Insights into Host–Pathogen Interactions in Biofilm-Infected Wounds Reveal Possibilities for New Treatment Strategies



Abstract
:1. Introduction
2. Host/Pathogen Interactions in Chronic Wounds and Implications for Wound Healing
2.1. Bacteriology
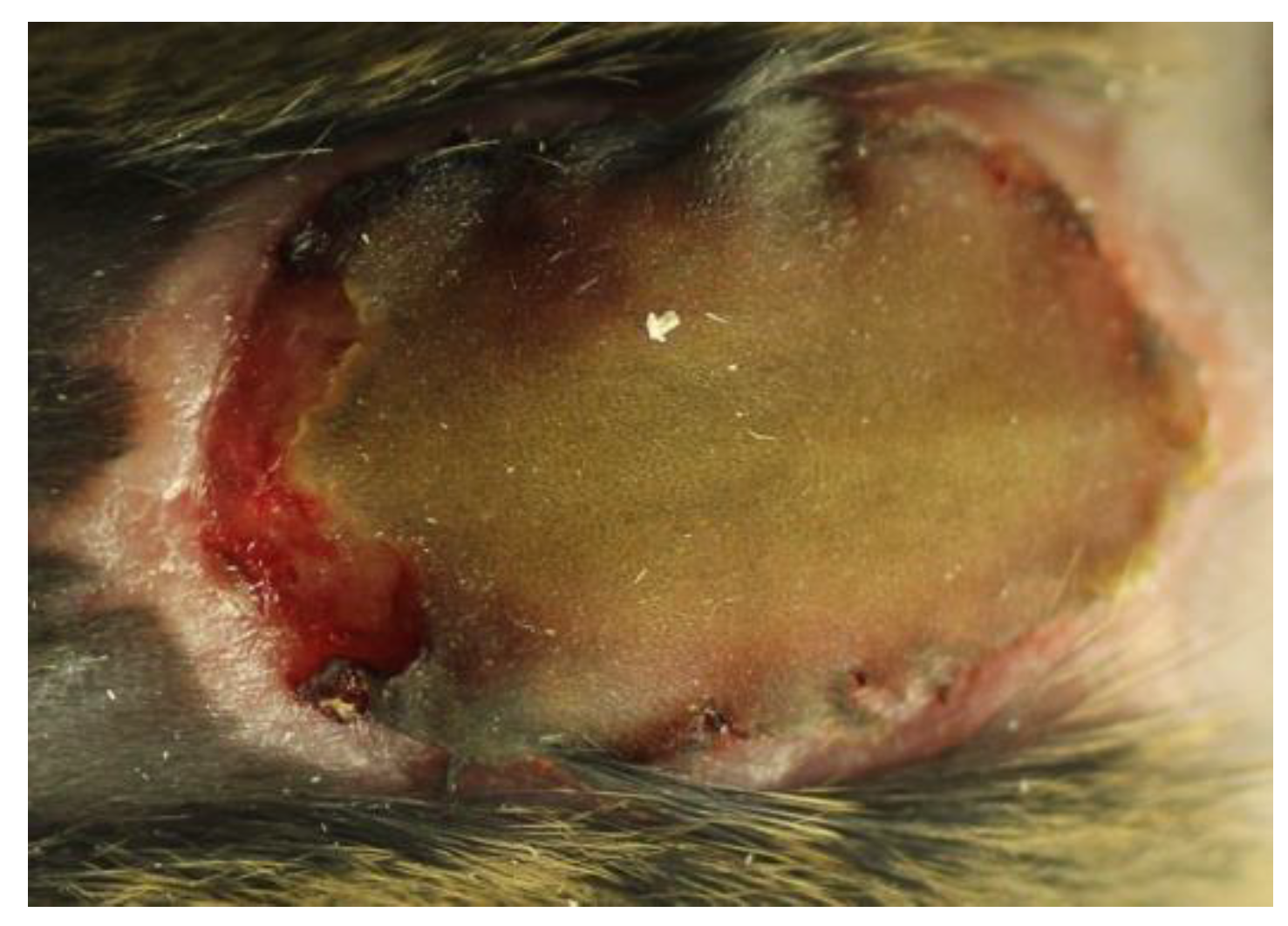
2.2. Experimental Models of Chronic Pseudomonas aeruginosa Biofilm Wound Infections
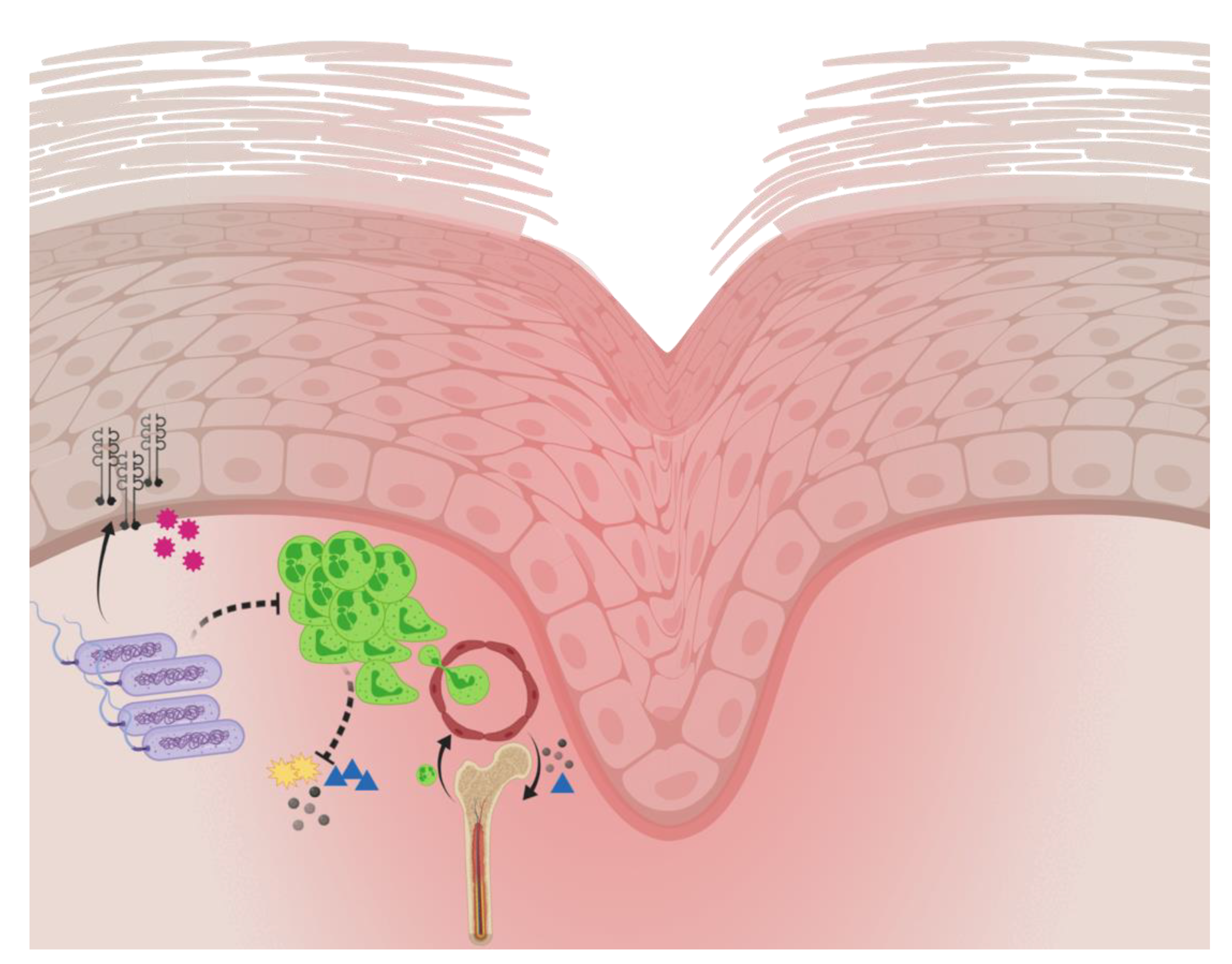
2.3. Host Response to Pseudomonas aeruginosa Biofilm Wound Infection
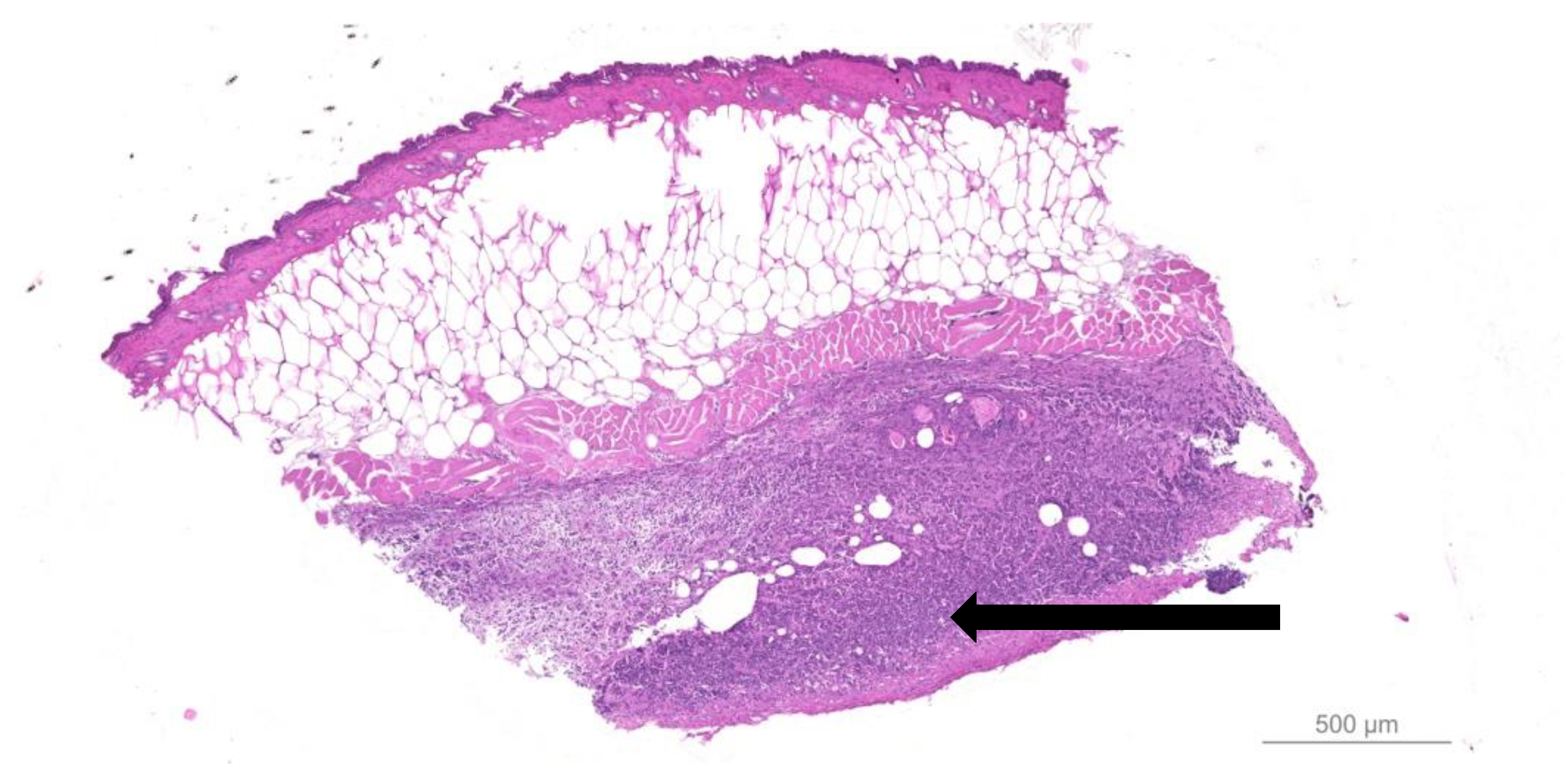
- Quantitative bacteriology and visualization of the bacteria and inflammatory cells in close proximity to the biofilm, located in the hypodermis.
- Proinflammatory cytokines and neutrophil chemoattractant profiling.
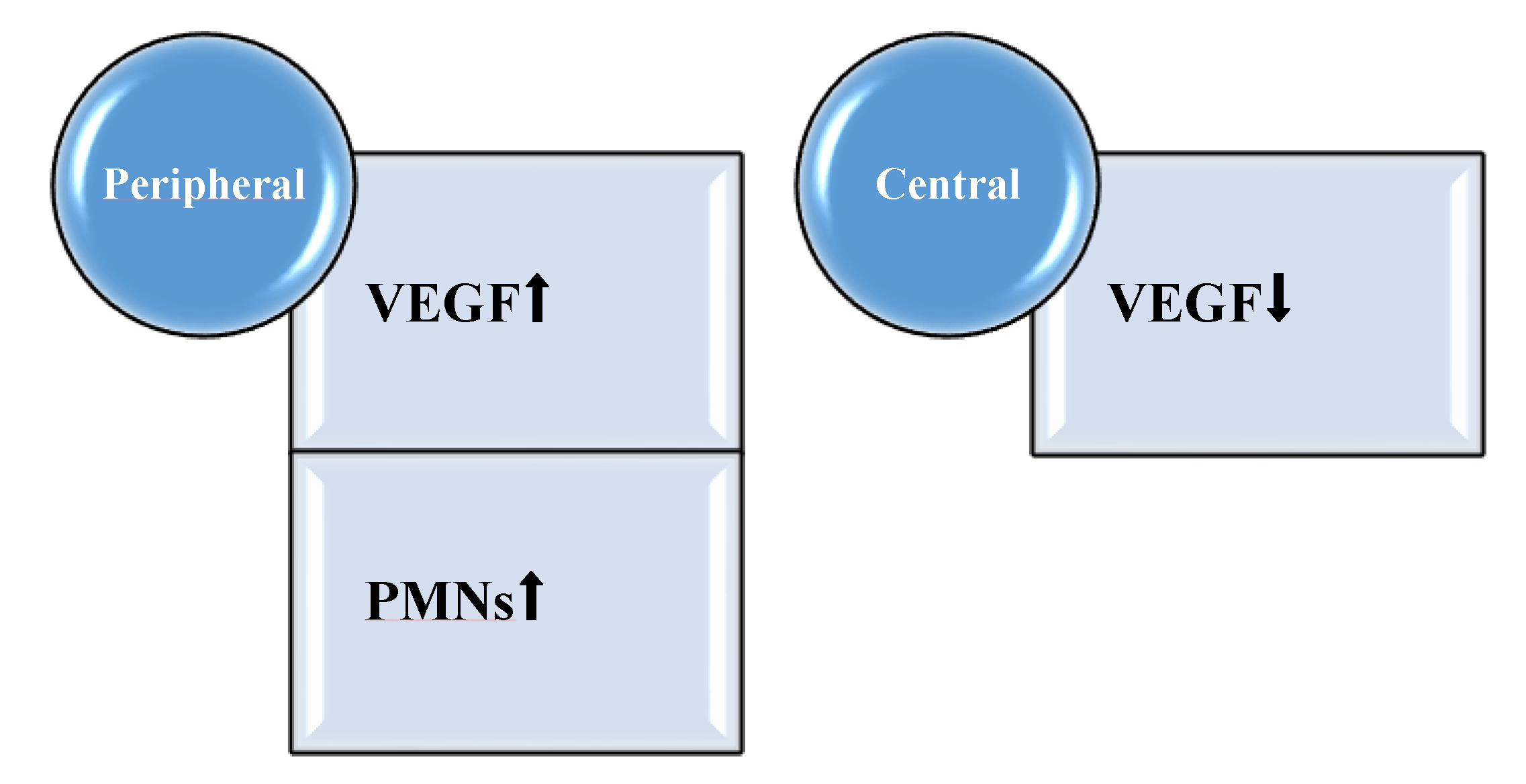
- Alterations in the growth factor profile in the proliferative state of healing.
- The impact of the biofilm-mediated recruitment of PMNs from the bone marrow to the blood.
2.4. Perturbation of Local Proinflammatory Cytokine and Growth Factor Profile by Pseudomonas aeruginosa Biofilm
2.5. Systemic Impact of Pseudomonas aeruginosa Biofilm Infection in Animal Models of Chronic Wounds
2.6. Impact of Pseudomonas aeruginosa Biofilm on Murine Wound Healing
3. Topical Intervention on Pseudomonas aeruginosa Biofilm-Infected Wounds through Mouse Models
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Saltmarche, A.E. Low level laser therapy for healing acute and chronic wounds—The extendicare experience. Int. Wound J. 2008, 5, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T.; Kirketerp-Møller, K.K.; Jensen, P.Ø.; Madsen, K.G.; Phipps, R.; Krogfeldt, K.; Høiby, N.; Givskov, M. Why chronic wounds will not heal: A novel hypothesis. Wound Repair Regen. 2008, 16, 2–10. [Google Scholar] [CrossRef]
- Trøstrup, H.; Bjarnsholt, T.; Kirketerp-Møller, K.; Høiby, N.; Moser, C. What is new in the understanding of nonhealing wounds. Epidemiology, pathophysiology and therapies. Ulcers 2013, 2013, 625934. [Google Scholar] [CrossRef] [Green Version]
- Kirketerp-Møller, K.; Jensen, P.Ø.; Fazli, M.; Madsen, K.G.; Pedersen, J.; Moser, C.; Tolker-Nielsen, T.; Høiby, N.; Givskov, M.; Bjarnsholt, T. Distribution, organization, and ecology of bacteria in chronic wounds. J. Clin. Microbiol. 2008, 46, 2717–2722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazli, M.; Bjarnsholt, T.; Kirketerp-Møller, K.; Jørgensen, B.; Andersen, A.S.; Krogfelt, K.A.; Givskov, M.; Tolker-Nielsen, T. Nonrandom distribution of Pseudomonas aeruginosa and Staphylococcus aureus in chronic wounds. J. Clin. Microbiol. 2009, 47, 4084–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Church, D.; Elsayed, S.; Reid, O.; Winston, B.; Lindsay, R. Burn Wound Infections. Clin. Microbiol. Rev. 2006, 19, 403–434. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, K.; Pech, C.M.; Stacey, M.C.; Wallace, H.J. Induction of MMP-1, MMP-3 and TIMP-1 in normal dermal fibroblasts by chronic venous leg ulcer wound fluid. Int. Wound J. 2008, 5, 79–86. [Google Scholar] [CrossRef]
- Høgsberg, T.; Bjarnsholt, T.; Thomsen, J.S.; Kirketerp-Møller, K. Succes rate of split-thickness skin grafting of chronic venous leg ulcers depends on the presence of Pseudomonas aeruginosa: A retrospective study. PLoS ONE 2011, 6, e20492. [Google Scholar] [CrossRef] [Green Version]
- Jeffcoate, W.J.; Harding, K.G. Diabetic foot ulcers. Lancet 2003, 361, 1545–1551. [Google Scholar] [CrossRef]
- Thomsen, K.; Trøstrup, H.; Christophersen, L.; Lundquist, R.; Høiby, N.; Moser, C. The phagocytic fitness of leukopatches may impact the healing of chronic wounds. Clin. Exp. Immunol. 2016, 184, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Game, F.; Jeffcoate, W.; Tarnow, L.; Jacobsen, J.L.; Whitham, D.J.; Harrison, E.F.; Ellender, S.J.; Fitzsimmons, D.; Löndahl, M. LeucoPatch system for the management of hard-to-heal diabetic foot ulcers in the UK, Denmark, and Sweden: An observer-masked, randomised controlled trial. Lancet Diabetes Endocrinol. 2018, 6, 870–878. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Liang, H.; Clarke, E.; Jackson, C.; Xue, M. Inflammation in chronic wounds. Int. J. Mol. Sci. 2016, 17, 2085. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.; Nadkarni, S.; Lele, J.; Sakhalkar, S.; Mokashi, P.; Kaushik, K.S. Bioengineered platforms for chronic wound infection studies: How can we make them more human-relevant? Front. Bioeng. Biotechnol. 2019, 7, 418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falanga, V. Classifications for wound bed preparation and stimulation of chronic wounds. Wound Repair Regen. 2000, 8, 347–352. [Google Scholar]
- Gjødsbøl, K.; Christensen, J.J.; Karlsmark, T.; Jørgensen, B.; Klein, B.M.; Krogfeldt, K.A. Multiple bacterial species reside in chronic wounds: A longitudinal study. Int. Wound J. 2006, 3, 225–231. [Google Scholar]
- Thomsen, T.R.; Aasholm, M.S.; Rudkjøbing, V.B.; Saunders, A.M.; Bjarnsholt, T.; Givskov, M.; Kirketerp-Møller, K.; Nielsen, P.H. The bacteriology of chronic venous leg ulcers examined by culture-independent molecular methods. Wound Repair Regen. 2010, 18, 38–49. [Google Scholar] [CrossRef]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [Green Version]
- Parrino, B.; Schillaci, D.; Carnevale, I.; Giovanetti, E.; Diana, P.; Cirrincione, G.; Cascioferro, S. Synthetic small molecules as anti-biofilm agents in the struggle against antibiotic resistance. Eur. J. Med. Chem. 2019, 161, 154–178. [Google Scholar] [CrossRef]
- Fazli, M.; Bjarnsholt, T.; Kirketerp-Møller, K.; Jørgensen, A.; Andersen, C.B.; Givskov, M.; Tolker-Nielsen, T. Quantitative analysis of the cellular inflammatory response against biofilm bacteria in chronic wounds. Wound Repair Regen. 2011, 19, 387–391. [Google Scholar] [CrossRef]
- Folkesson, A.; Jelsbak, L.; Yang, L.; Johansen, H.K.; Ciofu, O.; Høiby, N.; Molin, S. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: An evolutionary perspective. Nat. Rev. Microbiol. 2012, 10, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Van der Plas, M.J.; Bhongir, R.K.; Kjellström, S.; Siller, H.; Kasetty, G.; Morgelin, M.; Schmidtchen, A. Pseudomonas aeruginosa elastase cleaves a C-terminal peptide from human thrombin that inhibits host inflammatory responses. Nat. Commun. 2016, 7, 11567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dössel, J.; Meyer-Hoffert, U.; Schröder, J.-M.; Gerstel, U. Pseudomonas aeruginosa- derived rhamnolipids subvert the host inate immune response through manipulation of the human beta-defensin-2 expression. Cell. Microbiol. 2012, 14, 1364–1375. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Byrd, M.S.; Sergeant, S.; Azad, A.K.; Parsek, M.R.; McPhail, L.; Schlesinger, L.S.; Wozniak, D.J. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell. Microbiol. 2012, 14, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiller, N.L.; Joiner, K.A. Interaction of complement with serum-sensitive and serum-resistant strains of Pseudomonas aeruginosa. Infect. Immun. 1986, 54, 689–694. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southley-Pillig, C.J.; Davies, D.G.; Sauer, K. Characterization of temporal protein production in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2005, 187, 8114–8126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, G.; Flume, P.; Heijerman, H.; Elborn, J.S.; Consensus Study Group. Treatment of lung infection in patients with cystic fibrosis: Current and future strategies. J. Cyst. Fibros. 2012, 11, 461–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boles, B.R.; Thoendel, M.; Singh, P.K. Self-generated diversity produces ‘insurance effects’ in biofilm communities. Proc. Natl. Acad. Sci. USA 2004, 101, 16630–16635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, J. Animal models for wound repair. Arch. Dermatol. Res. 1998, 290, S1–S11. [Google Scholar] [CrossRef]
- Sullivan, T.P.; Eaglstein, W.H.; Davis, S.C.; Mertz, P. The pig as a model for human wound healing. Wound Repair Regen. 2001, 9, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Gloag, E.G.; Marshall, C.W.; Snyder, D.; Lewin, G.R.; Harris, J.S.; Santos-Lopez, A.; Chaney, S.B.; Whiteley, M.; Cooper, V.S.; Wozniak, D.J. Pseudomonas aeruginosa interstrain dynamics and selection of hyperbiofilm mutants during a chronic infection. mBio 2019, 10, e01698-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masella, P.C.; Balent, E.M.; Carlson, T.L.; Lee, K.W.; Pierce, L.M. Evaluation of six split-thickness skin graft donor-site dressing materials in a swine model. Plast. Reconstr. Surg. Glob. Open 2013, 1, e84. [Google Scholar] [CrossRef] [PubMed]
- Seaton, M.; Hocking, A.; Gibran, N.S. Porcine models of cutaneous wound healing. ILAR J. 2015, 56, 127–138. [Google Scholar] [CrossRef]
- Pletzer, D.; Mansour, S.C.; Wuerth, K.; Rahanjam, N.; Hancock, R.E.W. New mouse model for chronic infections by gram-negative bacteria enabling the study of anti-infective efficacy and host-microbe interactions. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweere, J.M.; Ishak, H.; Sunkari, V.; Bach, M.S.; Manasherob, R.; Yadava, K.; Ruppert, S.M.; Sen, C.K.; Balaji, S.; Keswani, S.G.; et al. The Immune Response to Chronic Pseudomonas aeruginosa Wound Infection in Immunocompetent Mice. Adv. Wound Care 2020, 9, 35–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalton, T.; Dowd, S.E.; Wolcott, R.D.; Sun, Y.; Watters, C.; Griswold, J.A.; Rumbaugh, K.P. An in vivo polymicrobial biofilm wound infection model to study interspecies interactions. PLoS ONE 2011, 6, e27317. [Google Scholar] [CrossRef] [Green Version]
- Asada, M.; Nakagami, G.; Minematsu, T.; Nagase, T.; Akase, T.; Huang, L.; Yoshimura, K.; Sanada, H. Novel models for bacterial colonization and infection of full-thickness wounds in rats. Wound Repair Regen. 2012, 20, 601–610. [Google Scholar] [CrossRef]
- Nakagami, G.; Morohoshi, T.; Ikeda, T.; Ohta, Y.; Sagara, H.; Huang, L.; Nagase, T.; Sugama, J.; Sanada, H. Contribution of quorum sensing to the virulence of Pseudomonas aeruginosa in pressure ulcer infection in rats. Wound Repair Regen. 2011, 19, 214–222. [Google Scholar] [CrossRef]
- Rashid, M.H.; Rumbaugh, K.; Passador, L.; Davies, D.G.; Hamood, A.N.; Iglewski, B.H.; Kornberg, A. Polyphosphate kinase is essential for biofilm development, quorum sensing, and virulence of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2000, 97, 9636–9641. [Google Scholar] [CrossRef] [Green Version]
- Calum, H.; Moser, C.; Jensen, P.Ø.; Christophersen, L.; Malling, D.S.; van Gennip, M.; Bjarnsholt, T.; Hougen, H.-P.; Givskov, M.; Jacobsen, G.K.; et al. Thermal injury induces impaired function in polymorphonuclear neutrophil granulocytes and reduced control of burn wound infection. Clin. Exp. Immunol. 2009, 156, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trøstrup, H.; Thomsen, K.; Christophersen, L.; Hougen, H.-P.; Bjarnsholt, T.; Jensen, P.Ø.; Kirkby, N.; Calum, H.; Høiby, N.; Moser, C. Pseudomonas aeruginosa Biofilm Aggravates Skin Inflammatory Response in BALB/c Mice in a Novel Chronic Wound Model. Wound Rep. Reg. 2013, 21, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Johansen, H.K.; Song, Z.; Hougen, H.-P.; Rygaard, J.; Høiby, N. Chronic Pseudomonas aeruginosa lung infection is more severe in Th2 responding BALB/c mice compared to Th1 responding C3H/HeN mice. APMIS 1997, 105, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Rumbaugh, K.P.; Griswold, J.A.; Iglewski, B.H.; Hamood, A.N. Contribution of quorum sensing to the virulence of Pseudomonas aeruginosa in bund wound infections. Infect. Immune. 1999, 67, 5854–5862. [Google Scholar] [CrossRef] [Green Version]
- Wolcott, R.D.; Rhoads, D.D.; Dowd, S.E. Biofilms and chronic wound inflammation. J Wound Care 2008, 17, 333–341. [Google Scholar] [CrossRef]
- Kharazmi, A.; Nielsen, H. Inhibition of human monocyte chemotaxis and chemiluminiscence by Pseudomonas aeruginosa elastase. APMIS 1991, 99, 93–95. [Google Scholar] [CrossRef]
- Trøstrup, H.; Lerche, C.J.; Christophersen, L.J.; Thomsen, K.; Jensen, P.Ø.; Hougen, H.-P.; Høiby, N.; Moser, C. Chronic Pseudomonas aeruginosa Biofilm Infection Impairs Murine S100A8/A9 and Neutrophil Effector Cytokines—Implications for Delayed Wound Closure? Pathog. Dis. 2017, 75, ftx068. [Google Scholar] [CrossRef]
- Jensen, P.Ø.; Bjarnsholt, T.; Phipps, R.; Rasmussen, T.B.; Calum, H.; Christoffersen, L.; Moser, C.; Williams, P.; Pressler, T.; Givskov, M.; et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 2007, 153 Pt 5, 1329–1338. [Google Scholar] [CrossRef] [Green Version]
- Hsi, E.D.; Remick, D.G. Monocytes are the major producers of interleukin-1 beta in an ex vivo model of local cytokine production. J. Interferon Cytokine Res. 1995, 15, 89–94. [Google Scholar] [CrossRef]
- Grazul-Bilska, A.T.; Johnson, M.L.; Bilski, J.J.; Redmer, D.A.; Reynolds, L.P.; Abdullah, A.; Abdullah, K.M. Wound healing: The role of growth factors. Drugs Today 2003, 39, 787–800. [Google Scholar] [CrossRef]
- Trøstrup, H.; Lerche, C.J.; Christophersen, L.J.; Thomsen, K.; Jensen, P.Ø.; Hougen, H.-P.; Høiby, N.; Moser, C. Pseudomonas aeruginosa Biofilm Hampers Murine Central Wound Healing by Suppression of Vascular Endothelial Growth Factor. Int. Wound J. 2018, 15, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.O.; Curry, F.E. Vascular endothelial growth factor increases hydraulic conductivity of isolated perfused microvessels. Am J Physiol. 1996, 271 Pt 2, H2520–H2528. [Google Scholar] [CrossRef]
- Lauer, G.; Sollberg, S.; Cole, M.; Flamme, I.; Stürzebecher, J.; Mann, K.; Krieg, T.; Eming, S.A. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J. Investig. Dermatol. 2000, 115, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrientos, S.; Brem, H.; Stojadinovic, O.; Tomic-Canic, M. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen. 2014, 22, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Birkenhauer, E.; Neethirajan, S. A double-edged sword: The role of VEGF in wound repair and chemoattraction of opportunist pathogens. Int. J. Mol. Sci. 2015, 16, 7159–7172. [Google Scholar] [CrossRef] [Green Version]
- Trøstrup, H.; Holstein, P.; Christophersen, L.; Jørgensen, B.; Karlsmark, T.; Høiby, N.; Moser, C.; Ågren, M.S. S100A8/A9 is an important host defence mediator in neuropathic foot ulcers in patients with type 2 diabetes mellitus. Arch. Dermatol. Res. 2016, 308, 347–355. [Google Scholar] [CrossRef]
- Detmar, M.; Brown, L.F.; Berse, B.; Jackman, R.W.; Elicker, B.M.; Dvorak, H.F.; Claffey, K.P. Hypoxia regulates the expresssion of vascular permeability factoe/vascular endothelial growth factor (VPF/VEGF) and its receptors in human skin. J. Investig. Derm. 1997, 108, 263–268. [Google Scholar] [CrossRef] [Green Version]
- James, G.A.; Zhao, A.G.; Usui, M.; Underwood, R.A.; Nguyen, H.; Beyenal, H.; deLancey Pulcini, E.; Agostinho Hunt, A.; Bernstein, H.C.; Fleckman, P.; et al. Microsensor and transcriptomic signatures of oxygen depletion in biofilms associated with chronic wounds. Wound Repair Regen. 2016, 24, 373–383. [Google Scholar] [CrossRef]
- Tibbles, P.M.; Edelsberg, J.S. Hyperbaric-oxygen therapy. N. Engl. J. Med. 1996, 334, 1642–1648. [Google Scholar] [CrossRef] [PubMed]
- Schreml, S.; Meier, R.J.; Kirschbaum, M.; Kong, S.C.; Gehmert, S.; Felthaus, O.; Küchler, S.; Sharpe, J.R.; Wöltje, K.; Weiß, K.T.; et al. Luminescent dual sensors reveal extracellular pH-gradients and hypoxia on chronic wounds that disrupt epidermal repair. Theranostics 2014, 4, 721–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, D.A.; Major, J.A.; Chudyk, A.; Hamilton, T.A. Neutrophil chemoattractant genes KC and MIP-2 are expressedin different cell populations at sites of surgical injury. J. Leukoc. Biol. 2004, 75, 641–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sroussi, H.Y.; Berline, J.; Palefsky, J.M. Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. J. Leukoc. Biol. 2007, 81, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Trøstrup, H.; Lerche, C.J.; Christophersen, L.; Jensen, P.Ø.; Høiby, N.; Moser, C. Immune Modulating Topical S100A8/A9 Inhibits Growth of Pseudomonas aeruginosa and Mitigates Biofilm Infection in Chronic Wounds. Int. J. Mol. Sci. 2017, 18, 1359. [Google Scholar] [CrossRef]
- Zhao, G.; Hochwalt, P.C.; Usui, M.L.; Underwood, R.A.; Singh, P.K.; James, G.A.; Stewart, P.S.; Fleckman, P.; Olerud, J.E. Delayed wound healing in diabetic (db/db) mice with Pseudomonas aeruginosa biofilm challenge: A model for the study of chronic wounds. Wound Repair Regen. 2010, 18, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Watters, C.; DeLeon, K.; Trivedi, U.; Griswold, J.A.; Lyte, M.; Hampel, K.J.; Wargo, M.J.; Rumbaugh, K.P. Pseudomonas aeruginosa biofilms perturb wound resolution and antibiotic tolerance in diabetic mice. Med. Microbiol. Immunol. 2013, 202, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.T.; Mustoe, T.A. effects of ischemia on ulcer wound healing: A new model in the rabbit ear. Ann. Plast. Surg. 1990, 24, 17. [Google Scholar] [CrossRef]
- Chang, A.C.; Dearman, B.; Greenwood, J.E. A comparison of wound area measurement techniques: Visitrak versus photography. Eplasty 2011, 11, e18. [Google Scholar]
- Jeffcoate, W.J.; Musgrove, A.J.; Lincoln, N.B. Using Image J to document healing in ulcers of the foot in diabetes. Int. Wound J. 2017, 14, 1137–1139. [Google Scholar] [CrossRef]
- Aragón- Sanchez, J.; Quintana-Marrero, Y.; Aragón-Hernandez, C.; Hernández-Herero, M.J. Image J: A free, easy, and reliable method to measure leg ulcers using digital pictures. Int. J. Low. Extrem. Wounds 2017, 16, 269–273. [Google Scholar] [CrossRef]
- Nunes, J.P.S.; Dias, A.A.M. ImageJ macros for the user-friendly analysis of soft-agar and wound healing assays. Biotechniques 2017, 62, 175–179. [Google Scholar] [PubMed] [Green Version]
- Li, X.; Gu, W.; Masinde, G.; Hamilton-Ulland, M.; Xu, S.; Mohan, S.; Baylink, D.J. Genetic control of the rate of wound healing in mice. Heredity 2001, 86 Pt 6, 668–674. [Google Scholar] [CrossRef]
- Moser, C.; Pedersen, H.T.; Lerche, C.J.; Kolpen, M.; Line, L.; Thomsen, K.; Høiby, N.; Jensen, P.Ø. Biofilms and host response—Helpful or harmful. APMIS 2017, 125, 320–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, D.; Rumbaugh, K. The consequences of biofilm dispersal on the host. Sci. Rep. 2018, 8, 10738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Percival, S.L.; Emanuel, C.; Cutting, K.F.; Williams, D.W. Microbiology of the skin and the role of biofilms in infection. Int. Wound J. 2012, 9, 14–32. [Google Scholar] [CrossRef] [PubMed]
- Thorey, I.S.; Roth, J.; Regenbogen, J.; Halle, J.P.; Bittner, M.; Vogl, T.; Kaesler, S.; Bugnon, P.; Reitmaier, B.; Durka, S.; et al. The Ca2+-binding proteins S100A8 and S100A9 are encoded by novel injury regulated genes. J. Biol. Chem. 2001, 276, 35818–35825. [Google Scholar] [CrossRef] [Green Version]
- Trøstrup, H.; Lundquist, R.; Christensen, L.H.; Jørgensen, L.N.; Karlsmark, T.; Haab, B.B.; Ågren, M.S. S100A8/A9 deficiency in nonhealing venous leg ulcers uncovered by multiplexed antibody microarray profiling. Br. J. Dermatol. 2011, 165, 292–301. [Google Scholar] [CrossRef]
- Chan, J.K.; Roth, J.; Oppenheim, J.J.; Tracey, K.J.; Vogl, T.; Feldmann, M.; Horwood, N.; Nanchahal, J. Alarmins: Awaiting a clinical response. J. Clin. Investig. 2012, 122, 2711–2719. [Google Scholar] [CrossRef]
- Basu, A.; Munir, S.; Mulaw, M.A.; Singh, K.; Crisan, D.; Sindrilaru, A.; Treiber, N.; Wlaschek, M.; Huber-Lang, M.; Gebhard, F.; et al. A Novel S100A8/A9 induced fingerprint of mesenchymal stem cells associated with enhanced wound healing. Sci. Rep. 2018, 8, 10214. [Google Scholar] [CrossRef]
- Gao, N.; Sang Yoon, G.; Liu, X.; Mi, X.; Chen, W.; Standiford, T.J.; Yu, F.S. Genome-wide transcriptional analysis of differentially expressed genes in flagellin-pretreated mouse corneal epithelial cells in response to Pseudomonas aeruginosa: Involvement of S100A8/A9. Mucosal Immunol. 2013, 6, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Clark, H.L.; Jhingran, A.; Sun, Y.; Vareechon, C.; de Jesus Carrion, S.; Skaar, E.P.; Chazin, W.J.; Calera, J.A.; Hohl, T.M.; Pearlman, E. Zinc and manganese chelation by neutrophil S100A8/A9 (Calprotectin) limits extracellular Aspergillus fumigatus hyphal growth and corneal infection. J. Immunol. 2016, 196, 336–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laulund, A.S.B.; Trøstrup, H.; Lerche, C.J.; Thomsen, K.; Christophersen, L.; Calum, H.; Høiby, N.; Moser, C. Synergistic effect of immunomodulatory S100A8/A9 and ciprofloxacin against Pseudomonas aeruginosa biofilm in a murine chronic wound model. Pathog. Dis. 2019, 22, ftz027. [Google Scholar] [CrossRef] [PubMed]
- Glaros, T.; Larsen, M.; Li, L. Macrophages and fibroblasts during inflammation, tissue damage and organ injury. Front. Biosci. 2009, 14, 3988–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C3H/HeN | BALB/c |
|---|---|
| Resistant towards infection Faster infection control | Susceptible to infection Aggravated inflammatory IL-1β response No infection control |
| Faster wound closure | Delayed wound closure |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trøstrup, H.; Laulund, A.S.B.; Moser, C. Insights into Host–Pathogen Interactions in Biofilm-Infected Wounds Reveal Possibilities for New Treatment Strategies. Antibiotics 2020, 9, 396. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9070396
Trøstrup H, Laulund ASB, Moser C. Insights into Host–Pathogen Interactions in Biofilm-Infected Wounds Reveal Possibilities for New Treatment Strategies. Antibiotics. 2020; 9(7):396. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9070396
Chicago/Turabian StyleTrøstrup, Hannah, Anne Sofie Boe Laulund, and Claus Moser. 2020. "Insights into Host–Pathogen Interactions in Biofilm-Infected Wounds Reveal Possibilities for New Treatment Strategies" Antibiotics 9, no. 7: 396. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9070396