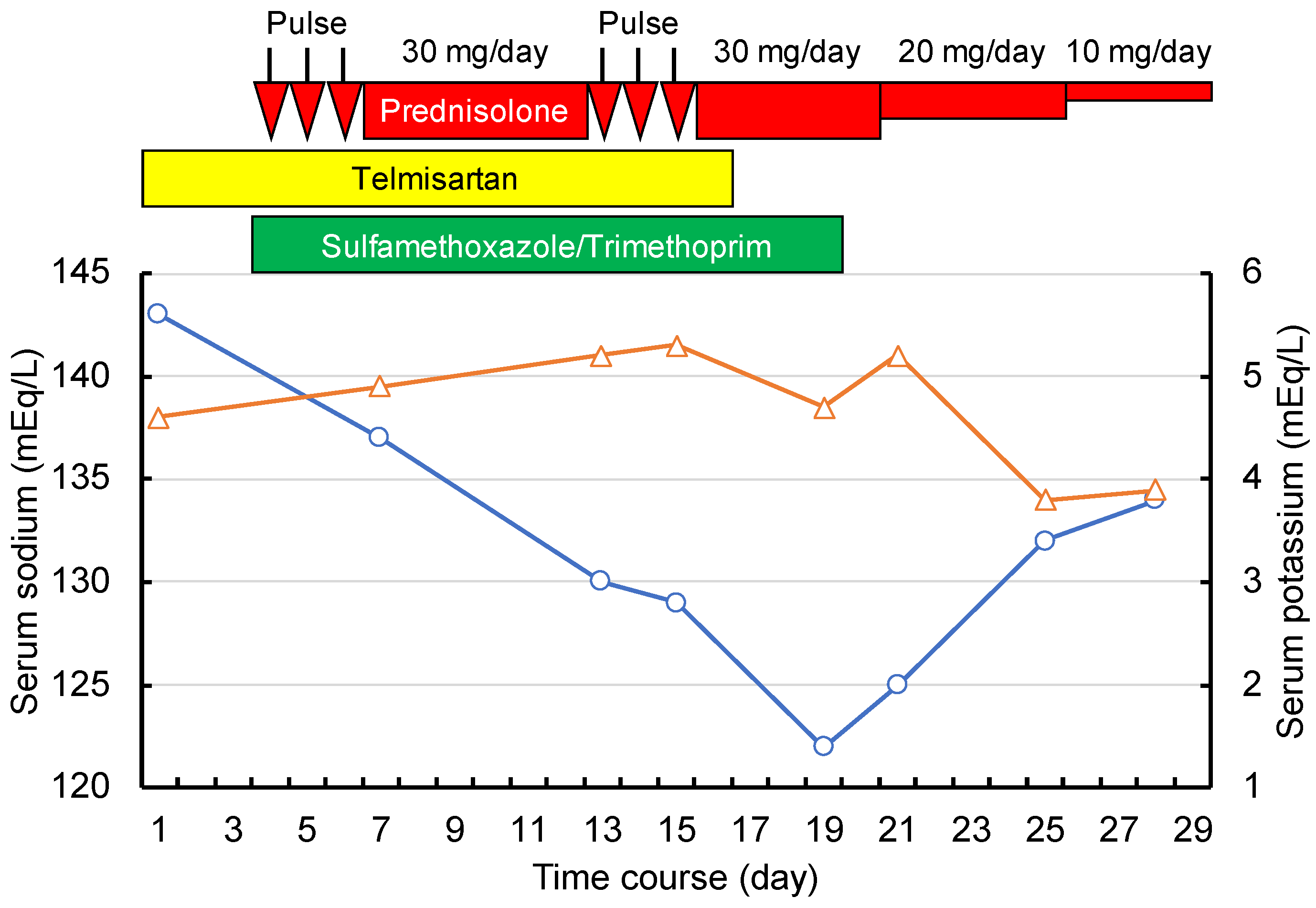{kind=link}
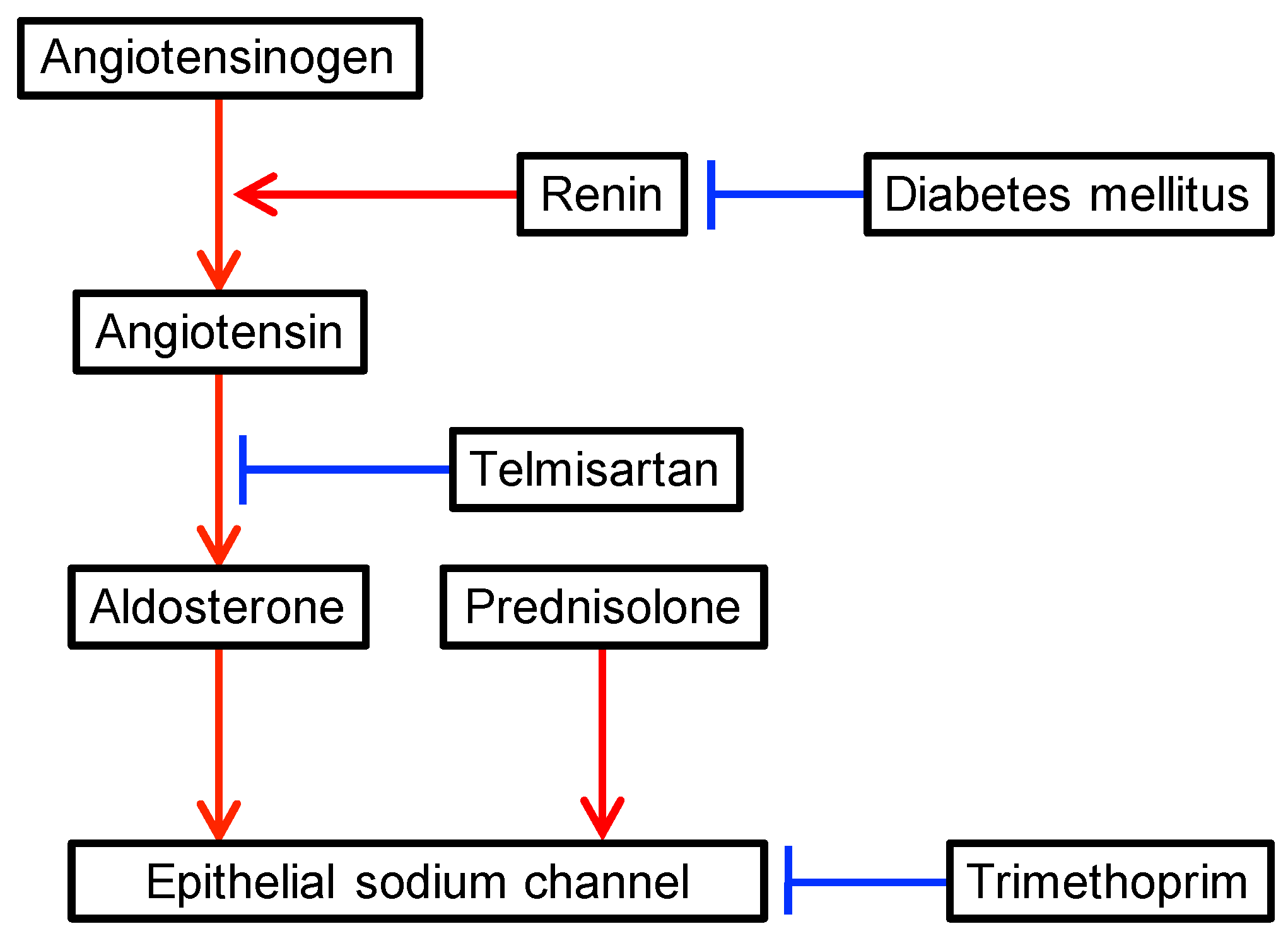
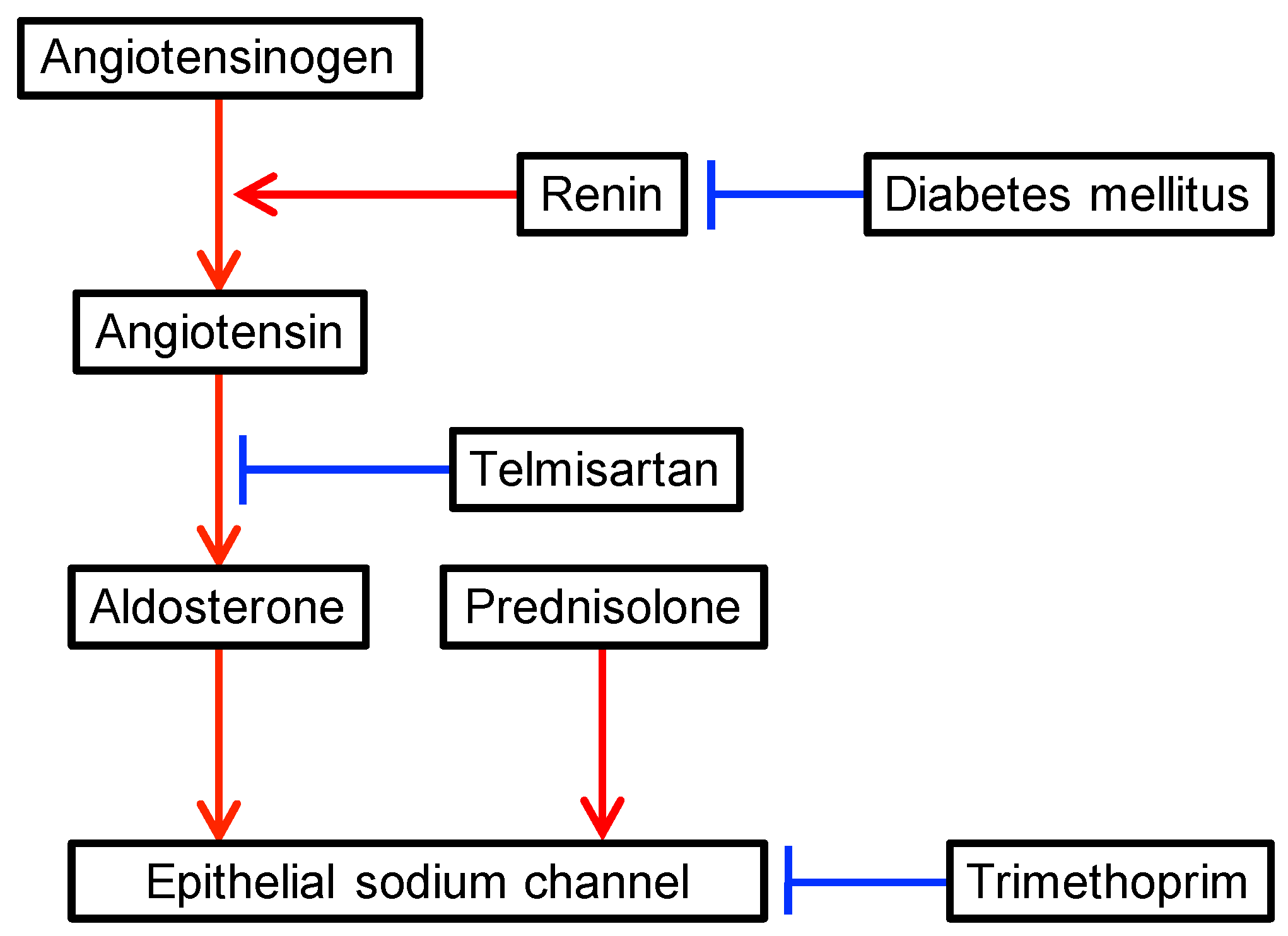{kind=link}
| Parameters | Values | Units | Reference Ranges |
|---|---|---|---|
| White blood cells | 6600 | /µL | (3300–8600) |
| Red blood cells | 4.31 | ×106/µL | (3.86–4.92) |
| Hemoglobin | 13.2 | g/dL | (11.6–14.8) |
| Platelets | 133,000 | /µL | (158,000–348,000) |
| Total bilirubin | 0.68 | mg/dL | (0.4–1.5) |
| Aspartate aminotransferase | 18 | U/L | (13–30) |
| Alanine aminotransferase | 16 | U/L | (7–23) |
| Gamma-glutamyl transpeptidase | 17 | U/L | (9–32) |
| Blood urea nitrogen | 11.2 | mg/dL | (8.0–20.0) |
| Creatinine | 0.49 | mg/dL | (0.46–0.79) |
| Estimated glomerular filtration rate | 98 | mL/min/1.73m2 | / |
| Uric acid | 3.8 | mg/dL | (2.6–7.0) |
| Sodium | 143 | mEq/L | (138–145) |
| Potassium | 4.6 | mEq/L | (3.6–4.8) |
| Chloride | 108 | mEq/L | (101–108) |
| C-reactive protein | 0.02 | mg/dL | (≤0.2) |
| Total protein | 7.8 | g/dL | (6.6-8.1) |
| Glucose | 81 | mg/dL | (73–109) |
| Glycated hemoglobin | 8.0 | % | (4.6–6.2) |
| Thyroid stimulating hormone | 0.84 | µIU/mL | (0.34–3.8) |
| Free thyroxine | 1.14 | ng/dL | (0.8–1.5) |
| Anti-AQP4 antibody | ≥40 | U/mL | (<3) |
| Urine pH | 5.5 | (5–7) | |
| Urine albumin | 20 | mg/day | / |
| Parameters | Values | Units | Reference Ranges | ||
|---|---|---|---|---|---|
| Day 21 | Day 25 | Day 28 | |||
| Body weight | 45.4 | 44.3 | 43.8 | kg | |
| Blood pressure | 128/59 | 119/66 | 129/63 | mmHg | |
| Pulse rate | 81 | 83 | 89 | beats/min | |
| Blood examination | |||||
| White blood cells | 11,100 | 8300 | 4600 | /µL | (3300–8600) |
| Red blood cells | 4.15 | 3.62 | 3.83 | ×106/µL | (3.86–4.92) |
| Hemoglobin | 12.7 | 11.2 | 11.9 | g/dL | (11.6–14.8) |
| Platelets | 181,000 | 156,000 | 161,000 | /µL | (158,000–348,000) |
| Total bilirubin | 0.85 | 0.65 | NM | mg/dL | (0.4–1.5) |
| Aspartate aminotransferase | 13 | 23 | 13 | U/L | (13–30) |
| Alanine aminotransferase | 18 | 31 | 20 | U/L | (7–23) |
| Gamma-glutamyl transpeptidase | 19 | 20 | 13 | U/L | (9–32) |
| Blood urea nitrogen | 11.9 | 8.0 | 6.7 | mg/dL | (8.0–20.0) |
| Creatinine | 0.47 | 0.40 | 0.41 | mg/dL | (0.46–0.79) |
| Estimated glomerular filtration rate | 102.6 | 122.4 | 119.2 | mL/min/1.73m2 | |
| Uric acid | 2.1 | 2.8 | 2.2 | mg/dL | (2.6–7.0) |
| C-reactive protein | 0.02 | 0.01 | 0.02 | mg/dL | (≤0.2) |
| Total protein | 5.9 | 5.3 | 5.7 | g/dL | (6.6–8.1) |
| Glucose | 149 | 96 | 113 | mg/dL | (73–109) |
| Sodium | 125 | 132 | 134 | mEq/L | (138–145) |
| Potassium | 5.2 | 3.8 | 3.9 | mEq/L | (3.6–4.8) |
| Chloride | 96 | 99 | 101 | mEq/L | (101–108) |
| Osmolality | 264 | 267 | 274 | mOsm/kg H2O | (276–292) |
| Plasma renin activity | 37 | 26 | 24 | ng/mL/hr | (0.3–2.9) |
| Plasma aldosterone | 43.2 | 38 | 167 | pg/mL | (29.9–159) |
| Brain natriuretic peptide | 7.7 | NM | NM | pg/mL | (≤18.4) |
| Arterial blood gas analysis | |||||
| pH | 7.438 | 7.465 | 7.469 | (7.35–7.45) | |
| CO2 | 26 | 32.3 | 30.4 | mmHg | (35–45) |
| HCO3- | 17.2 | 22.7 | 21.6 | mEq/L | (22–26) |
| Base excess | −5.6 | −0.4 | −1.2 | mEq/L | (–2–2) |
| Urine examination | |||||
| Sodium | 92 | 46 | 137 | mEq/L | (70–250) |
| Potassium | 17.7 | 24.5 | 42 | mEq/L | (25–100) |
| Chloride | 79 | 47 | 151 | mEq/L | (70–250) |
| Creatinine | 43.8 | 58 | 111.3 | mg/dL | (100–150) |
| Osmolality | 510 | 614 | 785 | mOsm/kg H2O | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takubo, M.; Tanaka, S.; Kushimoto, M.; Ikeda, J.; Ogawa, K.; Suzuki, Y.; Abe, M.; Ishihara, H.; Fujishiro, M. Hyponatremia Associated with Prophylactic Low-Dose Trimethoprim during Systemic Corticosteroid Therapy for AQP4-Positive Optic Neuritis in a Diabetic Patient. Antibiotics 2020, 9, 201. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040201
Takubo M, Tanaka S, Kushimoto M, Ikeda J, Ogawa K, Suzuki Y, Abe M, Ishihara H, Fujishiro M. Hyponatremia Associated with Prophylactic Low-Dose Trimethoprim during Systemic Corticosteroid Therapy for AQP4-Positive Optic Neuritis in a Diabetic Patient. Antibiotics. 2020; 9(4):201. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040201
Chicago/Turabian StyleTakubo, Masahiro, Sho Tanaka, Masaru Kushimoto, Jin Ikeda, Katsuhiko Ogawa, Yutaka Suzuki, Masanori Abe, Hisamitsu Ishihara, and Midori Fujishiro. 2020. "Hyponatremia Associated with Prophylactic Low-Dose Trimethoprim during Systemic Corticosteroid Therapy for AQP4-Positive Optic Neuritis in a Diabetic Patient" Antibiotics 9, no. 4: 201. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040201