Application of Rank Annihilation Factor Analysis for Antibacterial Drugs Determination by Means of pH Gradual Change-UV Spectral Data



Abstract
:1. Introduction
2. Materials and Methods
2.1. Instruments
2.2. Chemicals and Reagents
2.3. Standard Solutions
2.4. Treatment of Real Samples
2.5. Spectrophotometric Analysis of Sulfamethoxazole and Trimethoprim in Laboratory-Prepared Mixtures
2.6. Theory of Rank Annihilation Factor Analysis
2.7. Chemometrics Models
3. Results and Discussion
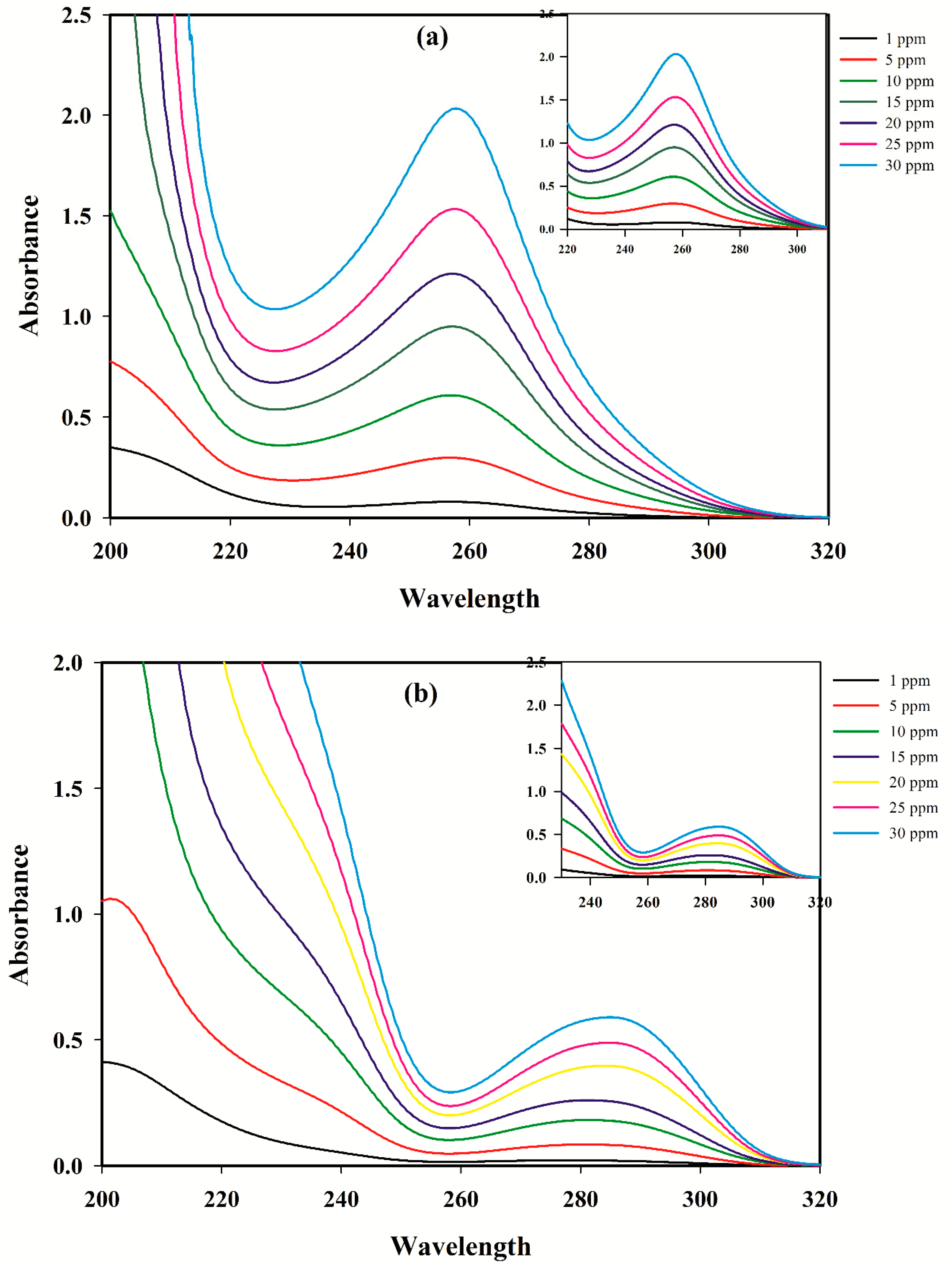
3.1. Linear Calibration Models for A Single Component
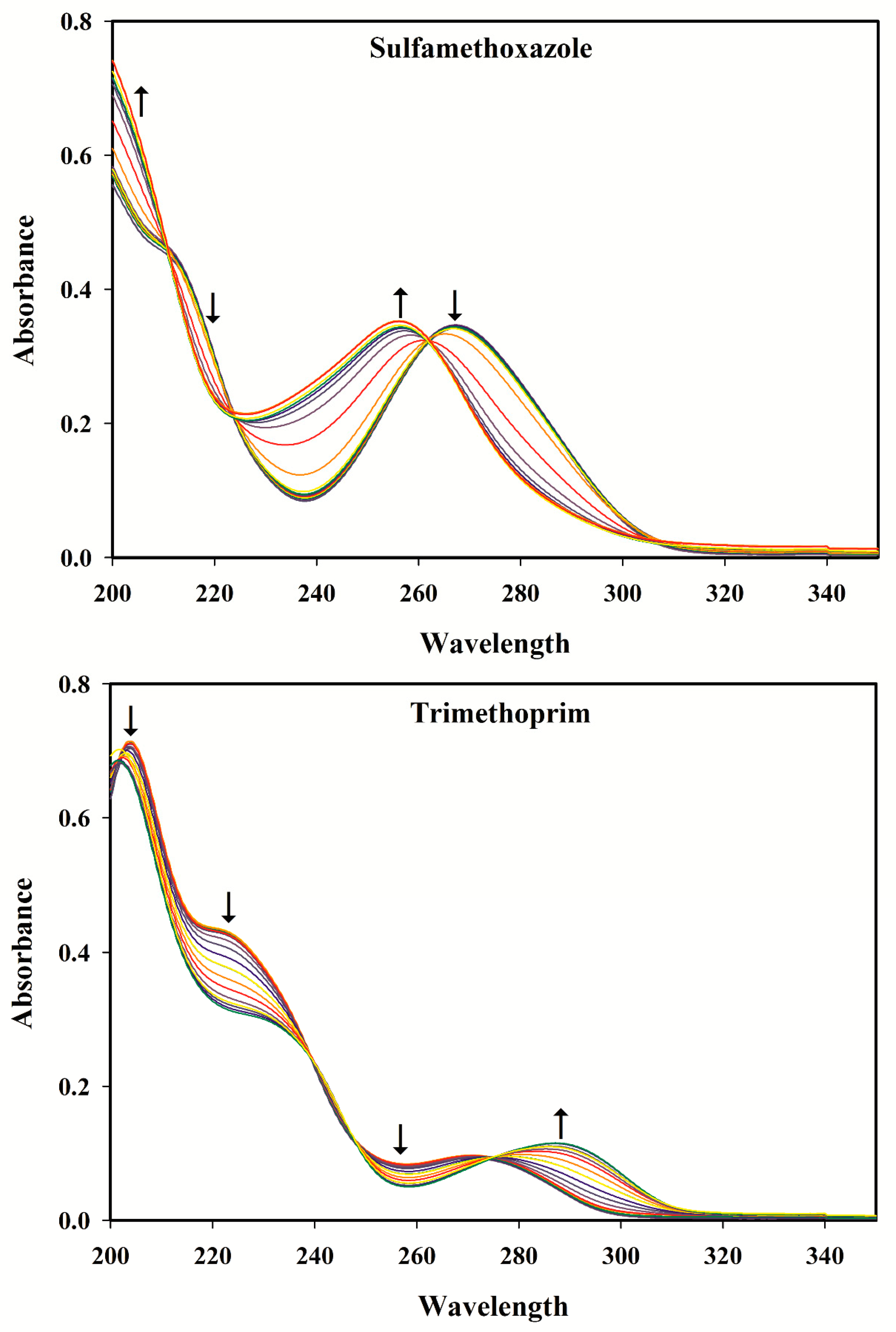
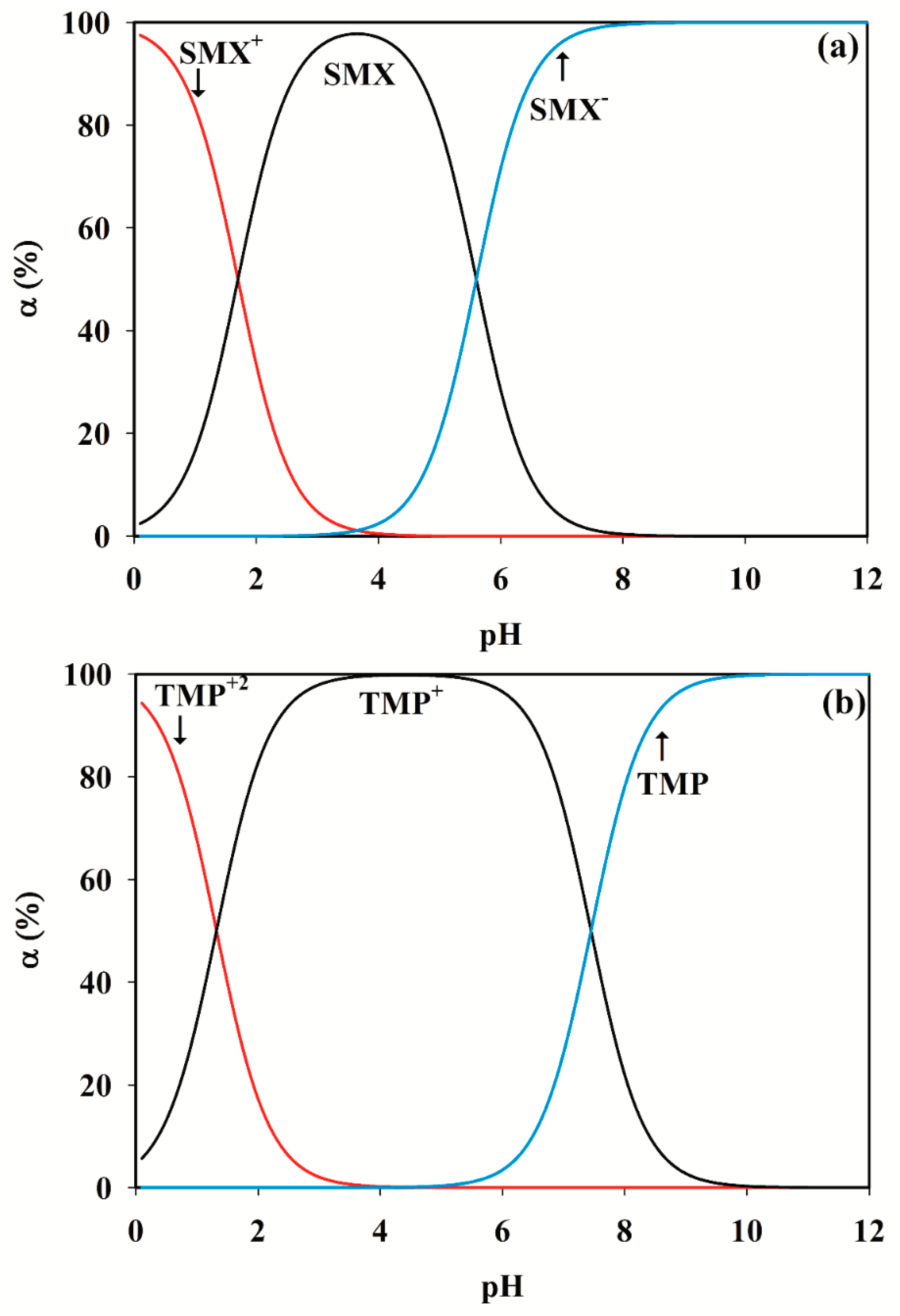
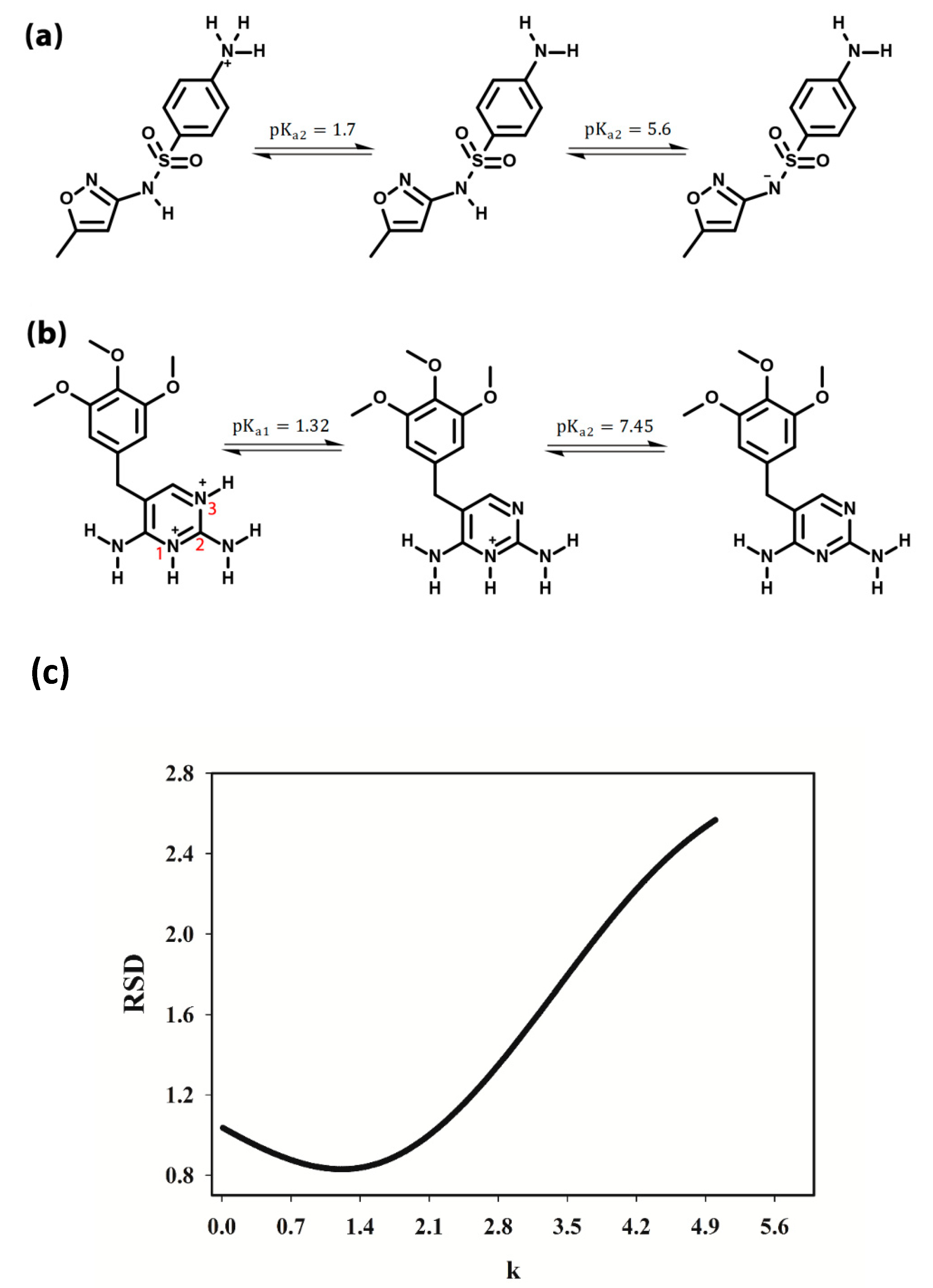
3.2. Selection of the pH Range
3.3. pH-Spectral Absorbance Data: Bilinearity, Trilinearity and Rank Deficiency
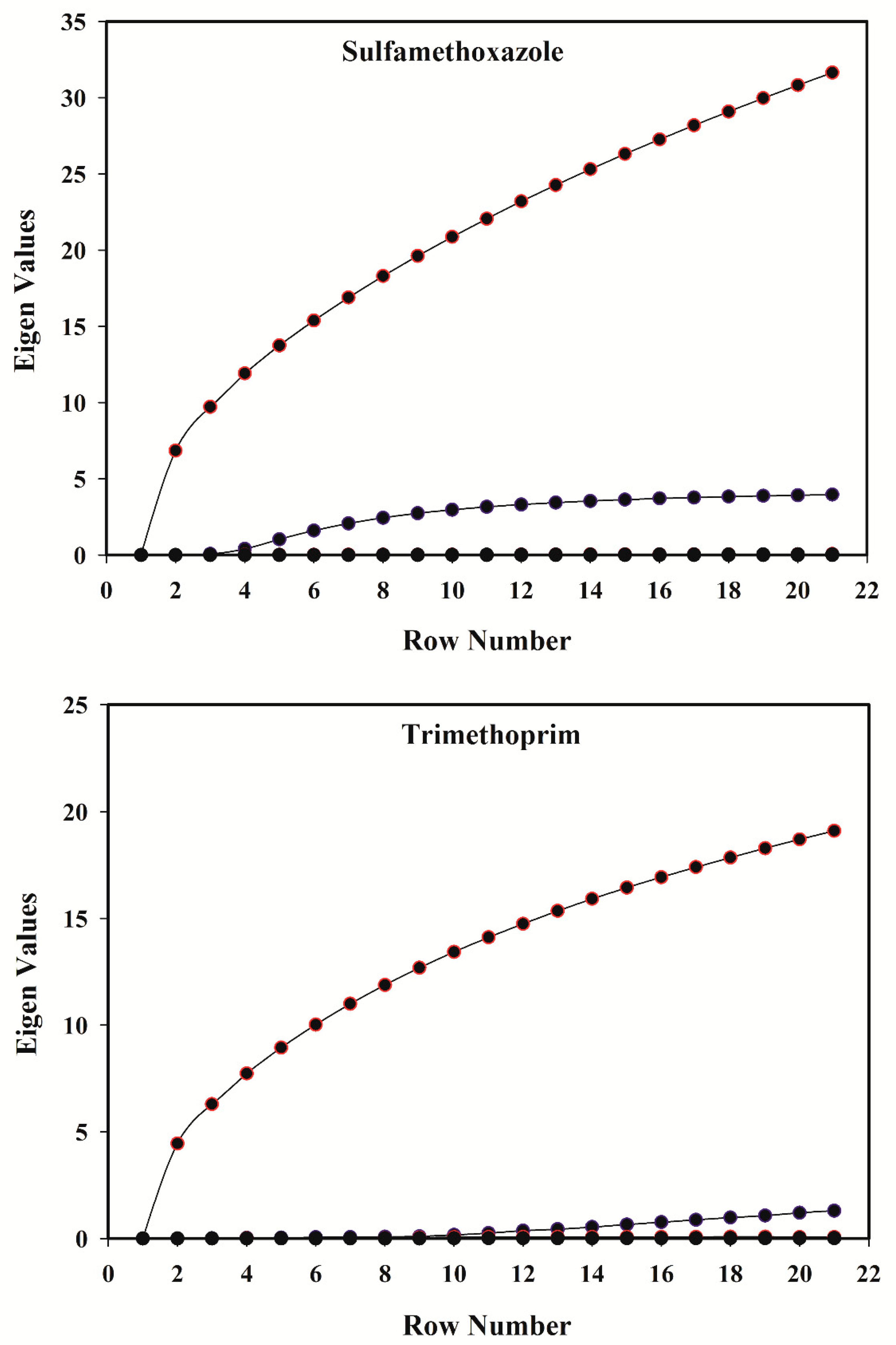
3.4. Rank Analysis
SMX + H2O ⇆ SMX− + H3O+
SMX+ + H2O ⇆ SMX + H3O+
TMP+ + H2O ⇆ TMP + H3O+ pKa2 = 7.45
3.5. Determination of SMX and TMP in Validation Samples
3.6. Determination of SMX and TMP in Real Samples
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ma, R.; Wang, Y.; Zou, X.; Hu, K.; Sun, B.; Fang, W. Pharmacokinetics of sulfamethoxazole and trimethoprim in Pacific white shrimp, Litopenaeus vannamei, after oral administration of single-dose and multiple-dose. Environ. Toxicol. Pharmacol. 2017, 52, 90–98. [Google Scholar] [CrossRef]
- Tungsanga, K.; Chongthaleong, A.; Udomsantisuk, N.; Petcharabutr, O.A.; Sitprija, V.; Wong, E.C.K. Norfloxacin versus co-trimoxazole for the treatment of upper urinary tract infections: A double blind trial. Scand. J. Infect. Dis. Suppl. 1988, 20, 28–34. [Google Scholar]
- Cooper, J.; McGillion, F.B. Treatment of acute exacerbations of chronic bronchitis. A double-blind trial of cotrimoxazole and cephalexin. Practitioner 1978, 221, 428–432. [Google Scholar]
- Feldman, W.; Richardson, H.; Rennie, B.; Dawson, P. A trial comparing cefaclor with co-trimoxazole in the treatment of acute otitis me. Arch. Dis. Child. 1982, 57, 594–596. [Google Scholar] [CrossRef]
- Sayar, E.; Sahin, S.; Cevheroglu, S.; Hincal, A. Development and validation of an HPLC method for simultaneous determination of trimethoprim and sulfamethoxazole in human plasma. Eur. J. Drug Metab. Pharmacokinet. 2010, 35, 41–46. [Google Scholar] [CrossRef]
- Ayejuyo, O.O.; Nwoko, C.; Hamed, M. Liquid Chromatographic Technique for the Simeltaneous Dtermination of Sulphamethoxazole and Trimethoprim in Pharmaceutical Formulations. UNILAG J. Med. Sci. Technol. 2016, 4, 120–132. [Google Scholar]
- Bedor, D.C.G.; Gonçalves, T.M.; Ferreira, M.L.L.; de Sousa, C.E.M.; Menezes, A.L.; Oliveira, E.J.; de Santana, D.P. Simultaneous determination of sulfamethoxazole and trimethoprim in biological fluids for high-throughput analysis: Comparison of HPLC with ultraviolet and tandem mass spectrometric detection. J. Chromatogr. B 2008, 863, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, I.S.; Vidal, D.T.R.; Do Lago, C.L.; Angnes, L. Fast simultaneous determination of trimethoprim and sulfamethoxazole by capillary zone electrophoresis with capacitively coupled contactless conductivity detection. J. Sep. Sci. 2013, 36, 1405–1409. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.F.; da Silva, W.P.; Muñoz, R.A.A.; Richter, E.M. A simple and fast batch injection analysis method for simultaneous determination of phenazopyridine, sulfamethoxazole, and trimethoprim on boron-doped diamond electrode. J. Electroanal. Chem. 2016, 766, 87–93. [Google Scholar] [CrossRef]
- Gonzalez, D.; Melloni, C.; Poindexter, B.B.; Yogev, R.; Atz, A.M.; Sullivan, J.E.; Mendley, S.R.; Delmore, P.; Delinsky, A.; Zimmerman, K.; et al. Simultaneous determination of trimethoprim and sulfamethoxazole in dried plasma and urine spots. Bioanalysis 2015, 7, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Sgobbi, L.F.; Razzino, C.A.; Machado, S.A.S. A disposable electrochemical sensor for simultaneous detection of sulfamethoxazole and trimethoprim antibiotics in urine based on multiwalled nanotubes decorated with Prussian blue nanocubes modified screen-printed electrode. Electrochim. Acta 2016, 191, 1010–1017. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Hu, X.L.; Bao, Y.F.; Yin, D.Q. Simultaneous determination of 29 pharmaceuticals in fish muscle and plasma by ultrasonic extraction followed by SPE–UHPLC–MS/MS. J. Sep. Sci. 2018, 41, 2139–2150. [Google Scholar] [CrossRef] [PubMed]
- Amini, H.; Ahmadiani, A. Rapid and simultaneous determination of sulfamethoxazole and trimethoprim in human plasma by high-performance liquid chromatography. J. Pharm. Biomed. Anal. 2007, 43, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- Teshima, D.; Otsubo, K.; Makino, K.; Itoh, Y.; Oishi, R. Simultaneous determination of sulfamethoxazole and trimethoprim in human plasma by capillary zone electrophoresis. Biomed. Chromatogr. 2004, 18, 51–54. [Google Scholar] [CrossRef]
- Afkhami, A.; Khalafi, L. Spectrophotometric investigation of the effect of β-cyclodextrin on the intramolecular cyclization reaction of catecholamines using rank annihilation factor analysis. Anal. Chim. Acta 2007, 599, 241–248. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Y.; Wang, Y.; Xu, M.; Hu, X. UV–Vis spectroscopy combined with chemometric study on the interactions of three dietary flavonoids with copper ions. Food Chem. 2018, 263, 208–215. [Google Scholar] [CrossRef]
- Esteki, M.; Nouroozi, S.; Amanifar, S.; Shahsavari, Z. A Simple and Highly Sensitive Method for Quantitative Detection of Methyl Paraben and Phenol in Cosmetics Using Derivative Spectrophotometry and Multivariate Chemometric Techniques. J. Chin. Chem. Soc. 2017, 64, 152–163. [Google Scholar] [CrossRef]
- Esteki, M.; Nouroozi, S.; Shahsavari, Z. A fast and direct spectrophotometric method for the simultaneous determination of methyl paraben and hydroquinone in cosmetic products using successive projections algorithm. Int. J. Cosmet. Sci. 2016, 38. [Google Scholar] [CrossRef]
- Bro, R. Multivariate calibration: What is in chemometrics for the analytical chemist? Anal. Chim. Acta 2003, 500, 185–194. [Google Scholar] [CrossRef]
- Othman, S. Multicomponent derivative spectroscopic analysis of sulfamethoxazole and trimethoprim. Int. J. Pharm. 1990, 63, 173–176. [Google Scholar] [CrossRef]
- Nevado, J.J.B.; Gallego, J.M.L.; Penalvo, G.C. Determination of sulfamethoxazole and trimethoprim by ratio spectra derivative spectrophotometry. Fresenius J. Anal. Chem. 1992, 342, 723–728. [Google Scholar] [CrossRef]
- López-Martínez, L.; López-de-Alba, P.L.; de-León-Rodríguez, L.M.; Yepez-Murrieta, M.L. Simultaneous determination of binary mixtures of trimethoprim and sulfamethoxazole or sulphamethoxypyridazine by the bivariate calibration spectrophotometric method. J. Pharm. Biomed. Anal. 2002, 30, 77–85. [Google Scholar] [CrossRef]
- Navarro, M.V.; Cabezón, M.A.; Damiani, P.C. Simultaneous Determination of Pesticides in Fruits by Using Second-Order Fluorescence Data Resolved by Unfolded Partial Least-Squares Coupled to Residual Bilinearization. J. Chem. 2018, 2018, 3217465. [Google Scholar] [CrossRef] [Green Version]
- Nikpour, H.; Mousavi, M.; Asadollahzadeh, H. Using inclusion complexes for achieving second-order advantage: A novel technique for cinnamic acid derivatives analysis with second-order calibration methods. J. Chemom. 2019. [Google Scholar] [CrossRef]
- Olivieri, A.C. On a versatile second-order multivariate calibration method based on partial least-squares and residual bilinearization: Second-order advantage and precision properties. J. Chemom. 2005, 19, 253–265. [Google Scholar] [CrossRef]
- El-Sheikh, A.H.; Al-Degs, Y.S. Spectrophotometric determination of food dyes in soft drinks by second order multivariate calibration of the absorbance spectra-pH data matrices. Dye Pigment. 2013, 97, 330–339. [Google Scholar] [CrossRef]
- Ho, C.N.; Christian, G.D.; Davidson, E.R. Application of the method of rank annihilation to quantitative analyses of multicomponent fluorescence data from the video fluorometer. Anal. Chem. 1978, 50, 1108–1113. [Google Scholar] [CrossRef]
- Hemmateenejad, B.; Yousefinejad, S. Multivariate standard addition method solved by net analyte signal calculation and rank annihilation factor analysis. Anal. Bioanal. Chem. 2009, 394, 1965–1975. [Google Scholar] [CrossRef]
- Etezadi, H.; Sajjadi, S.M.; Maleki, A. Crucial successes in drug delivery systems using multivariate chemometric approaches: Challenges and opportunities. New J. Chem. 2019. [Google Scholar] [CrossRef]
- Benvidi, A.; Dadras, A.; Abbasi, S.; Tezerjani, M.D.; Rezaeinasab, M.; Tabaraki, R. Experimental and computational study of the pKa of coumaric acid derivatives. J. Chin. Chem. Soc. 2019. [Google Scholar] [CrossRef]
- Alizadeh, S.; Moghtader, M.; Aliasgharlou, N. Rank Annihilation Factor Analysis for Spectrophotometric Study of Morphine Based on AuNPs Aggregation Using Multivariate Curve Resolution. Sens. Lett. 2019, 17, 337–343. [Google Scholar] [CrossRef]
- Mita, H.; Mansur, U.; Sartika, J. Optimation and Validation of Analytical Method of Cotrimoxazole in Tablet and Plasma In vitro by High Performance Liquid Chromatography. J. Bioanal. Biomed. 2012, 04, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Bahram, M.; Mabhooti, M. Rank annihilation factor analysis using mean centering of ratio spectra for kinetic–spectrophotometric analysis of unknown samples. Anal. Chim. Acta 2009, 639, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Meloun, M.; Čapek, J.; Mikšík, P.; Brereton, R.G. Critical comparison of methods predicting the number of components in spectroscopic data. Anal. Chim. Acta 2000, 423, 51–68. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi, H.; Safavi, A.; Zeinali, S. Model-based rank annihilation factor analysis for quantitative analysis of mixtures of monoprotic acids using multivariate spectrophotometric acid-base titrations. Chemom. Intell. Lab. Syst. 2008, 94, 112–117. [Google Scholar] [CrossRef]
- Zhou, W.; Moore, D.E. Photosensitizing activity of the anti-bacterial drugs sulfamethoxazole and trimethoprim. J. Photochem. Photobiol. B Biol. 1997, 39, 63–72. [Google Scholar] [CrossRef]
- Faber, N.M.; Buydens, L.M.C.; Kateman, G. Generalized rank annihilation method. I: Derivation of eigenvalue problems. J. Chemom. 1994, 8, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, A.C.; Wu, H.-L.; Yu, R.-Q. MVC2: A MATLAB graphical interface toolbox for second-order multivariate calibration. Chemom. Intell. Lab. Syst. 2009, 96, 246–251. [Google Scholar] [CrossRef]
- Smilde, A.K.; Tauler, R.; Saurina, J.; Bro, R. Calibration methods for complex second-order data. Anal. Chim. Acta 1999, 398, 237–251. [Google Scholar] [CrossRef]
- Rodríguez-Cuesta, M.; Boqué, R.; Xavier, R.F. Influence of selectivity and sensitivity parameters on detection limits in multivariate curve resolution of chromatographic second-order data. Anal. Chim. Acta 2003, 476, 111–122. [Google Scholar] [CrossRef]
- Goicoechea, H.C.; Olivieri, A.C. New Robust Bilinear Least Squares Method for the Analysis of Spectral-pH Matrix Data. Appl. Spectrosc. 2005, 59, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Amrhein, M.; Srinivasan, B.; Bonvin, D.; Schumacher, M.M. On the rank deficiency and rank augmentation of the spectral measurement matrix. Chemom. Intell. Lab. Syst. 1996, 33, 17–33. [Google Scholar] [CrossRef]
- Keller, H.R.; Massart, D.L. Evolving factor analysis. Chemom. Intell. Lab. Syst. 1991, 12, 209–224. [Google Scholar] [CrossRef]
- Babić, S.; Horvat, A.J.M.; Mutavdžić Pavlović, D.; Kaštelan-Macan, M. Determination of pKa values of active pharmaceutical ingredients. TrAC Trends Anal. Chem. 2007, 26, 1043–1061. [Google Scholar] [CrossRef]
- Chen, H.; Gao, B.; Li, H.; Ma, L.Q. Effects of pH and ionic strength on sulfamethoxazole and ciprofloxacin transport in saturated porous media. J. Contam. Hydrol. 2011, 126, 29–36. [Google Scholar] [CrossRef]
- Qiang, Z.; Adams, C. Potentiometric determination of acid dissociation constants (pKa) for human and veterinary antibiotics. Water Res. 2004, 38, 2874–2890. [Google Scholar] [CrossRef]
- Mikes, O.; Trapp, S. Acute Toxicity of the Dissociating Veterinary Antibiotics Trimethoprim to Willow Trees at Varying pH. Bull. Environ. Contam. Toxicol. 2010, 85, 556–561. [Google Scholar] [CrossRef]
- Effect of Water Quality on Rejection of Selected Human and Veterinary Antibiotics by Nanofiltration and Reverse Osmosis Membranes—Semantic Scholar. Membr. Technol. Conf.; Phoenix: International Water Association: London, UK, 2005; pp. 1–7.
- Cocco, L.; Roth, B.; Temple, C.; Montgomery, J.A.; London, R.E.; Blakley, R.L. Protonated state of methotrexate, trimethoprim, and pyrimethamine bound to dihydrofolate reductase. Arch. Biochem. Biophys. 1983, 226, 567–577. [Google Scholar] [CrossRef]
- Cao, J.; Cross, R.F. The separation of dihydrofolate reductase inhibitors and the determination of pKa,1 values by capillary zone electrophoresis. J. Chromatogr. A 1995, 695, 297–308. [Google Scholar] [CrossRef]
- Schwalbe, C.H.; Cody, V. Trimethoprim analysis. In Proceedings of the 7th International Symposium Structure Study on Antifolate Drugs; Blair, J.A., Ed.; Springer: Berlin/Heidelberg, Germany, 1983; pp. 511–515. [Google Scholar]
- Perrin, D. Dissociation Constants of Organic Bases in Aqueous Solution; Butterworths: London, UK, 1965. [Google Scholar]
- Javier, S.; Santiago, H.-C.; Romà, T.A.; Izquierdo-Ridorsa, A. Procedure for the Quantitative Determination of Mixtures of Nucleic Acid Components Based on Multivariate Spectrophotometric Acid−Base Titrations. Anal. Chem. 1999, 71, 126–134. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Sulfamethoxazole | Trimethoprim |
|---|---|---|
| Linear range (µg mL−1) | 1.0–30.0 | 1.0–30.0 |
| Correlation coefficient | 0.994 | 0.996 |
| Intercept | −0.0196 ± 0.0033 (n = 3) | −0.0121 ± 0.0025 (n = 3) |
| Slope (mL−1 µg) | 0.0632 ± 0.0028 (n = 3 ) | 0.0197 ± 0.0020 (n = 3) |
| Detection limit (µg mL−1) | 0.25 | 0.38 |
| Sample No. | SMX | TMP | ||||||
|---|---|---|---|---|---|---|---|---|
| Actual (µg mL−1) | Predicted (µg mL−1) | Error (µg mL−1) | RSD (%, n = 3) | Actual (µg mL−1) | Predicted (µg mL−1) | Error (µg mL−1) | RSD (%, n = 3) | |
| 1 | 1 | 0.91 | −0.09 | 3.1 | 1 | 1.03 | 0.03 | 2.78 |
| 2 | 6 | 5.65 | −0.35 | 2.32 | 1 | 0.98 | −0.02 | 2.12 |
| 3 | 13 | 14.59 | 1.59 | 1.51 | 1 | 0.95 | −0.05 | 2.98 |
| 4 | 20 | 20.18 | 0.18 | 3.20 | 1 | 1.08 | 0.08 | 3.15 |
| 5 | 30 | 30.55 | 0.55 | 0.55 | 1 | 0.96 | −0.04 | 3.19 |
| 6 | 1 | 0.89 | −0.11 | 1.68 | 6 | 5.66 | −0.34 | 1.59 |
| 7 | 6 | 6.58 | 0.58 | 2.32 | 6 | 6.12 | 0.12 | 2.31 |
| 8 | 13 | 12.77 | −0.23 | 1.98 | 6 | 6.23 | 0.23 | 1.66 |
| 9 | 20 | 19.32 | −0.68 | 1.33 | 6 | 5.89 | −0.11 | 1.45 |
| 10 | 30 | 28.51 | −1.49 | 2.52 | 6 | 6.08 | 0.08 | 1.98 |
| 11 | 1 | 1.05 | 0.05 | 2.31 | 13 | 13.78 | 0.78 | 1.32 |
| 12 | 6 | 6.87 | 0.87 | 1.56 | 13 | 12.32 | −0.68 | 1.75 |
| 13 | 13 | 13.88 | 0.88 | 1.23 | 13 | 12.42 | −0.58 | 2.13 |
| 14 | 20 | 21.89 | 1.89 | 1.32 | 13 | 13.88 | 0.88 | 1.55 |
| 15 | 30 | 29.12 | −0.88 | 1.45 | 13 | 13.59 | 0.59 | 1.32 |
| 16 | 1 | 1.10 | 0.1 | 1.05 | 20 | 20.13 | 0.13 | 2.62 |
| 17 | 6 | 5.64 | −0.36 | 1.23 | 20 | 21.16 | 1.16 | 1.78 |
| 18 | 13 | 14.02 | 1.02 | 2.12 | 20 | 19.55 | −0.45 | 3.21 |
| 19 | 20 | 22.21 | 2.21 | 1.21 | 20 | 19.17 | −0.83 | 2.17 |
| 20 | 30 | 28.65 | −1.35 | 1.59 | 20 | 21.21 | 1.21 | 2.79 |
| 21 | 1 | 0.99 | −0.01 | 2.32 | 30 | 32.42 | 2.42 | 1.98 |
| 22 | 6 | 5.22 | −0.78 | 1.48 | 30 | 30.67 | 0.67 | 3.31 |
| 23 | 13 | 14.65 | 1.65 | 1.75 | 30 | 29.10 | −0.9 | 2.78 |
| 24 | 20 | 20.03 | 0.03 | 0.75 | 30 | 31.78 | 1.78 | 1.44 |
| 25 | 30 | 31.22 | 1.22 | 0.89 | 30 | 32.52 | 2.52 | 1.32 |
| Drug | SMX | TMP | ||||
|---|---|---|---|---|---|---|
| Approximate Doses (mg) | Proposed Method (mg) | HPLC (mg) | Approximate Doses (mg) | Proposed Method (mg) | HPLC (mg) | |
| Co-trimoxazole adult tablet | 400 | 403 ± 4.3 | 402 ± 5.2 | 80 | 78.3 ± 2.6 | 79.6 ± 3.1 |
| Co-trimoxazole pediatric tablet | 100 | 98.23 ± 3.7 | 97.3 ± 4.1 | 20 | 22.3 ± 3.1 | 24.8 ± 4.1 |
| Co-trimoxazole oral suspension | 200 | 197.56 ± 4.9 | 198.4 ± 4.7 | 40 | 38.5 ± 4.5 | 38.1 ± 4.3 |
| Co-trimoxazole intravenous infusion | 400 | 398.91 ± 4.2 | 400.1 ± 5.1 | 80 | 82.8 ± 5.2 | 81.6 ± 6.1 |
| SMX | ||||||||
| Samples | Proposed Method | HPLC | ||||||
| Added (µg mL−1) | Found (µg mL−1) | Recovery (%) | RSD (%, n = 3) | Added (µg mL−1) | Found (µg mL−1) | Recovery (%) | RSD (%, n = 3) | |
| Serum | 10 | 10.98 | 109.8 | 5.54 | 10 | 9.3 | 93.0 | 6.5 |
| 20 | 19.10 | 95.5 | 6.32 | 20 | 19.05 | 95.2 | 6.95 | |
| 30 | 32.31 | 107.7 | 3.52 | 30 | 33.05 | 110.1 | 5.32 | |
| Plasma | 10 | 9.23 | 92.3 | 4.21 | 10 | 9.35 | 93.5 | 5.62 |
| 20 | 21.56 | 107.8 | 3.11 | 20 | 19.32 | 96.6 | 4.73 | |
| 30 | 29.11 | 97.0 | 4.63 | 30 | 32.15 | 107.2 | 6.72 | |
| TMP | ||||||||
| Samples | Proposed Method | HPLC | ||||||
| Added (µg mL−1) | Found (µg mL−1) | Recovery (%) | RSD (%, n = 3) | Added (µg mL−1) | Found (µg mL−1) | Recovery (%) | RSD (%, n = 3) | |
| Serum | 20 | 20.89 | 104.4 | 4.62 | 20 | 21.65 | 108.2 | 5.97 |
| 10 | 9.05 | 90.5 | 5.31 | 10 | 10.21 | 102.1 | 4.78 | |
| 25 | 24.32 | 97.3 | 4.65 | 25 | 23.65 | 94.6 | 5.64 | |
| Plasma | 20 | 19.52 | 97.6 | 3.12 | 20 | 19.23 | 96.15 | 4.68 |
| 10 | 10.23 | 102.3 | 2.65 | 10 | 9.53 | 95.3 | 6.28 | |
| 25 | 26.14 | 104.6 | 4.77 | 25 | 27.15 | 108.6 | 6.89 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteki, M.; Dashtaki, E.; Heyden, Y.V.; Simal-Gandara, J. Application of Rank Annihilation Factor Analysis for Antibacterial Drugs Determination by Means of pH Gradual Change-UV Spectral Data. Antibiotics 2020, 9, 383. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9070383
Esteki M, Dashtaki E, Heyden YV, Simal-Gandara J. Application of Rank Annihilation Factor Analysis for Antibacterial Drugs Determination by Means of pH Gradual Change-UV Spectral Data. Antibiotics. 2020; 9(7):383. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9070383
Chicago/Turabian StyleEsteki, Mahnaz, Elham Dashtaki, Yvan Vander Heyden, and Jesus Simal-Gandara. 2020. "Application of Rank Annihilation Factor Analysis for Antibacterial Drugs Determination by Means of pH Gradual Change-UV Spectral Data" Antibiotics 9, no. 7: 383. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9070383