Candidiasis and Mechanisms of Antifungal Resistance



1
Division of Infectious Diseases, Department of Medicine, Stony Brook University, Stony Brook, New York, NY 11794, USA
2
Department of Molecular Genetics and Microbiology, Stony Brook University, Stony Brook, New York, NY 11794, USA
3
Veterans Administration Medical Center, Northport, New York, NY 11768, USA
*
Author to whom correspondence should be addressed.
Antibiotics 2020, 9(6), 312; https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060312
Submission received: 4 May 2020
/
Revised: 6 June 2020
/
Accepted: 7 June 2020
/
Published: 9 June 2020
(This article belongs to the Special Issue Synopsis on Antimicrobials Challenges—from Dentistry to Environmental Visions)
Abstract
:Candidiasis can be present as a cutaneous, mucosal or deep-seated organ infection, which is caused by more than 20 types of Candida sp., with C. albicans being the most common. These are pathogenic yeast and are usually present in the normal microbiome. High-risk individuals are patients of human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS), organ transplant, and diabetes. During infection, pathogens can adhere to complement receptors and various extracellular matrix proteins in the oral and vaginal cavity. Oral and vaginal Candidiasis results from the overgrowth of Candida sp. in the hosts, causing penetration of the oral and vaginal tissues. Symptoms include white patches in the mouth, tongue, throat, and itchiness or burning of genitalia. Diagnosis involves visual examination, microscopic analysis, or culturing. These infections are treated with a variety of antifungals that target different biosynthetic pathways of the pathogen. For example, echinochandins target cell wall biosynthesis, while allylamines, azoles, and morpholines target ergosterol biosynthesis, and 5-Flucytosine (5FC) targets nucleic acid biosynthesis. Azoles are commonly used in therapeutics, however, because of its fungistatic nature, Candida sp. evolve azole resistance. Besides azoles, Candida sp. also acquire resistance to polyenes, echinochandins, and 5FC. This review discusses, in detail, the drug resistance mechanisms adapted by Candida sp.
1. Introduction
Candida species are commensals and thus are part of the normal human flora and are localized on skin and gastrointestinal and genital tracts. However, Candida can also cause various infections in susceptible patients that includes elderly, hospitalized, or immunosuppressed patients. Invasive Candida infection is one of the most common fungal infections globally [1]. In the United States, Candida sp. were reported to be one of the leading causes of healthcare-associated infections [1]. Amongst the different Candida sp., Candida albicans is the most commonly recovered (37%) from clinical species, followed by Candida glabrata (27%). Other clinically relevant species recovered from blood stream infections include C. parapsilosis (14%), C. krusei (2%), C. tropicalis (8%), C. dubliniensis (2%), C. lusitaniae (2%), and the most recent, C. auris. C. auris is an emergent multi-drug-resistant pathogen that is often misidentified and at present is a major concern in healthcare settings. C. auris reported cases increased by 318% between 2015 and 2018. As per the Centers for Disease Control and Prevention (CDC), an estimated 34,000 cases of Candidiasis were reported in hospitalized patients and about 1700 people died in 2017 [2]. Candidiasis has a diverse clinical spectrum ranging from non-life threatening superficial mucocutaneous infections to devastating invasive disease associated with candidemia. In fact, the attributable mortality observed with candidemia is between 30% and 47% [3]. Candida infection is also commonly associated with medical devices such as central venous catheters, cardiovascular devices, and urinary catheters [4]. An episode of candidemia can lead to seeding of any organs, including the liver, spleen, bones, joints, eyes, or brain.
Because of a lack of rapid diagnostic assays for invasive Candidiasis, most Candidiasis cases are still diagnosed by routine fungal cultures of blood, urine tissue, and other body fluids. This method can have a low sensitivity, and in some cases, it can also render false-positive results because of contamination, which can occur in the process. Commonly, empiric anti-fungal therapy is initiated in febrile or septic patients in the intensive care unit with an indwelling central venous catheter, recent abdominal surgery, or chemotherapy in the absence of response to anti-microbial therapy. This approach can lead to the unnecessary use of antifungal agents and promote the emergence of resistance in individuals without invasive Candidiasis or a delay in effective antifungal therapy for those who are infected.
The strategy for treatment of invasive Candidiasis depends on the patient’s immune status, location, and severity of the infection. In addition to an adequate source control, removal of infected medical devices and antifungal agents have been important therapeutic tools for invasive Candida infections [5]. At present, four main classes of antifungal drugs with activity against Candida species are available, including polyenes, azoles, echinocandins, and 5-Flucytosine (5FC). Similar to antibiotics for bacterial infection, an emergence of antifungal resistance among Candida species is a serious threat to public health worldwide. According to the US Center of Disease Control and Prevention (CDC) 2019 report of antibiotic resistance threat, more than 34,000 cases and 1700 deaths annually were due to drug-resistant Candida sp. In addition, 323 cases of emerging multidrug- resistant Candida auris infection were reported.
In the current review, we discuss the molecular mechanisms of action of these antifungal agents as well as mechanisms of drug resistance used by Candida sp.
2. Antifungals and Their Targets
Candidiasis can be treated with antifungals that belong to different classes of drugs and target different cellular processes, thereby either inhibiting (fungistatic) or killing (fungicidal) the growth of this pathogenic yeast. These cellular processes include the biosynthesis of the cell wall, cell membrane, and biosynthesis of RNA. Each of these biosynthesis processes involves a series of enzymes. The targets and the mechanisms of antifungals employed for treating Candidiasis are outlined below.
2.1. Antifungals that Target Ergosterol and Its Biosynthesis
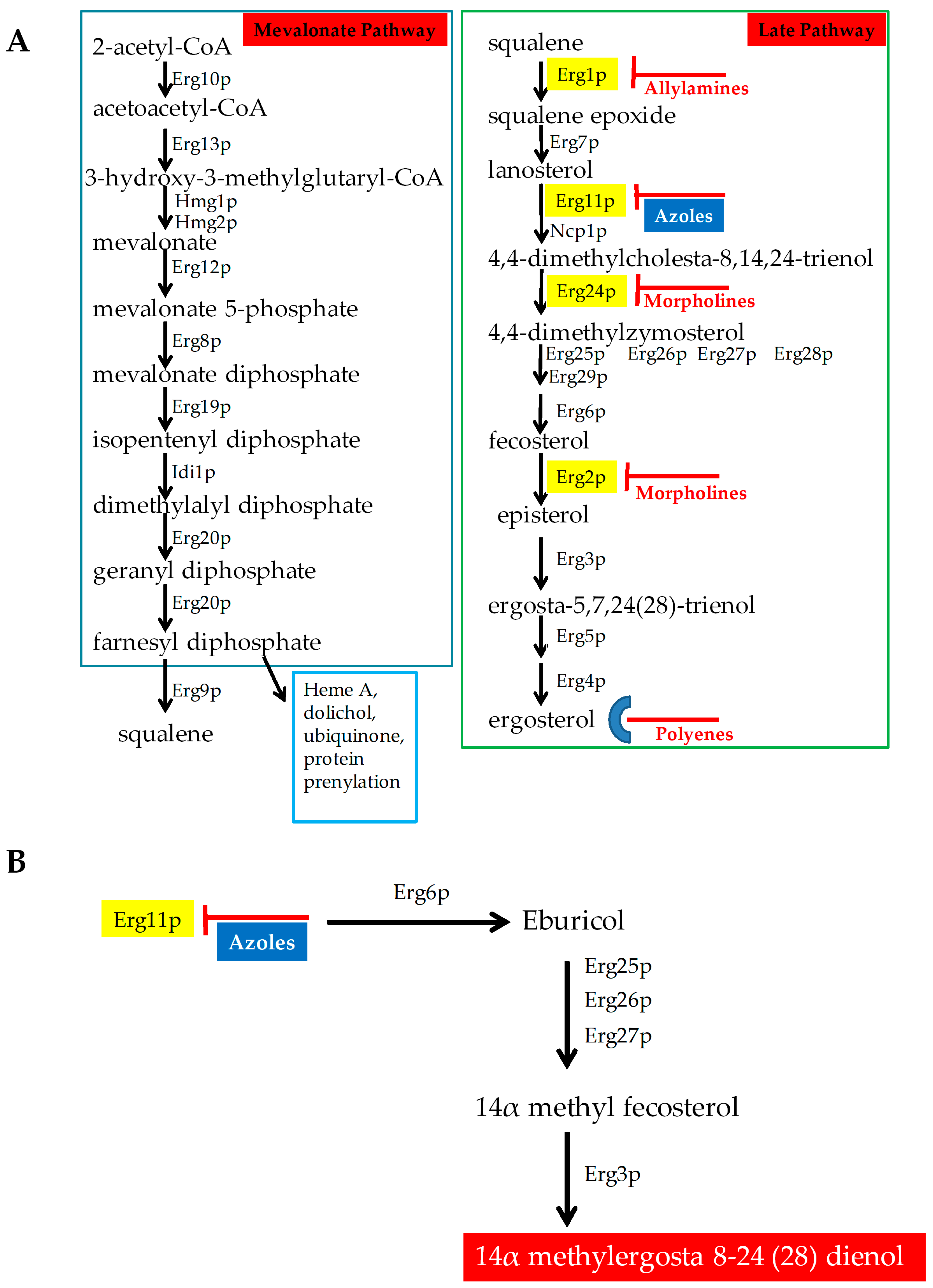
Ergosterol is the major sterol component of fungal cell membranes, including the plasma and mitochondrial membranes. It is vital for fungi to maintain the structure and function of these membranes. Together, sterols and sphingolipids form lipid rafts in the cell membrane. These lipid rafts contain many biologically important proteins, which are involved in signaling, response to stress, mating, and nutrient transport [6,7]. Ergosterol biosynthesis is catalyzed by a cascade of 25 different enzymes (Figure 1A). This biosynthetic pathway constitutes an ideal target for drugs because ergosterol is a very important lipid for fungi and plants but is not present in humans. Several drugs target either the biosynthesis of this lipid or ergosterol itself, and are described below (Figure 1).
Azoles: Azoles are five-membered heterocyclic compounds with antifungal properties. They are broadly classified into two groups that include imidazole and triazole. Imidazoles contain two nitrogen in the azole ring and include antifungals clotrimazole, econazole, ketoconazole, miconazole, and tioconazole. Triazoles contain three nitrogen in the azole ring and include fluconazole, itraconazole, voriconazole, isavuconazole, and posaconazole. Fluconazole is the most common azole used during therapy. Second generation of triazoles that include voriconazole, posaconazole, and isavuconazole are more potent against resistant pathogens. Isavuconazole is a novel azole, as effective as voriconazole but less toxic than voriconazole. Toxicity is lower in isavuconazole, since it is highly water soluble and does not require beta-cyclodextrin in its intravenous (IV) formulation, which is required for voriconazole, causing reduced nephrotoxicity [8,9]. Isavuconazole can be administered both intravenously (IV) and orally and its toxicity profile is similar to that of fluconazole but is more active [10,11].
Azoles are the most common antifungal drug class used for treating and preventing Candida infections. Azoles target the enzyme 14α–demethylase (Erg11p), an important enzyme in ergosterol biosynthesis (Figure 1) [12]. Azoles bind to Erg11p, thereby effectively lowering the ergosterol levels of the cell. When Erg11p is inhibited, other enzymes in the pathway (Erg6p, Erg25p, Erg26p, Erg27p, and Erg3p) synthesize a fungistatic toxic sterol (14α methylergosta 8-24 (28) dienol, Figure 1B) [7]. Besides this, azoles are also responsible for elevating the levels of reactive oxygen species (ROS) [13]. Both the elevated ROS levels and toxic sterol production inhibit the growth of the infecting fungus.
Polyenes: This class of drugs target ergosterol in the plasma membrane and are fungicidal. They bind to ergosterol and form pores [14]. Pore formation causes rapid leakage of monovalent ions (K+, Na+, H+, and Cl−) and subsequent fungal cell death. Polyene drugs include amphotericin B and nystatin, but only amphotericin B is used for systemic treatment. Ergosterol is more sensitive to amphotericin B than cholesterol, the common mammalian sterol. To decrease its toxicity, the conventional amphotericin B, which is complexed with sodium deoxycholate (ABD), has been modified as a cholesteryl sulfate complex (ABCD), as a lipid complex (ABLC), and as a liposomal formulation (LAMB). These formulations may exhibit different pharmacokinetic characteristics compared to conventional amphotericin B [15,16]. For the treatment of invasive Candidiasis, amphotericin B is rarely required. This drug is mostly chosen when the Candida sp. is resistant to other drug classes or the drugs do not penetrate into the relevant niche.
Other antifungals that inhibit growth of a large variety of fungi but are not used to treat Candidiasis are listed below:
Allylamines: These antifungals target Squalene epoxidase (Erg1p) in the ergosterol biosynthesis pathways [7]. These drugs include terbinafine (Lamisil), flunarizine, and naftifine. Terbinafine (Lamisil) is commonly used in treating dermatophyte infections [17].
Morpholines: This class of drugs include fenpropimorph, tridemorph, and amorpholine. They target the ergosterol biosynthetic enzyme C-14 sterol reductase (Erg24p) [7]. The morpholines are commonly used in agriculture, and exhibit high toxicity in humans [17]. Only 5% amorolfine hydrochloride containing nail lacquer solution is used for treating nail dermatophyte infections [18].
2.2. Inhibitors of Cell Wall Biosynthesis
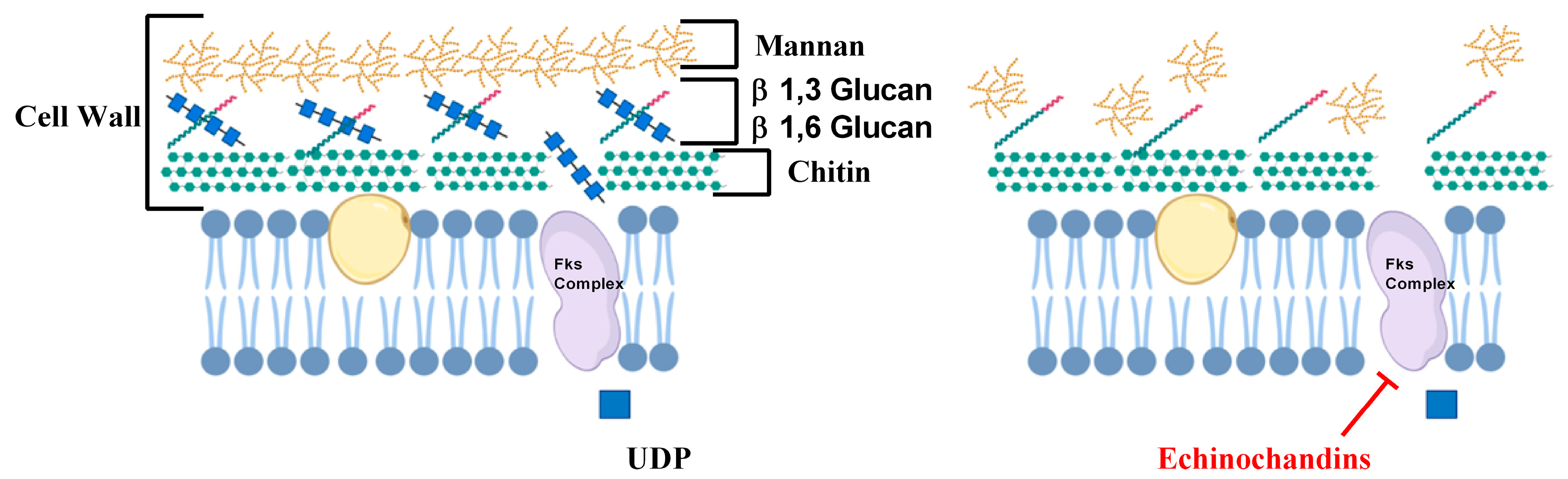
Besides ergosterol, cell wall biosynthesis of Candida sp. is also targeted by various antifungals. The cell wall is the rigid outermost layer of the fungal cell and is the first line of defense, protecting the fungal cells from osmotic stress. Mammalian cells do not have cell walls and therefore the enzymes of the cell wall biosynthetic pathways are important antifungal targets [19]. Echinocandins antifungal drugs target the cell wall and include caspofungin, micafungin, and anidulafungin. They target β1-3 glucan synthase enzyme, which is encoded by three genes FKS1, FKS2, and FKS3 [20]. β1-3 glucan synthase enzyme is a complex of three proteins, Fks1p, Fks2p, and Fks3p, that uses Uridine Diphosphate Glucose (UDP-glucose) to synthesize β1-3 glucan, an important component in the fungal cell wall (Figure 2) [21]. These drugs are usually fungicidal and commonly chosen because they exhibit low toxicities for humans [22]. Echinochandins are usually administered to patients with moderately severe to severe illness or to patients with prior exposure to azoles [23].
2.3. Inhibitors of Nucleic Acid Biosynthesis
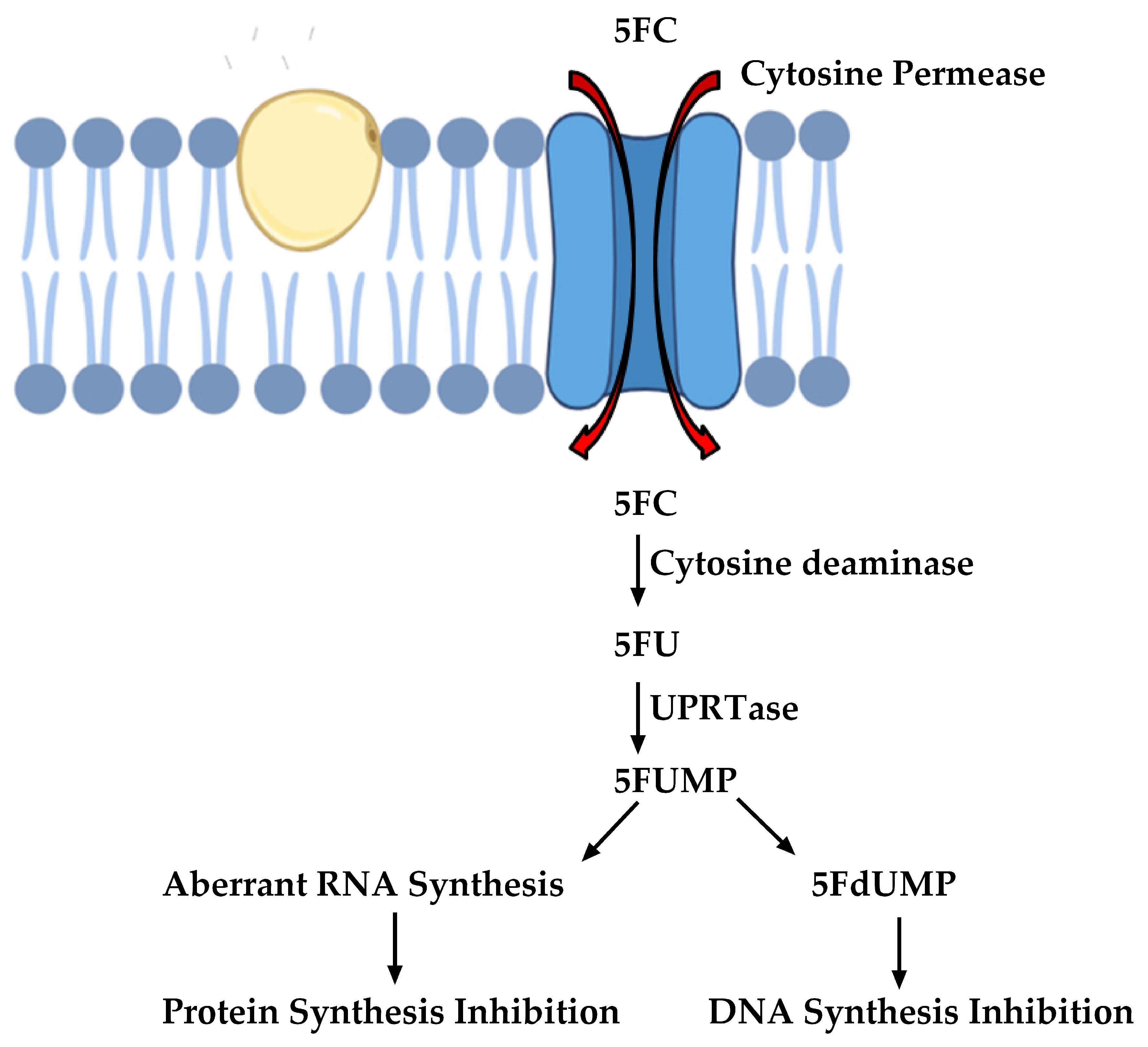
Other important antifungals are those that target nucleic acid biosynthesis. These were originally designed in the 1950s and are one of the oldest classes of antifungal drugs. 5-Flucytosine (5FC) is an antifungal that interferes with nucleic acid biosynthesis. The susceptible cells import 5FC via the cytosine permease enzyme [24]. 5FC is converted to 5-Fluorouracil (5FU), which is metabolized to 5-Fluorouridine triphosphate (5FUTP). 5FUTP is incorporated in the fungal RNA instead of uridine triphosphate (UTP), thereby affecting protein translation. Alternatively, 5FU can be converted to 5-Fuorodeoxyuridine monophosphate (5FdUMP) that inhibits thymidylate synthase, an important enzyme in DNA biosynthesis [24]. The pathway is summarized in Figure 3.
3. Drug Efficacies and Therapeutic Failure
Several important factors may contribute to treatment failure. These are listed below.
Bioavailability: Differences in drug bioavailability between different tissues can contribute to the effectiveness of antifungal agents in treating fungal infections. For example, bioavailability of azole drugs in vaginal tissues with a low pH is much lower than that in blood [25]. Also, infections in the central nervous system are particularly difficult to treat because echinocandins do not cross the blood–brain barrier sufficiently and therefore, adequate drug levels are not achieved. Furthermore, echinocandins are bound to albumin and are not excreted into the urine. Therefore, they may not penetrate at high levels to adequately treat Candida infections in the urinary tract. Importantly, different lipid formulations vary in their tissue distribution and toxicity. Specifically, although LAMB has a better overall therapeutic index, ABLC achieves higher lung and kidney tissue concentrations [16].
Host Immune system: The host immune system plays a critical role in successful therapeutics. Especially, static antifungal drugs such as the azoles require support from the host’s immune system for successful treatment of invasive Candida infections. In severely neutropenic patients, these drugs may be less effective and supportive treatment with growth factors to boost engraftment is required.
Successful azole therapy depends on the drug and on the host’s immune response to the fungus. Without a functional immune system, azoles cannot control the fungal infections. For example, Candida sp. can colonize the mouth of 64% to 84% of human immunodeficiency virus HIV-infected patients [26]. Development of azole-resistance in these patients has many contributing factors. Typically, oral candidiasis regularly relapses in HIV-infected patients with low CD4+ cell counts (<50/mm3) [27]. These patients typically receive a long-term, low-dose azole therapy, which can select for azole-resistant Candida sp. In one case, a series of 17 C. albicans clinical isolates were obtained from a single HIV patient receiving azole therapy over two years. Azole-resistance increased steadily as these isolates were exposed to increasing amounts of drugs [28].
Biofilm formation: Fungi can develop biofilms on the surface of medical equipment, such as catheters. These biofilms are not only resistant to the penetration of some antifungals but also the drug targets may be less expressed [29].
Drug pharmacokinetics, infection severity and side-effects: The pharmacokinetics of the drugs is an important factor that contribute to drug efficacies. These include drug metabolism, absorption, and distribution that can alter drug effectiveness. Additionally, drug efficacy can depend on the severity of the infection, and the population size of the infecting organisms [26]. Further, while administering an antifungal, one should consider its side effects. For example, 5FC was used in the past as monotherapy for treating candidiasis, aspergillosis, and cryptococcosis. Side effects of this drug are substantial and include, colitis, bone-marrow suppression, and liver toxicity. These drugs are now used mostly in combination with azoles for complicated fungal infections or in cases of infections with multidrug-resistant Candida sp. [24]. Toxicity is also a common side effect of amphotericin B [17]. Nephrotoxicity is commonly associated with amphotericin B that is caused by renal tubular acidosis, azotaemia, impaired renal concentrating ability, and electrolyte abnormalities [30,31]. Amphotericin B promotes inflammatory cytokines release by a Toll-like receptor- and CD14-dependent mechanism, which leads to systemic side effects (fever, nausea, hypotension, and shaking) [32]. Azoles and echinochandins have relatively low side effects. For example, in rare circumstances, long-term use of azoles, especially posaconazole and voriconazole, can cause liver injury [33]. Similarly, echinochandins also show minimum side effects, which include nausea, vomiting, and abdominal pain [34].
Antifungal drug resistance: Another important factor that can cause therapeutic failure is antifungal drug resistance of the infecting Candida sp. For example, prolonged use of a fungistatic azole drugs like fluconazole may cause the pathogenic yeasts to develop resistance, making the drug less effective. Fluconazole is the most common azole used for prophylaxis and treatment of Candida infections. However, several Candida sp. have evolved resistance to azoles, which is an emerging problem causing therapeutic failures [17]. For example, azole resistance in C. albicans became a severe problem in the 1990s, when 90% of acquired immunodeficiency syndrome (AIDS) patients had oral candidiasis, many received a long-term azole therapy and certain patient populations developed resistance [26].
4. Antifungal Drug Resistance
4.1. Azole Drug Resistance
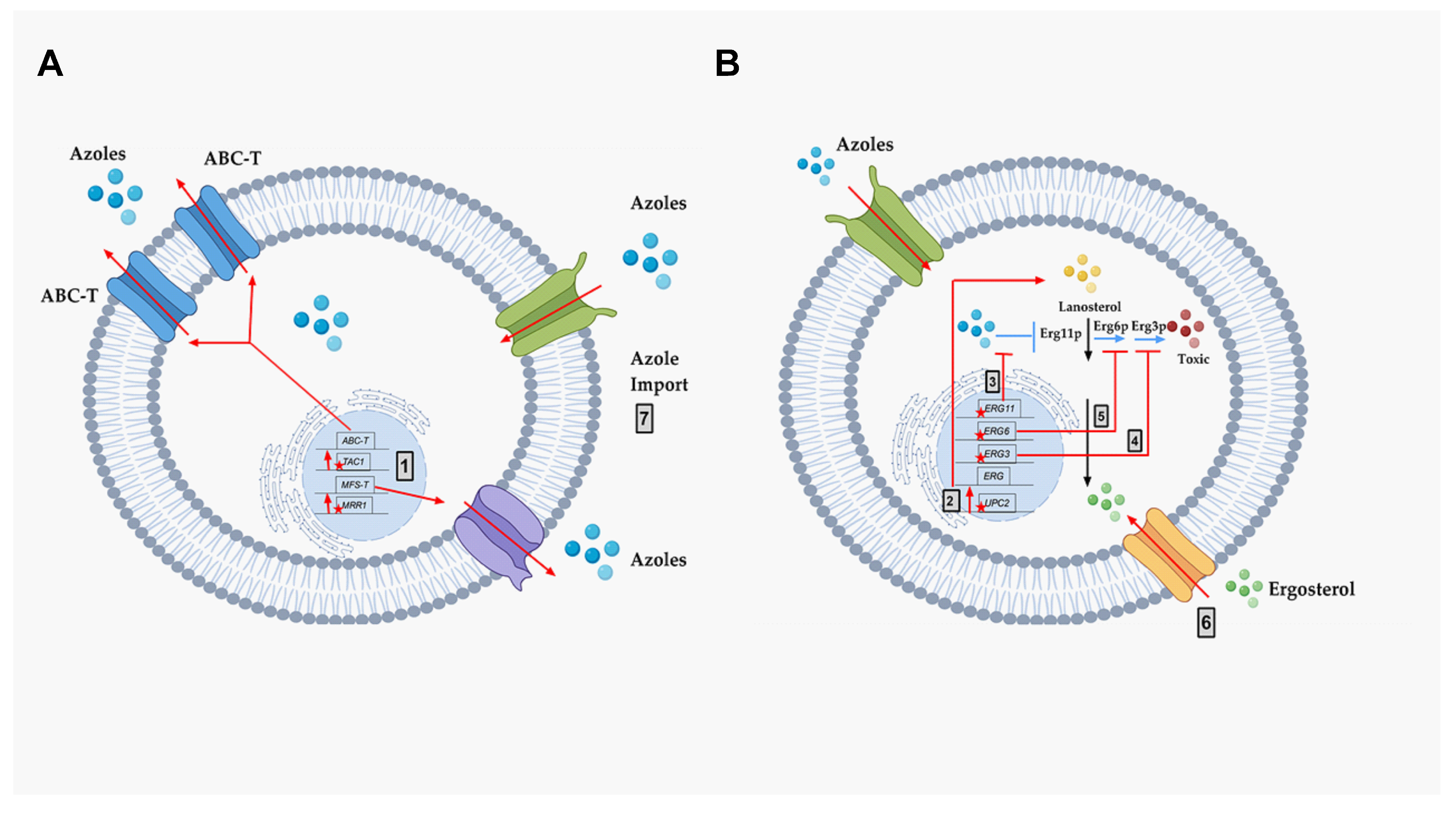
For decades, extensive research has analyzed the molecular mechanisms of azole resistance in pathogenic fungi. The known mechanisms are described below and summarized in Figure 4.
The different mechanisms of azole resistance are described below. These mechanisms are all well studied, are all equally important, and are frequently observed in several drug-resistant Candida clinical isolates.
4.1.1. Over-Expression of Membrane Transporters
A large number of membrane proteins are present in pathogenic yeast. These membrane proteins are present in the cell membrane [35], vacuolar membrane [36], and mitochondrial membrane [37]. They play various physiological roles that include environmental sensing, nutrient transport, signal transduction, drug efflux, drug modification, and drug detoxification [35]. For example, mitochondrial membrane-localized protein Atm1p, an ABC transporter, plays an important role in iron homeostasis [38], while vacuolar membrane-localized membrane transporter Mlt1p transports phosphatidyl choline (PC) [36]. Further, a single membrane transporter can have multiple physiological roles. For example, a recent study demonstrated that Mlt1p transporter is responsible for transporting PC as well as importing azoles into vacuoles. Deletion of Mlt1p caused susceptibilities to methotrexate and azoles in C. albicans [36].
Two types of membrane transporters have been identified that correlate to azole resistance in fungi. They are described below (Figure 4 Grey box 1):
- a)
- ABC-transporters. Adenosine Triphosphate (ATP)-binding cassette transporters (ABC-T) are active transporters requiring ATP as an energy source. Each ABC-T consists of two membrane-spanning domains (MSD), each containing six transmembrane segments, and two nucleotide binding domains (NBD). Each NBD consists of an ATP-binding cassette (ABC) that binds ATP [39]. Azoles are the substrates for the ABC-Ts that are listed in Table 1 [40].
- b)
- MFS-transporters. Major facilitator transporters (MFS-T) require a proton gradient of the plasma membrane as an energy source to transport xenobiotics. MFS-Ts do not have the NBDs that are characteristic of ABC-Ts, and they have 12 to 14 transmembrane segments [41]. Azoles are also the substrates of the MFS-Ts listed in Table 1.
In pathogenic fungi, increased expression of membrane transporters correlates with azole resistance. For example, overexpression of CaCDR1, CaCDR2, and CaMDR1 is commonly observed in azole-resistant oral, systemic, and vaginal C. albicans clinical isolates [25,42]. In vitro, the disruption of genes encoding these transporters causes hyper-susceptibility to specific antifungals, including azoles [43].
While molecular mechanisms can be characterized into randomly collected resistant clinical isolates, matched isolates provide a more specific analysis of drug resistance. One matched set of 16 C. albicans clinical isolates was obtained from one individual over a two-year period of antifungal therapy. Increased expressions of CaCDR1, CaCDR2, and CaMDR1 were observed in the matched resistant isolates when compared to their susceptible partners [25,28]. Analysis of random, non-matched resistant C. albicans isolates also shows increased efflux pump gene expression [25,42].
Besides CaCDR1, CaCDR2, and CaMDR1, increased expression of CaFLU1 (an MFS-T) and CaPDR16 (an ABC-T) has been observed in azole-resistant isolates [44]. CaPdr16p is a phosphatidylinositol transfer protein, disruption of which causes increased susceptibilities to various antifungals [45]. CaFlu1p is not considered a major azole transporter because in vitro overexpression does not confirm azole resistance [46]. The C. albicans genome contains many other membrane transporters. However, to date, none have been shown to effect azole resistance [47].
The efflux pumps CaCDR1 and CaCDR2 are regulated by transcription factor CaTac1p, while CaMDR1 is regulated by transcription factor CaMrr1p [48,49]. Both transcription factors are zinc-cluster transcription factors (Zn2-Cys6). Gain of function (GOF) mutations in CaTAC1 and in CaMRR1 result in constitutive overexpression of their respective pumps, and azole resistance in many clinical isolates (Figure 4, grey box 1). Specifically, several GOF mutations in CaTAC1, including T225A, V736A, N972D, N977D, G980E, and G980W, have been correlated with resistance in clinical isolates [50]. GOF mutations in CaMRR1, including P683S and P683H, have also been correlated with resistance in clinical isolates [51,52]. GOFs in MRR1 are also observed in C. dubliniensis isolates resistant to fluconazole. These mutations include T374I, S595Y, C866Y, T965Δ, and (D987-I998)Δ [53].
There are other transcription factors that regulate the expression of CaCDR1 and CaCDR2, including the transcription factors, CaNDT80, CaFCR1, and CaFCR3 [54,55]. To date, no studies have shown the association of CaNDT80, CaFCR1, and CaFCR3 with resistance in clinical isolates. CaMDR1 is transcriptionally regulated by CaMRR1 as well as CaCAP1 and CaMCM1 [56]. In vitro, a truncated C-terminus of CaCap1p correlated with increased azole resistance [57].
In C. glabrata, overexpression of the ABC-T efflux pumps CgCDR1, CgSNQ2, and CgPDH1 has been shown to contribute to azole resistance in clinical isolates [58,59]. Overexpression or disruption of ABC-T encoded by CgPDR16 has also been associated with the azole resistance mechanism in C. glabrata [60,61]. Amongst the MFS-Ts, increased expression of CgFLR1 and CgQDR2 is also observed in azole-resistant clinical isolates [62,63].
GOF mutations in the transcription factor CgPdr1p activates CgPdr1p, which in turn induces the ABC-T pumps CgCDR1, CgSNQ2, and CgPDH1 [64]. These GOFs include L328F, R376W, D1082G, T588A, T607S, E1083Q, Y584C, D876Y, L280F, N691D, S316I, D261G, R293I, R592S, G583S, S343F, and R376G. These GOFs significantly increased resistance to fluconazole [64]. GOF mutations in CgPDR1 also regulate virulence. For example, GOF L280F in PDR1 regulates adherence to macrophages and macrophage-mediated phagocytosis [65]. GOF in CgPDR1 results in increased expression of adhesin proteins like Epa1, causing increased adherence [66]. CgPDR1 also regulates MFS-T CgQDR2, which upregulates in response to azoles [67]. This upregulation of CgQDR2 is caused by GOF L946S in CgPDR1. Deletion of CgQDR2 showed susceptibility to azole, miconazole, suggesting the role of CgQDR2 in azole resistance [65].
Mitochondria also plays an important role in efflux pump-mediated azole resistance in C. glabrata. Loss of mitochondria leads to azole resistance, which correlated with upregulation of CgCDR1, CgPDH1, and other genes [59].
Membrane transporter overexpression can also affect intrinsically azole-resistant C. krusei. In clinical isolates of C. krusei, overexpression of the ABC-T pumps CkAbc1p and CkAbc2p correlates with drug resistance, expression of both pumps can be induced by azoles, and expression of the pumps in a hyper-susceptible S. cerevisiae strain results in resistance [68]. However, azole resistance was not affected by efflux pump inhibitors, suggesting that efflux-mediated resistance is not due to efflux pumps alone in C. krusei [69]. Increased efflux pump expression also correlates with increased azole resistance in C. parapsilosis and C. dubliniensis [70,71].
Membrane transporter-based azole resistance was also recently observed in C. auris, an emerging multidrug-resistant fungal pathogen [72,73]. C. auris contains C. albicans homolog CDR1, which was found to be overexpressed in generationally aged C. auris that showed increased tolerance to fluconazole [72]. Recently, it was observed that increased expression of C. albicans homolog of CDR1 contributed in the evolution of azole-resistant C. auris isolates [74]. This increased expression correlated with increased copy number of the transcription factor TAC1 in the evolved strain when compared to its parent [74].
4.1.2. Altered Ergosterol Biosynthesis
Besides membrane transporters, azole resistance via altered ergosterol biosynthesis was also demonstrated previously. These are summarized below.
- Mutation and/or overexpression of ergosterol pathway genes (Figure 4; Grey box 3)
- ERG11/CYP51: Erg11p is a cytochrome p450 enzyme that regulates a rate-limiting step in the ergosterol biosynthetic pathway [12]. It is an essential enzyme in the pathway, and cellular ergosterol levels decrease when Erg11p is inhibited by azoles [75]. ERG11 overexpression is linked with azole resistance in many fungi. Many azole-resistant C. albicans clinical isolates show increased CaERG11 expression [25,42,76]. CgERG11 overexpression is observed in azole-resistant C. glabrata isolates [77]. Overexpression of ERG11 was also recently observed in generationally aging C. auris that showed increased resilience to azoles [72]. Several point mutations in ERG11 have been identified in resistant clinical isolates. For example, CaErg11p mutations A61V, A114S, Y132F, Y132H, K143Q, K143R, Y257H, S405F, G448E, F449S, G464S, R467K, and I471T contribute to azole resistance in C. albicans [78]. Many of these point mutations lower azole binding in the active site of the enzyme. Point mutations in ERG11 associated with resistance have been identified in other azole-resistant Candida sp. These include, C. glabrata mutations C108G, C423T, and A1581G, and C. krusei mutations A497C and G1570A [79].
- ERG3: Besides ERG11, ERG3 is also linked with azole resistance. Erg3p is a C5 sterol desaturase enzyme, and converts episterol to ergosta-5,7,24 (28)- trienol during ergosterol biosynthesis (Figure 4; Grey box 4). When Erg11p is inhibited, Erg3p and Erg6p synthesize a toxic sterol, 14α methyl ergosta 8,24 (28)-dien-3β, 6α-diol, in both C. albicans and C. glabrata, [80,81,82]. Disruption of ERG3 results in increased azole resistance in C. albicans [83]. Q139A in Erg3p is reported in azole-resistant C. glabrata isolates [84].
- ERG6: In the presence of azoles, ERG6 contributes to the formation of the toxic diol from lanosterol [80,81,82] (Figure 4; Grey box 5). Erg6p is a Δ24 sterol C-methyl transferase, a non-essential enzyme in the ergosterol biosynthetic pathway. Significant azole resistance is observed in the heterozygous ERG6 deletion in C. albicans [85]. Similarly, Δerg6 in S. cerevisiae showed increased azole resistance [86], increased membrane permeability, and low Pdr5p efflux activity [87]. This suggests that ERG6-dependent azole resistance is the result of toxic sterol formation, and not due to efflux pump overexpression.
- Gain of Function Mutation (GOF) in UPC2 (Figure 4; Grey box 2)
Upc2p is a transcription factor that regulates the majority of ergosterol biosynthetic genes. For example, it regulates the expression of CaERG2 and CaERG11 in C. albicans [88]. Upc2p is auto-regulatory [89], and is induced by azoles, anaerobic growth, and low levels of ergosterol [90]. Upc2p is a Zn2-Cys6 zinc cluster transcription factor [88], and is well characterized in C. albicans. The N-terminus of Upc2p is the DNA binding domain, while the C-terminus is an activation/regulatory domain [91]. Ergosterol binds to the C-terminus of Upc2p and negatively regulates the transcription of UPC2 [89]. Several mutations in CaUpc2p, including G648D, G648S, A643T, A643V, Y642F, G304R, A646V, and W478C are described in azole-resistant clinical isolates [76]. Seven of these mutations increased the expression of CaERG11 [76]. Besides CaERG11, GOF mutations in CaUPC2 increase the expression of other ergosterol pathway genes. For example, A643V GOF mutation in CaUPC2 induces the expression of CaERG2, CaERG3, CaERG5, CaERG6, CaERG9, and CaERG10 [92]. However, this mutation only causes a two-fold increase in azole resistance [92]. In vitro, GOF mutations A643T and A648D in CaUPC2 significantly increased azole resistance [93]. Since C. albicans is diploid, GOF mutations in both CaUPC2 alleles will cause more resistance than a GOF mutation in one allele [93].
The C. glabrata genome contains two homologs of CaUPC2, CgUPC2A and CgUPC2B [94]. CgUPC2A is an important regulator of the ergosterol pathway in C. glabrata, and is required for azole resistance [94]. Deletion of CgUPC2A in C. glabrata leads to azole susceptibility, whereas no effect is observed in the CgUPC2B deleted strain [94]. To date, no GOF mutations have been reported in CgUPC2A or CgUPC2B.
4.1.3. Altered Sterol Import
Sterol import as a potential mechanism of azole resistance has been identified recently (Figure 4 Grey box 6). Azoles lower the ergosterol levels of the cell, which can be compensated by exogenous sterol import. Sterol import has been well characterized in S. cerevisiae, C. albicans, and C. glabrata [95]. Both S. cerevisiae and C. glabrata import sterols under anaerobic or microaerophilic conditions using the sterol importers Aus1p and Pdr11p [96,97,98], and show increased azole resistance [95]. Mutations in UPC2, as well as defects in the biosynthesis of ergosterol, and heme, can cause increased sterol import [99,100]. For example, S. cerevisiae strains with mutated ScERG1 and ScERG7 have increased sterol import [101].
Unlike C. glabrata and S. cerevisiae, C. albicans imports sterols aerobically, increasing azole resistance in the presence of both serum and cholesterol [95]. Thus, C. albicans may develop resistance by importing cholesterol and serum from the blood.
4.1.4. Genome Plasticity
Genomic variations including loss of heterozygosity (LOH) and aneuploidy can correlate with azole resistance in fungi. If one allele of a gene is mutated, LOH can copy the mutation to the second allele. In clinical isolates of C. albicans, LOH has been observed in CaTAC1, CaERG11, and CaMRR1, correlating with increased resistance [102]. Clinical isolates of C. albicans can also include segmental aneuploidy, in which two copies of the left arm of chromosome 5 containing CaERG11 and CaTAC1 form an isochromosome that correlates with azole resistance [103]. Additionally, trisomy in chromosome 4 also caused an increase in azole resistance [104]. However, this observed increased resistance was neither attributed to increased efflux pump activities nor attributed to altered ergosterol biosynthesis in the clinical isolate. Thus, the azole resistance mechanisms by chromosome 4 trisomy are still unknown and need to be studied in the future. In another study, loss of one homologue of chromosome 4 contributed significantly with increased azole resistance [105]. Further, trisomy in chromosome 3 and chromosome R in C. albicans correlates with increased triazole resistance [105,106]. Trisomy of chromosome 3 develops under prolonged azole exposure. Chromosome 3 hosts efflux pump encoding genes CDR1 and CDR2 [105], and increased chromosome 3 copy number caused increased CDR1 and CDR2 expression [105]. Aneuploidy in another chromosome, chromosome 6, also corelates with azole resistance [105]. Chromosome 6 hosts efflux pump-encoding gene MDR1 and increased expression of MDR1 corelated with chromosome 6 trisomy under fluconazole exposure [105].
Chromosomal alterations are also observed in C. glabrata. For example, segmental rearrangements were previously observed in chromosome M and F in azole-resistant C. glabrata clinical isolates. Chromosome M harbors efflux pump-encoding gene CgCDR1, which was duplicated in these isolates, while chromosome F harbors efflux pump-encoding gene CgPDH1 [107]. Besides aneuploidy, alteration in gene copy numbers is also associated with increased azole resistance. For example, increased copy number of ERG11 in C. glabrata caused increased azole resistance [108]. Recently, duplication in genes CDR1 and ERG11 was also observed in generationally aging C. auris cells that rendered increased resilience to fluconazole [72].
4.1.5. Other Hypothetical Azole Resistance Mechanism—Altered Azole Import
Several studies have hypothesized that defective azole import may contribute to drug resistance (Figure 4 Grey box 7). Azoles are imported by facilitative diffusion (FD) in C. albicans [109], and the import is energy-independent. Clinical isolates of many pathogenic fungi vary in azole import [109]. However, to date, no correlation has been observed between azole import and resistance. Further studies are required to identify the azole importers and characterize their roles in azole resistance.
4.2. Resistance to Other Drugs
Polyene resistance is also linked to changes in both ERG3 and ERG6. For example, in vitro, disruption of ERG3 and ERG6 causes decreased ergosterol levels, and amphotericin B resistance in C. albicans and C. glabrata [110]. However, polyene resistance in clinical isolates has not been well characterized.
Echinocandins are the newest antifungal category, targeting β 1-3 glucan synthase, an enzyme important for cell wall biosynthesis localized to the plasma membrane [111]. The enzyme is encoded by the homologs CaFKS1 and CaFKS2. Mutations in CaFKS1 are observed in echinocandin-resistant clinical isolates of C. albicans [111], mutations clustered in two regions (amino acid regions 637–654 and 1345–1365) [22,112]. Mutations in CaFKS2 can cause echinocandin resistance in C. albicans in vitro, but has not been observed in clinical isolates [113]. FKS1 point mutations have also been observed in C. glabrata- and C. krusei-resistant isolates [113]. FKS1 and FKS2 point mutations spontaneously arise in the presence of echinocandin selection pressure in C. glabrata, with FKS2 point mutation outnumbering FKS1 point mutation by 2 to 1 [113,114]. FKS1 point mutations involved in echinocandin resistance include S629P, F625Δ, and F625C. FKS2 point mutations associated with increased echinocandin resistance are F659Δ, S663F, R1378S, R1378G, S663P, P667H, P667T, E655G, and E655K [113,114]. Besides point mutations in FKS, genome plasticity also causes increased echinocandin resistance. For example, chromosome 2 trisomy in C. albicans causes caspofungin resistance [115]. However, the specific mechanism behind this needs to be studied.
The other medically important drug 5FC is metabolized in the pyrimidine salvage pathway [116], and is used in treating candidiasis in combination with other antifungals. 5FC resistance is observed in Candida sp. In C. albicans., 5FC is imported by cytosine permease, CaFcy2p, and is then deaminated to 5FU by cytosine deaminase, CaFcy1p. 5FU is then converted to 5FUMP by phosphoribosyl transferase, CaFur1p. Inactivation of these enzymes causes increased 5FC resistance [117].
Clinical isolates can develop resistance to multiple antifungals, as previously described in clinical isolates of C. albicans, C. glabrata, and C. auris [25,72,118]. Resistance to antifungals can be intrinsic (primary resistance) or acquired (secondary resistance) [119]. Intrinsic resistance is highly stable and is predictive of therapeutic failure. For example, C. krusei is intrinsically resistant to fluconazole. Further, intrinsic resistance to several antifungals including fluconazole and polyenes are also observed in C. auris, an emerging pathogen [119]. Recently, C. auris has proliferated at an alarming rate, causing severe problems in hospital settings. Mortality rates range from 30% to 60% and infections occur within weeks after hospital admission. Infection control is quite difficult because of high rates of colonization and environmental contamination by C. auris [119].
Besides intrinsic resistance, resistance to antifungals can be acquired. Acquired resistance can be either stable or transient. The acquired resistance develops due to long exposure of antifungals. For example, in studies with matched clinical isolates, where one set of C. albicans was isolated before azole treatment and one set after 6 months of azole treatment, C. albicans acquired azole resistance after azole therapy [25]. These resistance mechanisms were highly stable even after removal of azole pressure. Additionally, drug-induced aneuploidy can provide fitness advantages, as observed in C. albicans [120]. Aneuploidy, as described previously in this review, plays an important role in acquired drug resistance. These aneuploidies can be stable for several passages in the absence of antifungals, as observed in previous studies [104,120].
5. Conclusions
Candidiasis is caused by pathogenic yeasts, Candida sp., which are opportunistic pathogens present in the normal microbiome. The pathogens’ growth in the microbiome are checked by the host’s immune system. However, these pathogens overgrow in immune-compromised individuals, causing disseminated infections. Yeasts cannot be avoided in the day-to-day diet and some yeasts are useful in maintaining the host microbiome. Hence, total elimination of yeast from the body is neither desirable nor feasible. These infections are treated by different classes of antifungals that include azoles, polyenes, and echinocandins. These drugs target different biochemical pathways that are unique to yeasts. For example, azoles target the biosynthesis of ergosterol, a unique cell membrane component, present only in fungi. These antifungals are either fungistatic (azoles) or fungicidal (echinocandins). The efficacies of these drugs depend on the quality of the hosts’ immune system, site and severity of infection, and pharmacokinetics of the drugs. Besides drug efficacies, therapeutic failures may result from evolution of drug resistance in the infecting Candida sp. The molecular mechanisms associated with antifungal resistance were summarized in this review. These mechanisms include overexpression of membrane transporters, altered cell wall and ergosterol biosynthesis, gain of function mutations in the transcription factors regulating membrane transporters, and ergosterol biosynthesis. The ongoing research on better understanding of these mechanisms may aid in detecting resistant isolates, identify novel drug targets, and inhibiting the rise of drug resistance.
Author Contributions
S.B. contributed significantly in writing the review. S.B., S.S.-T., and B.C.F. all wrote the article. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health (NIH 1R01AI127704- 01A1 to B.C.F.). B.C.F. is an attending at the US Department of Veterans Affairs, Northport VA Medical Center, Northport, NY. She is supported by the US Veterans Affairs Merit Review Award 5I01 BX003741 and NIAID R01 AI127704. The contents of this review do not represent the views of VA or the United States Government.
Acknowledgments
We thank BCFs Lab for their input in this manuscript. We especially thank Theodore C. White for providing his invaluable advice while writing this review. We also thank biorender.com for providing the necessary tools for some of the illustrations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pfaller, M.A.; Diekema, D. Epidemiology of Invasive Candidiasis: A Persistent Public Health Problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [Green Version]
- Preventions, C.f.D.C.a. DRUG-RESISTANT CANDIDA SPECIES. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/candida-508.pdf (accessed on 9 June 2020).
- Pfaller, M.; Neofytos, D.; Diekema, D.; Azie, N.; Meier-Kriesche, H.-U.; Quan, S.-P.; Horn, D. Epidemiology and outcomes of candidemia in 3648 patients: Data from the Prospective Antifungal Therapy (PATH Alliance®) registry, 2004–2008. Diagn. Microbiol. Infect. Dis. 2012, 74, 323–331. [Google Scholar] [CrossRef]
- Kojic, E.M.; Darouiche, R.O. Candida Infections of Medical Devices. Clin. Microbiol. Rev. 2004, 17, 255–267. [Google Scholar] [CrossRef] [Green Version]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.R.; Clancy, C.J.; A Marr, K.; Ostrosky-Zeichner, L.L.; Reboli, A.; Schuster, M.G.; A Vazquez, J.; Walsh, T.J.; et al. Executive Summary: Clinical Practice Guideline for the Management of Candidiasis: 2016 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2016, 62, 409–417. [Google Scholar] [CrossRef] [Green Version]
- Kodedová, M.; Sychrová, H. Changes in the Sterol Composition of the Plasma Membrane Affect Membrane Potential, Salt Tolerance and the Activity of Multidrug Resistance Pumps in Saccharomyces cerevisiae. PLoS ONE 2015, 10, e0139306. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Esquivel, B.D.; White, T.C. Overexpression or Deletion of Ergosterol Biosynthesis Genes Alters Doubling Time, Response to Stress Agents, and Drug Susceptibility inSaccharomyces cerevisiae. mBio 2018, 9, e01291-18. [Google Scholar] [CrossRef] [Green Version]
- Yasu, T.; Konuma, T.; Kuroda, S.; Takahashi, S.; Tojo, A. Effect of Cumulative Intravenous Voriconazole Dose on Renal Function in Hematological Patients. Antimicrob. Agents Chemother. 2018, 62, e00507-18. [Google Scholar] [CrossRef] [Green Version]
- Jenks, J.D.; Salzer, H.J.; Prattes, J.; Krause, R.; Buchheidt, D.; Hoenigl, M. Spotlight on isavuconazole in the treatment of invasive aspergillosis and mucormycosis: Design, development, and place in therapy. Drug Des. Dev. Ther. 2018, 12, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Wilson, D.; Dimondi, V.P.; Johnson, S.W.; Jones, T.M.; Drew, R.H. Role of isavuconazole in the treatment of invasive fungal infections. Ther. Clin. Risk Manag. 2016, 12, 1197–1206. [Google Scholar] [CrossRef] [Green Version]
- Pasqualotto, A.C.; Falci, D.R. Profile of isavuconazole and its potential in the treatment of severe invasive fungal infections. Infect. Drug Resist. 2013, 6, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Veen, M.; Stahl, U.; Lang, C. Combined overexpression of genes of the ergosterol biosynthetic pathway leads to accumulation of sterols in Saccharomyces cerevisiae. FEMS Yeast Res. 2003, 4, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Delattin, N.; Cammue, B.P.; Thevissen, K. Reactive oxygen species-inducing antifungal agents and their activity against fungal biofilms. Future Med. Chem. 2014, 6, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Efimova, S.S.; Schagina, L.V.; Ostroumova, O.S. Investigation of Channel-Forming Activity of Polyene Macrolide Antibiotics in Planar Lipid Bilayers in the Presence of Dipole Modifiers. Acta Nat. 2014, 6, 67–79. [Google Scholar] [CrossRef]
- Hamill, R.J. Amphotericin B Formulations: A Comparative Review of Efficacy and Toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef]
- Vogelsinger, H.; Weiler, S.; Djanani, A.; Kountchev, J.; Bellmann-Weiler, R.; Wiedermann, C.J.; Bellmann, R. Amphotericin B tissue distribution in autopsy material after treatment with liposomal amphotericin B and amphotericin B colloidal dispersion. J. Antimicrob. Chemother. 2006, 57, 1153–1160. [Google Scholar] [CrossRef]
- Sanglard, D.; Coste, A.T.; Ferrari, S. Antifungal drug resistance mechanisms in fungal pathogens from the perspective of transcriptional gene regulation. FEMS Yeast Res. 2009, 9, 1029–1050. [Google Scholar] [CrossRef] [Green Version]
- Shirwaikar, A.A.; Thomas, T.; Shirwaikar, A.; Lobo, R.; Prabhu, K.S. Treatment of Onychomycosis: An Update. Indian J. Pharm. Sci. 2008, 70, 710–714. [Google Scholar] [CrossRef] [Green Version]
- Popolo, L.; Gualtieri, T.; Ragni, E. The yeast cell-wall salvage pathway. Med. Mycol. 2001, 39, 111–121. [Google Scholar] [CrossRef]
- Perlin, D.S. Current perspectives on echinocandin class drugs. Future Microbiol. 2011, 6, 441–457. [Google Scholar] [CrossRef] [Green Version]
- Douglas, C.M. Fungal beta(1,3)-D-glucan synthesis. Med. Mycol. 2001, 39, 55–66. [Google Scholar] [CrossRef]
- Munro, C. Fungal echinocandin resistance. F1000 Boil. Rep. 2010, 2, 66. [Google Scholar] [CrossRef] [Green Version]
- Pappas, P.G.; Kauffman, C.A.; Andes, D.; Benjamin, D.K.; Calandra, T.; Edwards, J.J.E.; Filler, S.G.; Fisher, J.F.; Kullberg, B.-J.; Ostrosky-Zeichner, L.L.; et al. Clinical Practice Guidelines for the Management of Candidiasis: 2009 Update by the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 503–535. [Google Scholar] [CrossRef] [Green Version]
- Vermes, A.; Guchelaar, H.-J.; Dankert, J. Flucytosine: A review of its pharmacology, clinical indications, pharmacokinetics, toxicity and drug interactions. J. Antimicrob. Chemother. 2000, 46, 171–179. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Sobel, J.D.; White, T.C. A Combination Fluorescence Assay Demonstrates Increased Efflux Pump Activity as a Resistance Mechanism in Azole-Resistant Vaginal Candida albicans Isolates. Antimicrob. Agents Chemother. 2016, 60, 5858–5866. [Google Scholar] [CrossRef] [Green Version]
- White, T.C.; Marr, K.A.; Bowden, R.A. Clinical, Cellular, and Molecular Factors That Contribute to Antifungal Drug Resistance. Clin. Microbiol. Rev. 1998, 11, 382–402. [Google Scholar] [CrossRef] [Green Version]
- Rex, J.H.; Rinaldi, M.G.; Pfaller, M.A. Resistance of Candida species to fluconazole. Antimicrob. Agents Chemother. 1995, 39, 1–8. [Google Scholar] [CrossRef] [Green Version]
- White, T.C. Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 1997, 41, 1482–1487. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.F.; Henriques, M. Oral mucositis caused byCandida glabratabiofilms: Failure of the concomitant use of fluconazole and ascorbic acid. Ther. Adv. Infect. Dis. 2017, 4, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Sabra, R.; Branch, R.A. Amphotericin B Nephrotoxicity. Drug Saf. 1990, 5, 94–108. [Google Scholar] [CrossRef]
- Faustino, C.; Pinheiro, L. Lipid Systems for the Delivery of Amphotericin B in Antifungal Therapy. Pharmaceutics 2020, 12, 29. [Google Scholar] [CrossRef] [Green Version]
- Sau, K.; Mambula, S.S.; Latz, E.; Henneke, P.; Golenbock, D.T.; Levitz, S.M. The Antifungal Drug Amphotericin B Promotes Inflammatory Cytokine Release by a Toll-like Receptor- and CD14-dependent Mechanism. J. Boil. Chem. 2003, 278, 37561–37568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Re, V.L.; Carbonari, D.; Goldberg, D.; Forde, K.A.; Goldberg, D.S.; Reddy, K.R.; Haynes, K.; Roy, J.A.; Sha, D.; Marks, A.R.; et al. Oral Azole Antifungal Medications and Risk of Acute Liver Injury, Overall and by Chronic Liver Disease Status. Am. J. Med. 2015, 129, 283–291.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Zapata, D.; Petraitiene, R.; Petraitis, V. Echinocandins: The Expanding Antifungal Armamentarium. Clin. Infect. Dis. 2015, 61, S604–S611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamping, E.; Monk, B.C.; Niimi, K.; Holmes, A.; Tsao, S.; Tanabe, K.; Niimi, M.; Uehara, Y.; Cannon, R.D. Characterization of Three Classes of Membrane Proteins Involved in Fungal Azole Resistance by Functional Hyperexpression in Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 1150–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandelwal, N.K.; Wasi, M.; Nair, R.; Gupta, M.; Kumar, M.; Mondal, A.K.; Gaur, N.A.; Prasad, R. Vacuolar Sequestration of Azoles, a Novel Strategy of Azole Antifungal Resistance Conserved across Pathogenic and Nonpathogenic Yeast. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Leighton, J.; Schatz, G. An ABC transporter in the mitochondrial inner membrane is required for normal growth of yeast. EMBO J. 1995, 14, 188–195. [Google Scholar] [CrossRef]
- Kispal, G.; Csere, P.; Guiard, B.; Lill, R. The ABC transporter Atm1p is required for mitochondrial iron homeostasis. FEBS Lett. 1997, 418, 346–350. [Google Scholar] [CrossRef] [Green Version]
- Michealis, S.; Berkower, C. Sequence Comparison of Yeast ATP-binding Cassette Proteins. Cold Spring Harb. Symp. Quant. Boil. 1995, 60, 291–307. [Google Scholar] [CrossRef]
- Cannon, R.D.; Lamping, E.; Holmes, A.; Niimi, K.; Baret, P.V.; Keniya, M.V.; Tanabe, K.; Niimi, M.; Goffeau, A.; Monk, B.C. Efflux-Mediated Antifungal Drug Resistance. Clin. Microbiol. Rev. 2009, 22, 291–321. [Google Scholar] [CrossRef] [Green Version]
- Marger, M.D.; Saier, M.H. A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem. Sci. 1993, 18, 13–20. [Google Scholar] [CrossRef]
- White, T.C.; Holleman, S.; Dy, F.; Mirels, L.F.; Stevens, D.A. Resistance Mechanisms in Clinical Isolates of Candida albicans. Antimicrob. Agents Chemother. 2002, 46, 1704–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsao, S.; Rahkhoodaee, F.; Raymond, M. Relative Contributions of the Candida albicans ABC Transporters Cdr1p and Cdr2p to Clinical Azole Resistance. Antimicrob. Agents Chemother. 2009, 53, 1344–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, D.; Bille, J.; Sanglard, D. A novel multidrug efflux transporter gene of the major facilitator superfamily from Candida albicans (FLU1) conferring resistance to fluconazole. Microbiology 2000, 146, 2743–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Yeater, K.M.; Hoyer, L.L. Cellular and Molecular Biology of Candida albicans Estrogen Response. Eukaryot. Cell 2006, 5, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panwar, S.L.; Pasrija, R.; Prasad, R. Membrane homoeostasis and multidrug resistance in yeast. Biosci. Rep. 2008, 28, 217–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saidane, S.; Weber, S.; De Deken, X.; St-Germain, G.; Raymond, M. PDR16-mediated azole resistance in Candida albicans. Mol. Microbiol. 2006, 60, 1546–1562. [Google Scholar] [CrossRef]
- Lohberger, A.; Coste, A.T.; Sanglard, D. Distinct Roles of Candida albicans Drug Resistance Transcription FactorsTAC1,MRR1, andUPC2in Virulence. Eukaryot. Cell 2013, 13, 127–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morschhäuser, J.; Barker, K.S.; Liu, T.T.; Blaß-Warmuth, J.; Homayouni, R.; Rogers, P.D. The Transcription Factor Mrr1p Controls Expression of the MDR1 Efflux Pump and Mediates Multidrug Resistance in Candida albicans. PLoS Pathog. 2007, 3, e164. [Google Scholar] [CrossRef] [Green Version]
- Coste, A.T.; Turner, V.; Ischer, F.; Morschhäuser, J.; Forche, A.; Selmecki, A.; Berman, J.; Bille, J.; Sanglard, D. A Mutation in Tac1p, a Transcription Factor Regulating CDR1 and CDR2, Is Coupled With Loss of Heterozygosity at Chromosome 5 to Mediate Antifungal Resistance in Candida albicans. Genetics 2006, 172, 2139–2156. [Google Scholar] [CrossRef] [Green Version]
- Dunkel, N.; Blass, J.; Rogers, P.D.; Morschhäuser, J. Mutations in the multi-drug resistance regulatorMRR1, followed by loss of heterozygosity, are the main cause ofMDR1overexpression in fluconazole-resistantCandida albicansstrains. Mol. Microbiol. 2008, 69, 827–840. [Google Scholar] [CrossRef] [Green Version]
- Kalkandelen, K.T.; Dereli, M.D. [Investigation of mutations in transcription factors of efflux pump genes in fluconazole-resistant Candida albicans strains overexpressing the efflux pumps]. Mikrobiyoloji Bulteni 2015, 49, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.; Rogers, P.D.; Morschhäuser, J. Gain-of-Function Mutations in the Transcription Factor MRR1 Are Responsible for Overexpression of the MDR1 Efflux Pump in Fluconazole-Resistant Candida dubliniensis Strains. Antimicrob. Agents Chemother. 2008, 52, 4274–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-G.; Yang, Y.-L.; Shih, H.-I.; Su, C.-L.; Lo, H.-J. CaNdt80 Is Involved in Drug Resistance in Candida albicans by Regulating CDR1. Antimicrob. Agents Chemother. 2004, 48, 4505–4512. [Google Scholar] [CrossRef] [Green Version]
- Talibi, D.; Raymond, M. Isolation of a Putative Candida albicansTranscriptional Regulator Involved in Pleiotropic Drug Resistance by Functional Complementation of a pdr1 pdr3 Mutation inSaccharomyces cerevisiae. J. Bacteriol. 1999, 181, 231–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogavero, S.; Tavanti, A.; Senesi, S.; Rogers, P.D.; Morschhäuser, J. Differential Requirement of the Transcription Factor Mcm1 for Activation of the Candida albicans Multidrug Efflux PumpMDR1by Its Regulators Mrr1 and Cap1. Antimicrob. Agents Chemother. 2011, 55, 2061–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alarco, A.-M.; Raymond, M. The bZip Transcription Factor Cap1p Is Involved in Multidrug Resistance and Oxidative Stress Response inCandida albicans. J. Bacteriol. 1999, 181, 700–708. [Google Scholar] [CrossRef] [Green Version]
- Sanguinetti, M.; Posteraro, B.; Fiori, B.; Ranno, S.; Torelli, R.; Fadda, G. Mechanisms of Azole Resistance in Clinical Isolates of Candida glabrata Collected during a Hospital Survey of Antifungal Resistance. Antimicrob. Agents Chemother. 2005, 49, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Vermitsky, J.-P.; Edlind, T.D. Azole Resistance in Candida glabrata: Coordinate Upregulation of Multidrug Transporters and Evidence for a Pdr1-Like Transcription Factor. Antimicrob. Agents Chemother. 2004, 48, 3773–3781. [Google Scholar] [CrossRef] [Green Version]
- Culakova, H.; Dzugasova, V.; Valencikova, R.; Gbelska, Y.; Šubík, J. Stress response and expression of fluconazole resistance associated genes in the pathogenic yeast Candida glabrata deleted in the CgPDR16 gene. Microbiol. Res. 2015, 174, 17–23. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Fries, B.C. Enhanced Efflux Pump Activity in OldCandida glabrataCells. Antimicrob. Agents Chemother. 2018, 62, e02227-17. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Dias, P.; Sá-Correia, I.; Teixeira, M. MFS multidrug transporters in pathogenic fungi: Do they have real clinical impact? Front. Physiol. 2014, 5, 197. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Ribeiro, J.; Miranda, I.M.; Silva-Dias, A.; Cavalheiro, M.; Costa-De-Oliveira, S.; Rodrigues, A.G.; Teixeira, M. Clotrimazole Drug Resistance in Candida glabrata Clinical Isolates Correlates with Increased Expression of the Drug:H+ Antiporters CgAqr1, CgTpo1_1, CgTpo3, and CgQdr2. Front. Microbiol. 2016, 7, 1773. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ischer, F.; Calabrese, D.; Posteraro, B.; Sanguinetti, M.; Fadda, G.; Rohde, B.; Bauser, C.; Bader, O.; Sanglard, D. Gain of Function Mutations in CgPDR1 of Candida glabrata Not Only Mediate Antifungal Resistance but Also Enhance Virulence. PLoS Pathog. 2009, 5, e1000268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Q.; Wang, C.; Tian, Y.; Dong, D.; Jiang, C.; Mao, E.; Peng, Y. CgPDR1 gain-of-function mutations lead to azole-resistance and increased adhesion in clinical Candida glabrata strains. Mycoses 2018, 61, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Vale-Silva, L.A.; Moeckli, B.; Torelli, R.; Posteraro, B.; Sanguinetti, M.; Sanglard, D. Upregulation of the Adhesin Gene EPA1 Mediated by PDR1 in Candida glabrata Leads to Enhanced Host Colonization. mSphere 2016, 1, e00065-15. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Pires, C.; Cabrito, T.R.; Renaudin, A.; Ohno, M.; Chibana, H.; Sá-Correia, I.; Teixeira, M. Candida glabrata Drug:H+Antiporter CgQdr2 Confers Imidazole Drug Resistance, Being Activated by Transcription Factor CgPdr1. Antimicrob. Agents Chemother. 2013, 57, 3159–3167. [Google Scholar] [CrossRef] [Green Version]
- Lamping, E.; Ranchod, A.; Nakamura, K.; Tyndall, J.D.A.; Niimi, K.; Holmes, A.; Niimi, M.; Cannon, R.D. Abc1p is a Multidrug Efflux Transporter That Tips the Balance in Favor of Innate Azole Resistance in Candida krusei. Antimicrob. Agents Chemother. 2008, 53, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Guinea, J.; Sánchez-Somolinos, M.; Cuevas, O.; Peláez, T.; Bouza, E. Fluconazole resistance mechanisms inCandida krusei: The contribution of efflux-pumps. Med. Mycol. 2006, 44, 575–578. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xiao, M.; Watts, M.R.; Wang, H.; Fan, X.; Kong, F.; Xu, Y.-C. Development of fluconazole resistance in a series of Candida parapsilosis isolates from a persistent candidemia patient with prolonged antifungal therapy. BMC Infect. Dis. 2015, 15, 340. [Google Scholar] [CrossRef] [Green Version]
- Moran, G.P.; Pinjon, E.; Coleman, D.; Sullivan, D. Azole susceptibility and resistance in Candida dubliniensis. Biochem. Soc. Trans. 2005, 33, 1210. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Holowka, T.; Orner, E.P.; Fries, B.C. Gene Duplication Associated with Increased Fluconazole Tolerance in Candida auris cells of Advanced Generational Age. Sci. Rep. 2019, 9, 5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaabane, F.; Graf, A.; Jequier, L.; Coste, A.T. Review on Antifungal Resistance Mechanisms in the Emerging Pathogen Candida auris. Front. Microbiol. 2019, 10, 2788. [Google Scholar] [CrossRef] [PubMed]
- Rybak, J.M.; Muñoz, J.F.; Barker, K.S.; Parker, J.E.; Esquivel, B.D.; Berkow, E.L.; Lockhart, S.R.; Gade, L.; Palmer, G.E.; White, T.C.; et al. Mutations in TAC1B: A Novel Genetic Determinant of Clinical Fluconazole Resistance in Candida auris. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Arthington-Skaggs, B.A.; Jradi, H.; Desai, T.; Morrison, C.J. Quantitation of Ergosterol Content: Novel Method for Determination of Fluconazole Susceptibility of Candida albicans. J. Clin. Microbiol. 1999, 37, 3332–3337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flowers, S.A.; Barker, K.S.; Berkow, E.L.; Toner, G.; Chadwick, S.G.; Gygax, S.E.; Morschhäuser, J.; Rogers, P.D. Gain-of-Function Mutations inUPC2Are a Frequent Cause ofERG11Upregulation in Azole-Resistant Clinical Isolates of Candida albicans. Eukaryot. Cell 2012, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pam, V.K.; Akpan, J.U.; O Oduyebo, O.; O Nwaokorie, F.; A Fowora, M.; O Oladele, R.; Ogunsola, F.T.; I Smith, S. Fluconazole susceptibility and ERG11 gene expression in vaginal candida species isolated from lagos Nigeria. Int. J. Mol. Epidemiol. Genet. 2012, 3, 84–90. [Google Scholar]
- Xiang, M.-J.; Liu, J.-Y.; Ni, P.-H.; Wang, S.; Shi, C.; Wei, B.; Ni, Y.-X.; Ge, H.-L. Erg11mutations associated with azole resistance in clinical isolates ofCandida albicans. FEMS Yeast Res. 2013, 13, 386–393. [Google Scholar] [CrossRef] [Green Version]
- Silva, D.B.D.S.; Rodrigues, L.M.C.; De Almeida, A.A.; De Oliveira, K.M.P.; Grisolia/, A.B. Novel point mutations in the ERG11 gene in clinical isolates of azole resistant Candida species. Memórias do Instituto Oswaldo Cruz 2016, 111, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Hull, C.M.; Parker, J.E.; Bader, O.; Weig, M.; Gross, U.; Warrilow, A.; Kelly, D.E.; Kelly, S.L. Facultative Sterol Uptake in an Ergosterol-Deficient Clinical Isolate of Candida glabrata Harboring a Missense Mutation inERG11and Exhibiting Cross-Resistance to Azoles and Amphotericin B. Antimicrob. Agents Chemother. 2012, 56, 4223–4232. [Google Scholar] [CrossRef] [Green Version]
- Chau, A.S.; Gurnani, M.; Hawkinson, R.; Laverdiere, M.; Cacciapuoti, A.; McNicholas, P.M. Inactivation of Sterol Δ5,6-Desaturase Attenuates Virulence in Candida albicans. Antimicrob. Agents Chemother. 2005, 49, 3646–3651. [Google Scholar] [CrossRef] [Green Version]
- Kelly, S.L.; Lamb, D.C.; Kelly, D.E.; Manning, N.J.; Loeffler, J.; Hebart, H.; Schumacher, U.; Einsele, H. Resistance to fluconazole and cross-resistance to amphotericin B in Candida albicans from AIDS patients caused by defective sterol delta5,6-desaturation. FEBS Lett. 1997, 400, 80–82. [Google Scholar] [CrossRef] [Green Version]
- Sanglard, D.; Ischer, F.; Parkinson, T.; Falconer, D.; Bille, J. Candida albicans Mutations in the Ergosterol Biosynthetic Pathway and Resistance to Several Antifungal Agents. Antimicrob. Agents Chemother. 2003, 47, 2404–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.I.; Choi, C.W.; Lee, K.M.; Lee, Y.S. Gene Expression and Identification Related to Fluconazole Resistance of Candida glabrata Strains. Osong Public Heal. Res. Perspect. 2010, 1, 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Jiang, B.; Ketela, T.; Lemieux, S.; Veillette, K.; Martel, N.; Davison, J.; Sillaots, S.; Trosok, S.; Bachewich, C.; et al. Genome-Wide Fitness Test and Mechanism-of-Action Studies of Inhibitory Compounds in Candida albicans. PLoS Pathog. 2007, 3, e92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.B.; Sirjusingh, C.; Parsons, A.B.; Boone, C.; Wickens, C.; E Cowen, L.; Kohn, L.M. Mode of selection and experimental evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 2003, 163, 1287–1298. [Google Scholar]
- Akins, R. An update on antifungal targets and mechanisms of resistance inCandidaalbicans. Med Mycol. 2005, 43, 285–318. [Google Scholar] [CrossRef] [Green Version]
- Silver, P.M.; Oliver, B.G.; White, T.C. Role of Candida albicans Transcription Factor Upc2p in Drug Resistance and Sterol Metabolism. Eukaryot. Cell 2004, 3, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Tong, J.; Lee, C.W.; Ha, S.; Eom, S.H.; Im, Y.J. Structural mechanism of ergosterol regulation by fungal sterol transcription factor Upc2. Nat. Commun. 2015, 6, 6129. [Google Scholar] [CrossRef] [Green Version]
- Hoot, S.J.; Oliver, B.G.; White, T.C. Candida albicans UPC2 is transcriptionally induced in response to antifungal drugs and anaerobicity through Upc2p-dependent and -independent mechanisms. Microbiology 2008, 154, 2748–2756. [Google Scholar] [CrossRef] [Green Version]
- Davies, B.S.; Wang, H.S.; Rine, J. Dual Activators of the Sterol Biosynthetic Pathway of Saccharomyces cerevisiae: Similar Activation/Regulatory Domains but Different Response Mechanisms. Mol. Cell. Boil. 2005, 25, 7375–7385. [Google Scholar] [CrossRef] [Green Version]
- Hoot, S.J.; Smith, A.R.; Brown, R.P.; White, T.C. An A643V Amino Acid Substitution in Upc2p Contributes to Azole Resistance in Well-Characterized Clinical Isolates ofCandida albicans. Antimicrob. Agents Chemother. 2010, 55, 940–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilmann, C.J.; Schneider, S.; Barker, K.S.; Rogers, P.D.; Morschhäuser, J. An A643T Mutation in the Transcription Factor Upc2p Causes Constitutive ERG11 Upregulation and Increased Fluconazole Resistance in Candida albicans. Antimicrob. Agents Chemother. 2009, 54, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagi, M.; Nakayama, H.; Tanabe, K.; Bard, M.; Aoyama, T.; Okano, M.; Higashi, S.; Ueno, K.; Chibana, H.; Niimi, M.; et al. Transcription factors CgUPC2A and CgUPC2B regulate ergosterol biosynthetic genes in Candida glabrata. Genes Cells 2010, 16, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Zavrel, M.; Hoot, S.J.; White, T.C. Comparison of Sterol Import under Aerobic and Anaerobic Conditions in Three Fungal Species, Candida albicans, Candida glabrata, and Saccharomyces cerevisiae. Eukaryot. Cell 2013, 12, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Kuo, D.; Tan, K.; Zinman, G.; Ravasi, T.; Bar-Joseph, Z.; Ideker, T. Evolutionary divergence in the fungal response to fluconazole revealed by soft clustering. Genome Boil. 2010, 11, R77. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, L.J.; Balderes, D.A.; Wharton, B.; Tinkelenberg, A.H.; Rao, G.; Sturley, S.L. Transcriptional Profiling Identifies Two Members of the ATP-binding Cassette Transporter Superfamily Required for Sterol Uptake in Yeast. J. Boil. Chem. 2002, 277, 32466–32472. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, H.; Tanabe, K.; Bard, M.; Hodgson, W.; Wu, S.; Takemori, D.; Aoyama, T.; Kumaraswami, N.S.; Metzler, L.; Takano, Y.; et al. The Candida glabrata putative sterol transporter gene CgAUS1 protects cells against azoles in the presence of serum. J. Antimicrob. Chemother. 2007, 60, 1264–1272. [Google Scholar] [CrossRef]
- A Lewis, T.; Taylor, F.R.; Parks, L.W. Involvement of heme biosynthesis in control of sterol uptake by Saccharomyces cerevisiae. J. Bacteriol. 1985, 163, 199–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, J.H.; Leak, F.W.; Shianna, K.V.; Tove, S.; Parks, L.W. A Mutation in a Purported Regulatory Gene Affects Control of Sterol Uptake in Saccharomyces cerevisiae. J. Bacteriol. 1998, 180, 4177–4183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, F.; Achstetter, T.; Duport, C.; Karst, F.; Spagnoli, R.; Degryse, E. Sterol Uptake in Saccharomyces cerevisiae Heme Auxotrophic Mutants is Affected by Ergosterol and Oleate but Not by Palmitoleate or by Sterol Esterification. J. Bacteriol. 1998, 180, 1913–1919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, C.B.; Funt, J.M.; Abbey, D.; Issi, L.; Guiducci, C.; A Martinez, D.; DeLorey, T.; Li, B.Y.; White, T.C.; Cuomo, C.A.; et al. The evolution of drug resistance in clinical isolates of Candida albicans. eLife 2015, 4, 00662. [Google Scholar] [CrossRef] [Green Version]
- Selmecki, A.; Forche, A.; Berman, J. Aneuploidy and Isochromosome Formation in Drug-Resistant Candida albicans. Science 2006, 313, 367–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.Z.; Saha, A.; Haseeb, A.; Bennett, R.J. A chromosome 4 trisomy contributes to increased fluconazole resistance in a clinical isolate of Candida albicans. Microbiology 2017, 163, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Perepnikhatka, V.; Fischer, F.J.; Niimi, M.; Baker, R.A.; Cannon, R.D.; Wang, Y.-K.; Sherman, F.; Rustchenko, E. Specific Chromosome Alterations in Fluconazole-Resistant Mutants of Candida albicans. J. Bacteriol. 1999, 181, 4041–4049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, F.; Li, D.; Zhou, M.; Wang, X.; Xu, Q.; Zhang, Y.; Yan, L.; Jiang, Y. Trisomy of chromosome R confers resistance to triazoles in Candida albicans. Med. Mycol. 2015, 53, 302–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poláková, S.; Blume, C.; Zárate, J.; Mentel, M.; Jørck-Ramberg, D.; Stenderup, J.; Piškur, J. Formation of new chromosomes as a virulence mechanism in yeastCandida glabrata. Proc. Natl. Acad. Sci. USA 2009, 106, 2688–2693. [Google Scholar] [CrossRef] [Green Version]
- Marichal, P.; Bossche, H.V.; Odds, F.C.; Nobels, G.; Warnock, D.W.; Timmerman, V.; Van Broeckhoven, C.; Fay, S.; Mose-Larsen, P. Molecular biological characterization of an azole-resistant Candida glabrata isolate. Antimicrob. Agents Chemother. 1997, 41, 2229–2237. [Google Scholar] [CrossRef] [Green Version]
- Mansfield, B.E.; Oltean, H.N.; Oliver, B.G.; Hoot, S.J.; Leyde, S.E.; Hedstrom, L.; White, T.C. Azole Drugs Are Imported By Facilitated Diffusion in Candida albicans and Other Pathogenic Fungi. PLoS Pathog. 2010, 6, e1001126. [Google Scholar] [CrossRef] [Green Version]
- Vandeputte, P.; Ferrari, S.; Coste, A.T. Antifungal Resistance and New Strategies to Control Fungal Infections. Int. J. Microbiol. 2011, 2012, 1–26. [Google Scholar] [CrossRef]
- Kahn, J.N.; Garcia-Effron, G.; Hsu, M.-J.; Park, S.; Marr, K.A.; Perlin, D.S. Acquired Echinocandin Resistance in a Candida krusei Isolate Due to Modification of Glucan Synthase. Antimicrob. Agents Chemother. 2007, 51, 1876–1878. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.A.; Gow, N.A.; Munro, C.A. Fungal echinocandin resistance. Fungal Genet. Biol. 2010, 2, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katiyar, S.K.; Alastruey-Izquierdo, A.; Healey, K.; Johnson, M.E.; Perlin, D.S.; Edlind, T.D. Fks1 and Fks2 Are Functionally Redundant but Differentially Regulated in Candida glabrata: Implications for Echinocandin Resistance. Antimicrob. Agents Chemother. 2012, 56, 6304–6309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, R.K.; Kline, E.G.; Healey, K.; Kordalewska, M.; Perlin, D.S.; Nguyen, M.H.; Clancy, C.J. Spontaneous Mutational Frequency and FKS Mutation Rates Vary by Echinocandin Agent against Candida glabrata. Antimicrob. Agents Chemother. 2018, 63, e01692-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, H.-J.; Nelliat, A. A Double-Edged Sword: Aneuploidy is a Prevalent Strategy in Fungal Adaptation. Genes 2019, 10, 787. [Google Scholar] [CrossRef] [Green Version]
- Hope, W.; Tabernero, L.; Denning, D.; Anderson, M.J. Molecular Mechanisms of Primary Resistance to Flucytosine in Candida albicans. Antimicrob. Agents Chemother. 2004, 48, 4377–4386. [Google Scholar] [CrossRef] [Green Version]
- Chapeland-Leclerc, F.; Bouchoux, J.; Goumar, A.; Chastin, C.; Villard, J.; Noël, T. Inactivation of the FCY2 Gene Encoding Purine-Cytosine Permease Promotes Cross-Resistance to Flucytosine and Fluconazole in Candida lusitaniae. Antimicrob. Agents Chemother. 2005, 49, 3101–3108. [Google Scholar] [CrossRef] [Green Version]
- Chapeland-Leclerc, F.; Hennequin, C.; Papon, N.; Girard, A.; Socié, G.; Ribaud, P.; Lacroix, C. Acquisition of Flucytosine, Azole, and Caspofungin Resistance in Candida glabrata Bloodstream Isolates Serially Obtained from a Hematopoietic Stem Cell Transplant Recipient. Antimicrob. Agents Chemother. 2009, 54, 1360–1362. [Google Scholar] [CrossRef] [Green Version]
- Arendrup, M.C.; Patterson, T.F. Multidrug-Resistant Candida: Epidemiology, Molecular Mechanisms, and Treatment. J. Infect. Dis. 2017, 216, S445–S451. [Google Scholar] [CrossRef] [Green Version]
- Kwon-Chung, K.J.; Chang, Y.C. Aneuploidy and Drug Resistance in Pathogenic Fungi. PLoS Pathog. 2012, 8, e1003022. [Google Scholar] [CrossRef]
Figure 1.
Antifungals that target ergosterol and its biosynthesis. (A) Ergosterol biosynthetic Pathway with different antifungals targeting different enzymes in the pathway. (B) Mechanism of action of azole antifungals.
Figure 1.
Antifungals that target ergosterol and its biosynthesis. (A) Ergosterol biosynthetic Pathway with different antifungals targeting different enzymes in the pathway. (B) Mechanism of action of azole antifungals.
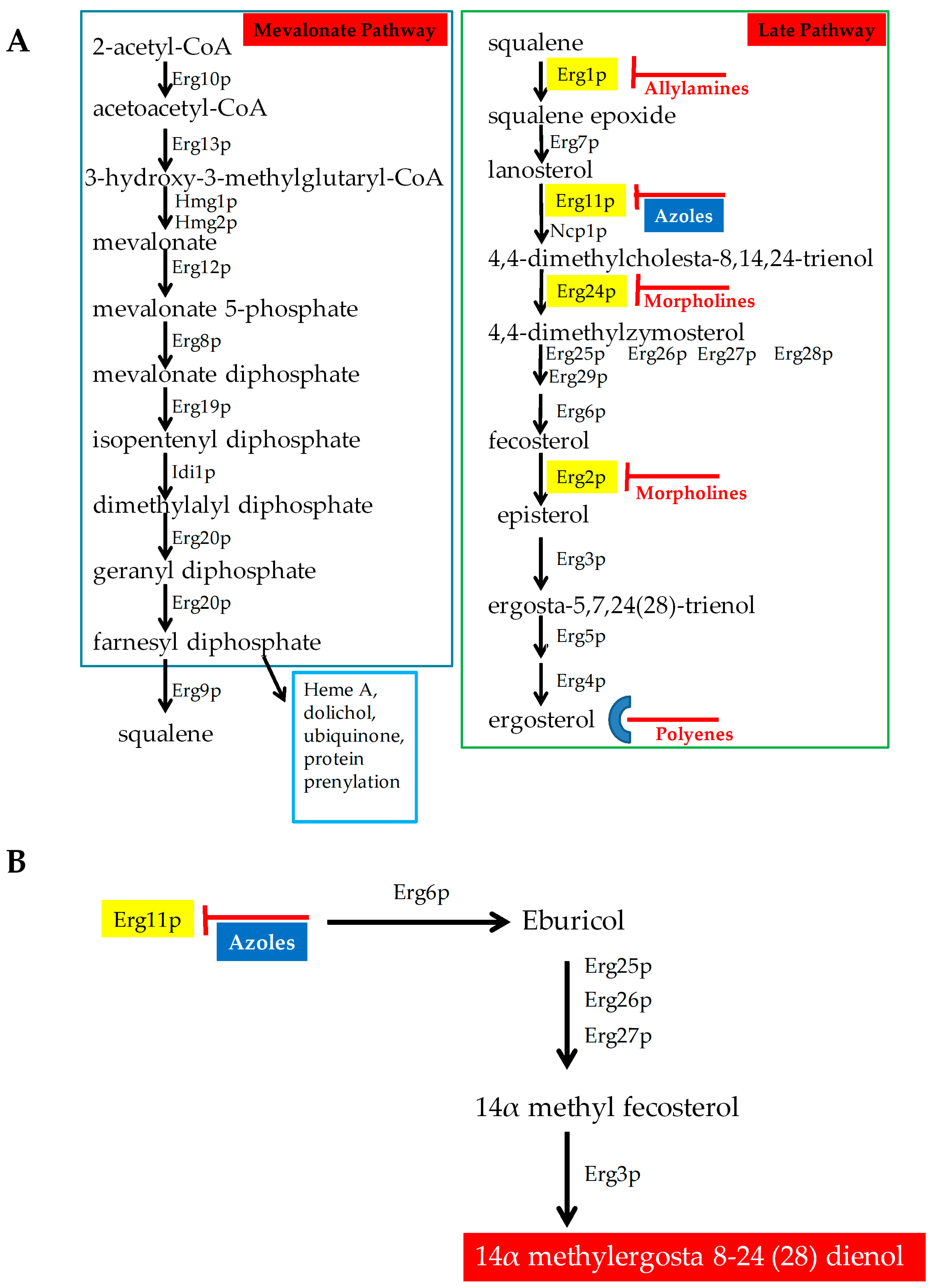
Figure 2.
Role of echinochandin in inhibiting cell wall biosynthesis. Fks complex comprises of three proteins Fks1p, Fks2p and Fks3p that utilizes UDP-glucose (UDP) to synthesize β1-3 glucan. (Images were created with the help of biorender.com).
Figure 2.
Role of echinochandin in inhibiting cell wall biosynthesis. Fks complex comprises of three proteins Fks1p, Fks2p and Fks3p that utilizes UDP-glucose (UDP) to synthesize β1-3 glucan. (Images were created with the help of biorender.com).
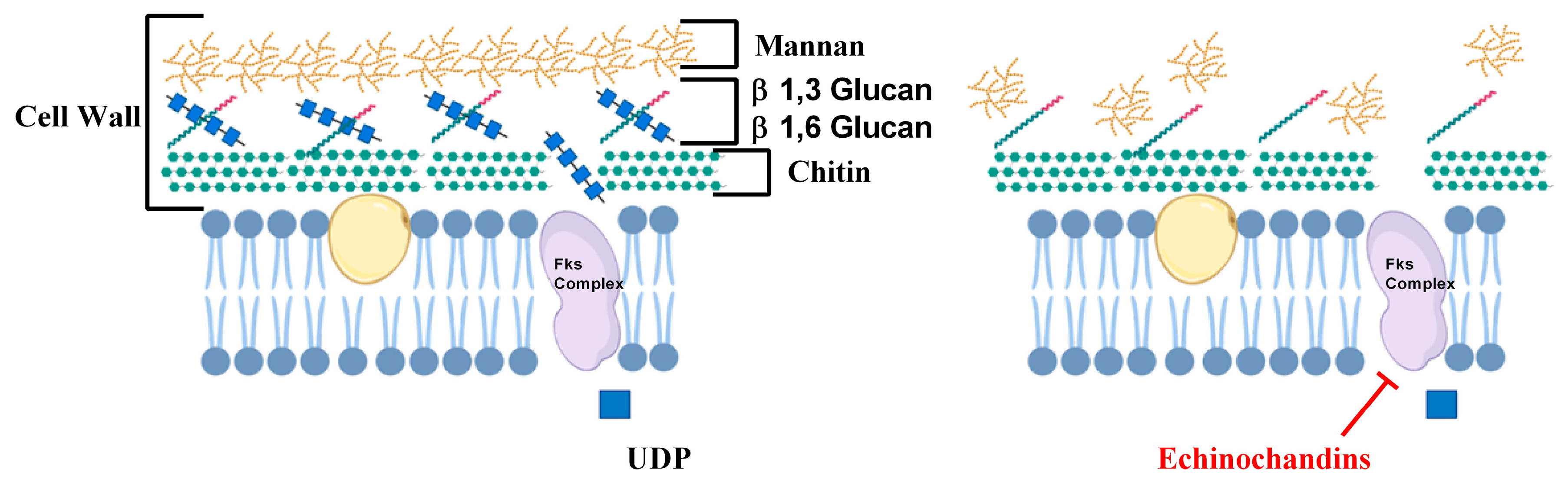
Figure 3.
Mode of action of 5-Flucytosine (5FC) (Images were created with the help of biorender.com).
Figure 3.
Mode of action of 5-Flucytosine (5FC) (Images were created with the help of biorender.com).
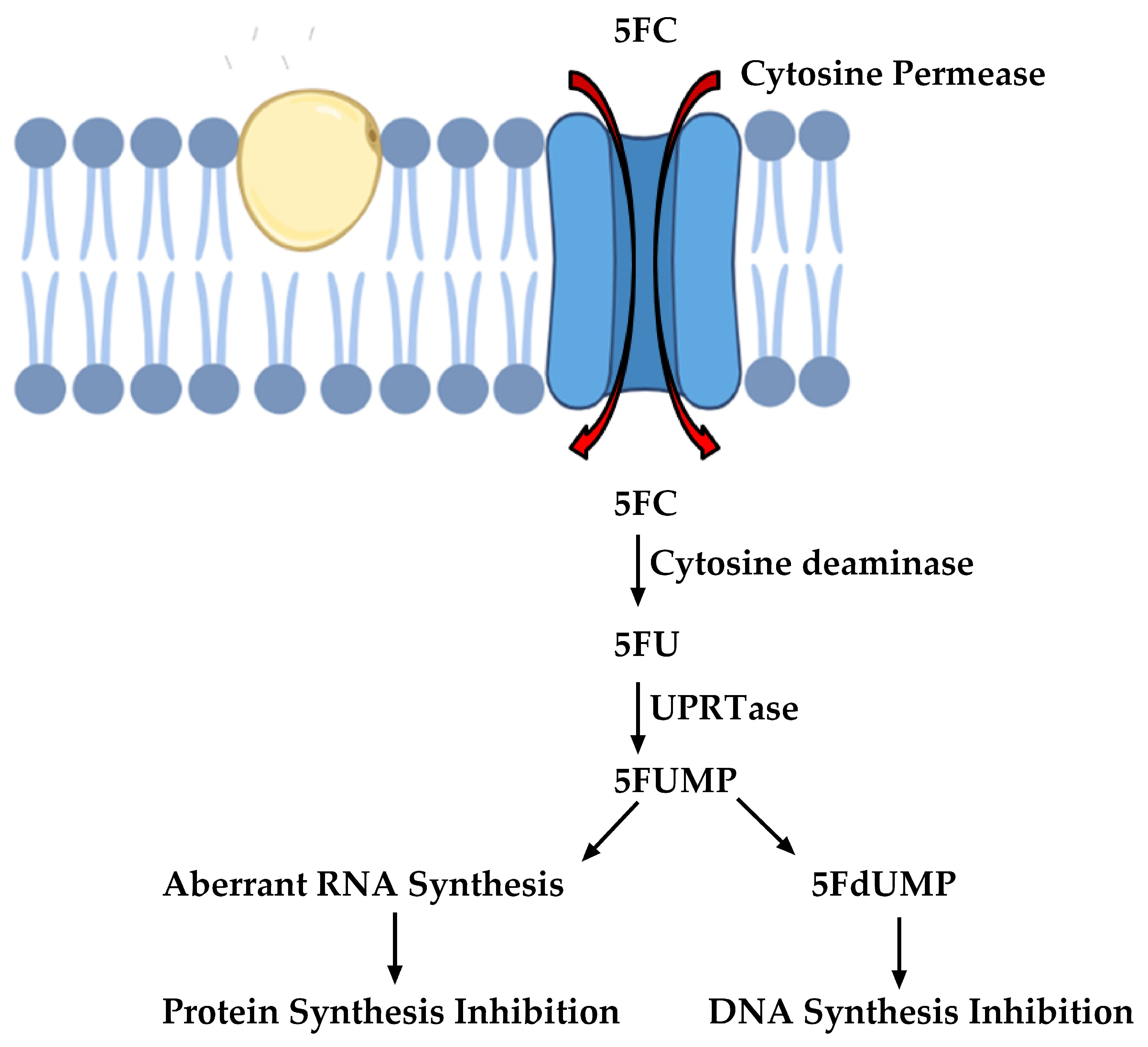
Figure 4.
The molecular mechanisms of azole resistance. A schematic representation of known molecular mechanisms of azole resistance. Black arrows represent the normal ergosterol pathway, including lanosterol (Yellow Circles), ergosterol (Green Circles), and ergosterol inhibition of UPC2. Blue arrows represent the mechanism of action of azole drugs (Blue Circles), including the inhibition of target Erg11p, and production of toxic diol (Red Circles) through several steps, including Erg6p and Erg3p. The genes important for resistance are diagrammed within the nucleus as black rectangles in their chromosomal locations (black line). Red stars (mutations) and red arrows represent the different known molecular mechanisms of drug resistance in pathogenic fungi: (A) Azole resistance mechanism via efflux pump activities. (B) Azole resistance mechanism via altered ergosterol biosynthesis. Grey box 1—Increased efflux of azoles due to point mutations in MRR1 and TAC1 (stars), resulting in over-expression of MFS-Ts and ABC-Ts, respectively. (B) Grey box 2—Point mutations in UPC2 causing increased expression throughout the ergosterol (ERG) pathway. Grey box 3—Point mutations in ERG11 that prevent azole binding. Grey box 4—Point mutations in ERG3 that prevent the formation of toxic sterol. Grey box 5—Point mutations in ERG6 that prevent the formation of toxic sterol. Grey box 6—Sterol import that reduces the need for sterol biosynthesis. Grey box 7—Altered azole import that reduces intracellular azoles. This figure was created with the help of biorender.com.
Figure 4.
The molecular mechanisms of azole resistance. A schematic representation of known molecular mechanisms of azole resistance. Black arrows represent the normal ergosterol pathway, including lanosterol (Yellow Circles), ergosterol (Green Circles), and ergosterol inhibition of UPC2. Blue arrows represent the mechanism of action of azole drugs (Blue Circles), including the inhibition of target Erg11p, and production of toxic diol (Red Circles) through several steps, including Erg6p and Erg3p. The genes important for resistance are diagrammed within the nucleus as black rectangles in their chromosomal locations (black line). Red stars (mutations) and red arrows represent the different known molecular mechanisms of drug resistance in pathogenic fungi: (A) Azole resistance mechanism via efflux pump activities. (B) Azole resistance mechanism via altered ergosterol biosynthesis. Grey box 1—Increased efflux of azoles due to point mutations in MRR1 and TAC1 (stars), resulting in over-expression of MFS-Ts and ABC-Ts, respectively. (B) Grey box 2—Point mutations in UPC2 causing increased expression throughout the ergosterol (ERG) pathway. Grey box 3—Point mutations in ERG11 that prevent azole binding. Grey box 4—Point mutations in ERG3 that prevent the formation of toxic sterol. Grey box 5—Point mutations in ERG6 that prevent the formation of toxic sterol. Grey box 6—Sterol import that reduces the need for sterol biosynthesis. Grey box 7—Altered azole import that reduces intracellular azoles. This figure was created with the help of biorender.com.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
List of known multi-drug resistance-causing efflux pumps.
| Efflux Pump | Pump Type | Organism |
|---|---|---|
| Cdr1p, Cdr2p | ABC-T | Candida albicans |
| CgCdr1p | ABC-T | Candida glabrata |
| CgFlr1p | MFS-T | Candida glabrata |
| CgPdh1p | ABC-T | Candida glabrata |
| CgQdr2p | MFS-T | Candida glabrata |
| CgSnq2p | ABC-T | Candida glabrata |
| CkAbc1p | ABC-T | Candida krusei |
| CkAbc2p | ABC-T | Candida krusei |
| Mdr1p | MFS-T | Candida albicans |
| Cdr1p | ABC-T | Candida auris |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bhattacharya, S.; Sae-Tia, S.; Fries, B.C. Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics 2020, 9, 312. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060312
AMA Style
Bhattacharya S, Sae-Tia S, Fries BC. Candidiasis and Mechanisms of Antifungal Resistance. Antibiotics. 2020; 9(6):312. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060312
Chicago/Turabian StyleBhattacharya, Somanon, Sutthichai Sae-Tia, and Bettina C. Fries. 2020. "Candidiasis and Mechanisms of Antifungal Resistance" Antibiotics 9, no. 6: 312. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9060312
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.