
Insight into the Effects of Solvent Treatment of Natural Fibers Prior to Structural Composite Casting: Chemical, Physical and Mechanical Evaluation

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Fibers and Solvents
2.2. Samples Casting and Washing Protocols
2.3. Physical Properties Characterization (Density and Linear Density)
2.4. Chemical Analysis via Attenuated Total Reflectance–Fourier Transform Infrared Spectroscopy
2.5. Wettability Evaluation via Static Sessile Drop Contact Angle
2.6. Tensile Testing
3. Results and Discussion
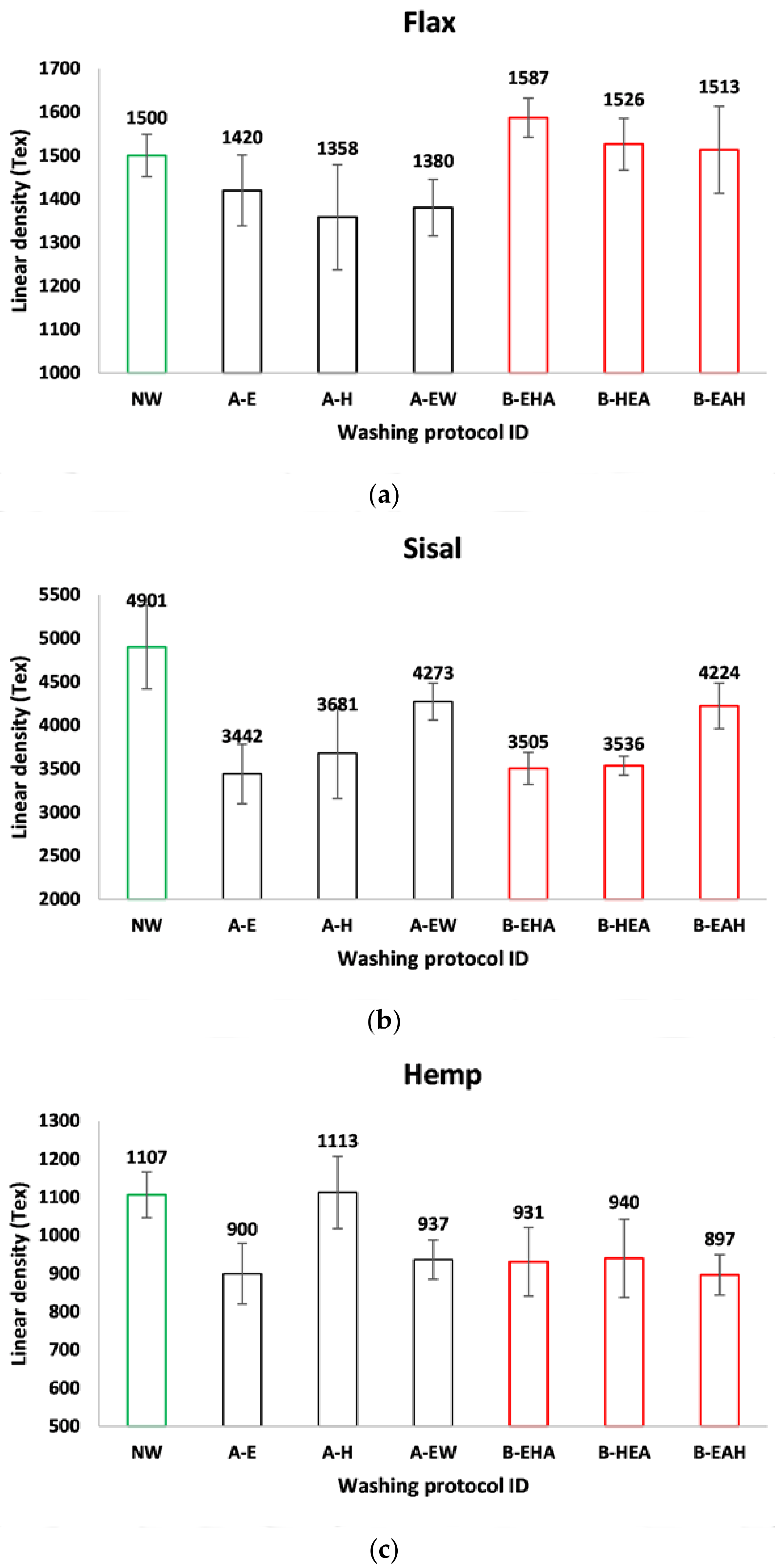
3.1. Physical Characterization
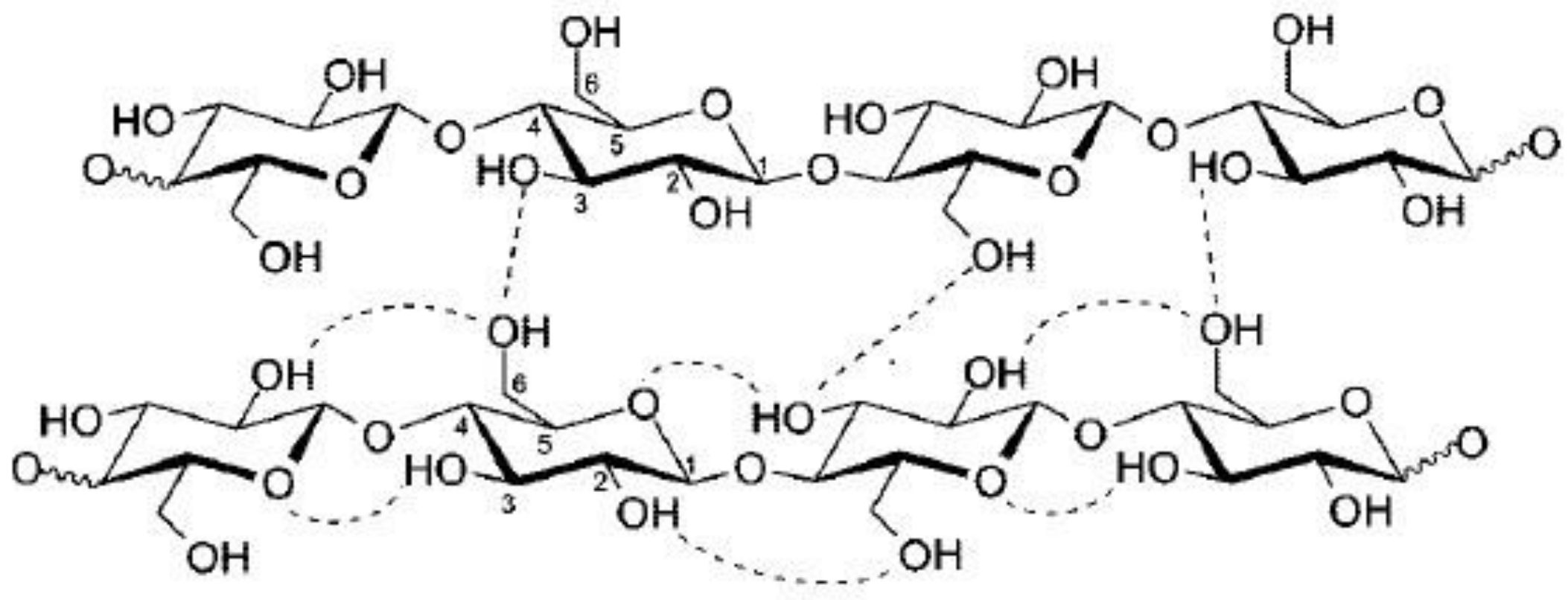
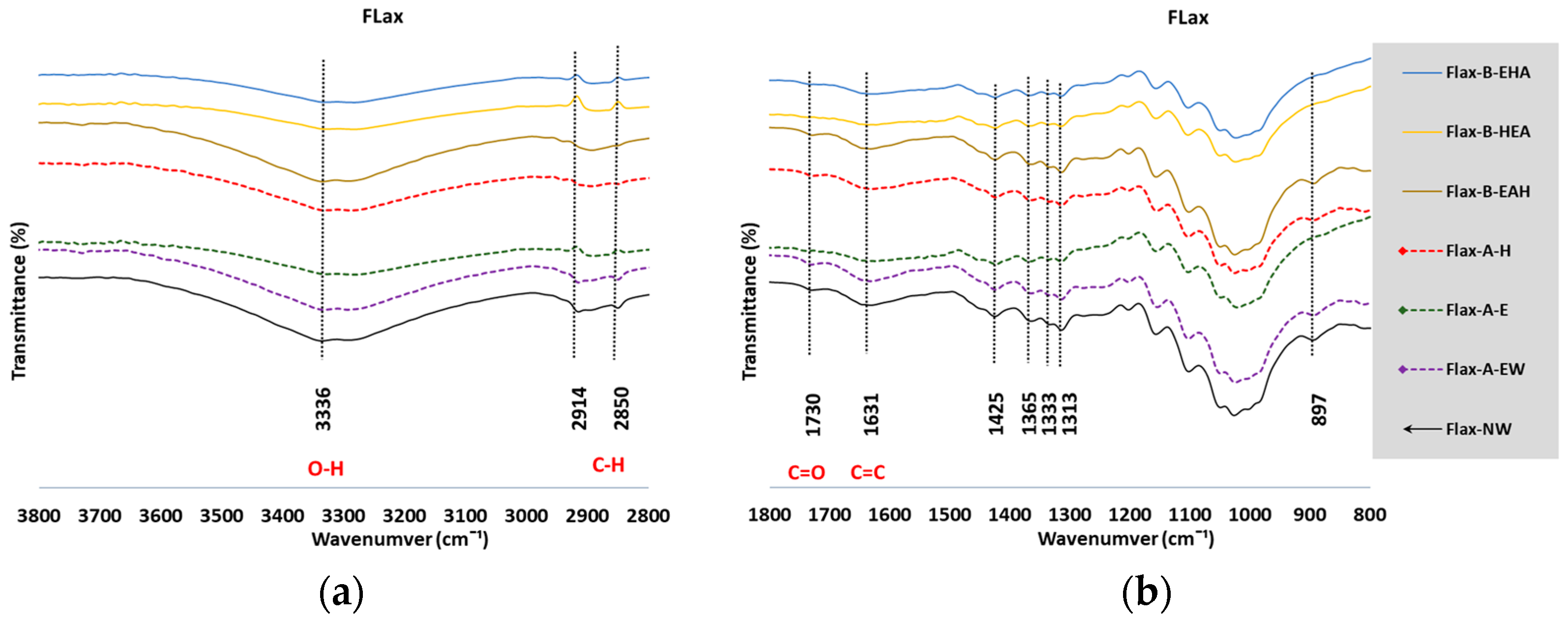
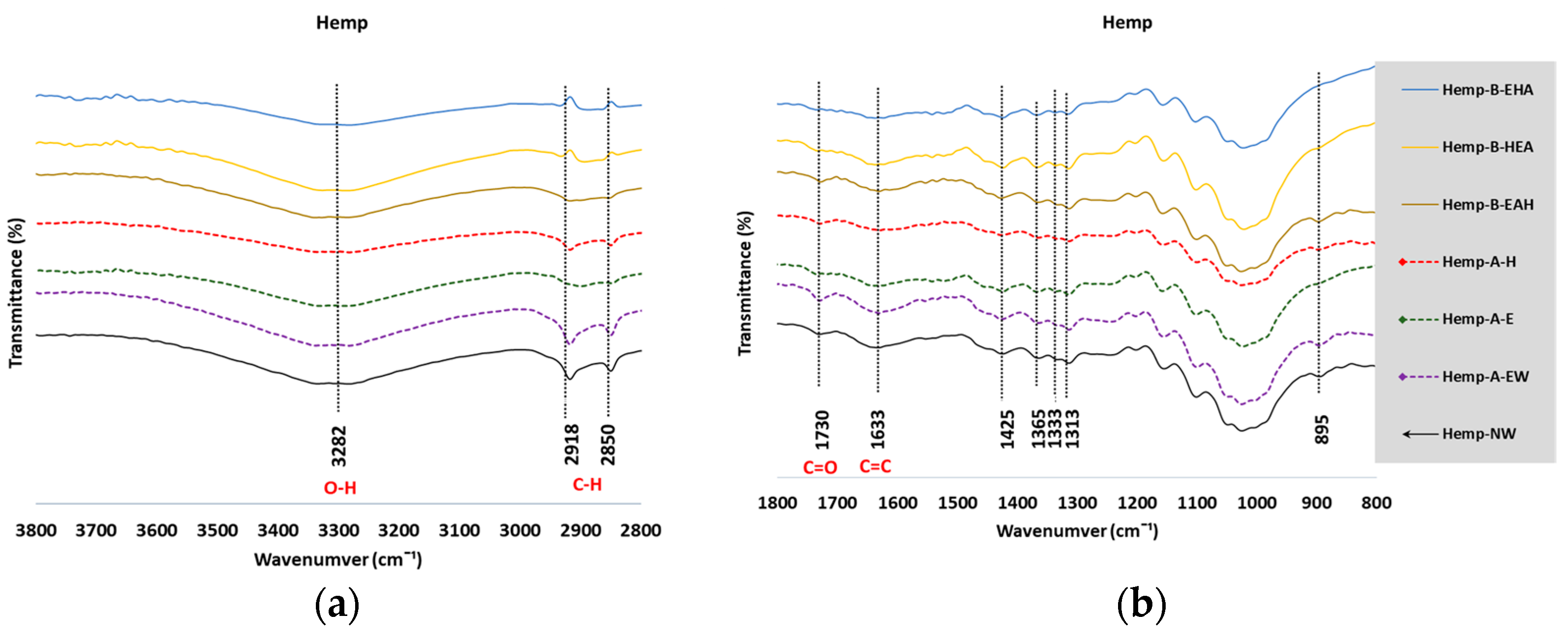
3.2. Fourier-Transform Infrared Spectroscopy
3.2.1. Analysis of the FTIR Spectra
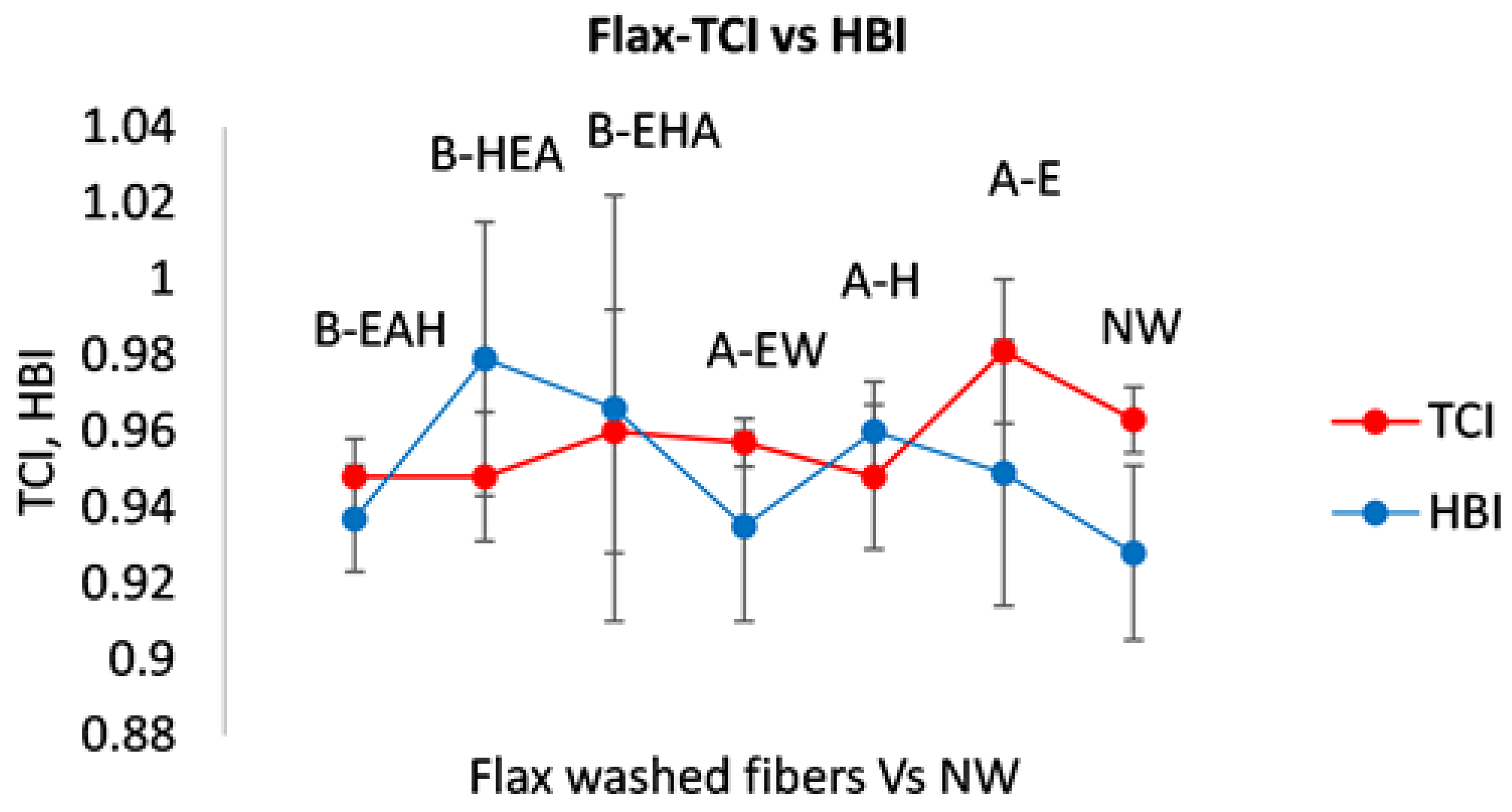
3.2.2. Estimate of Crystallinity Indexes
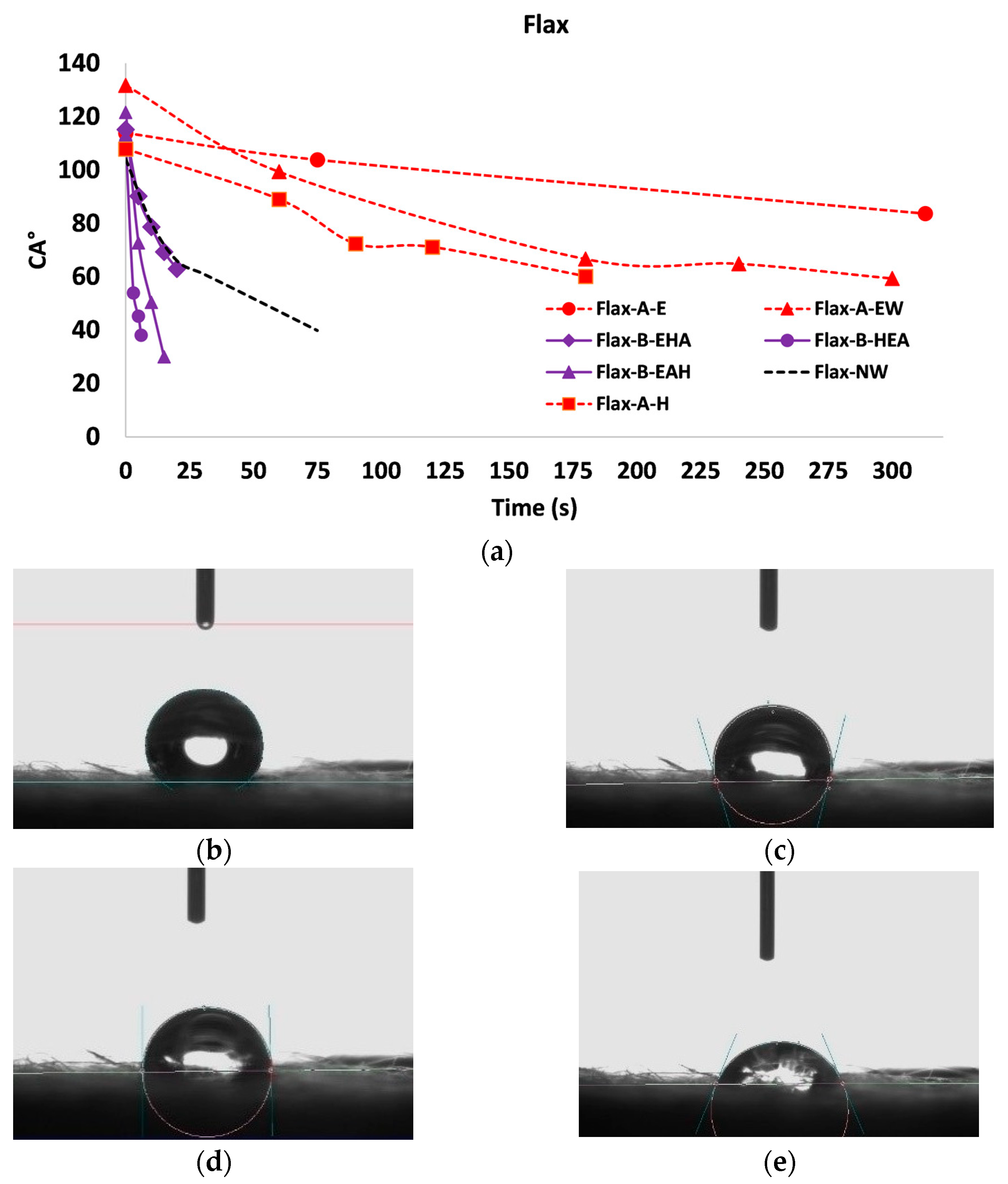
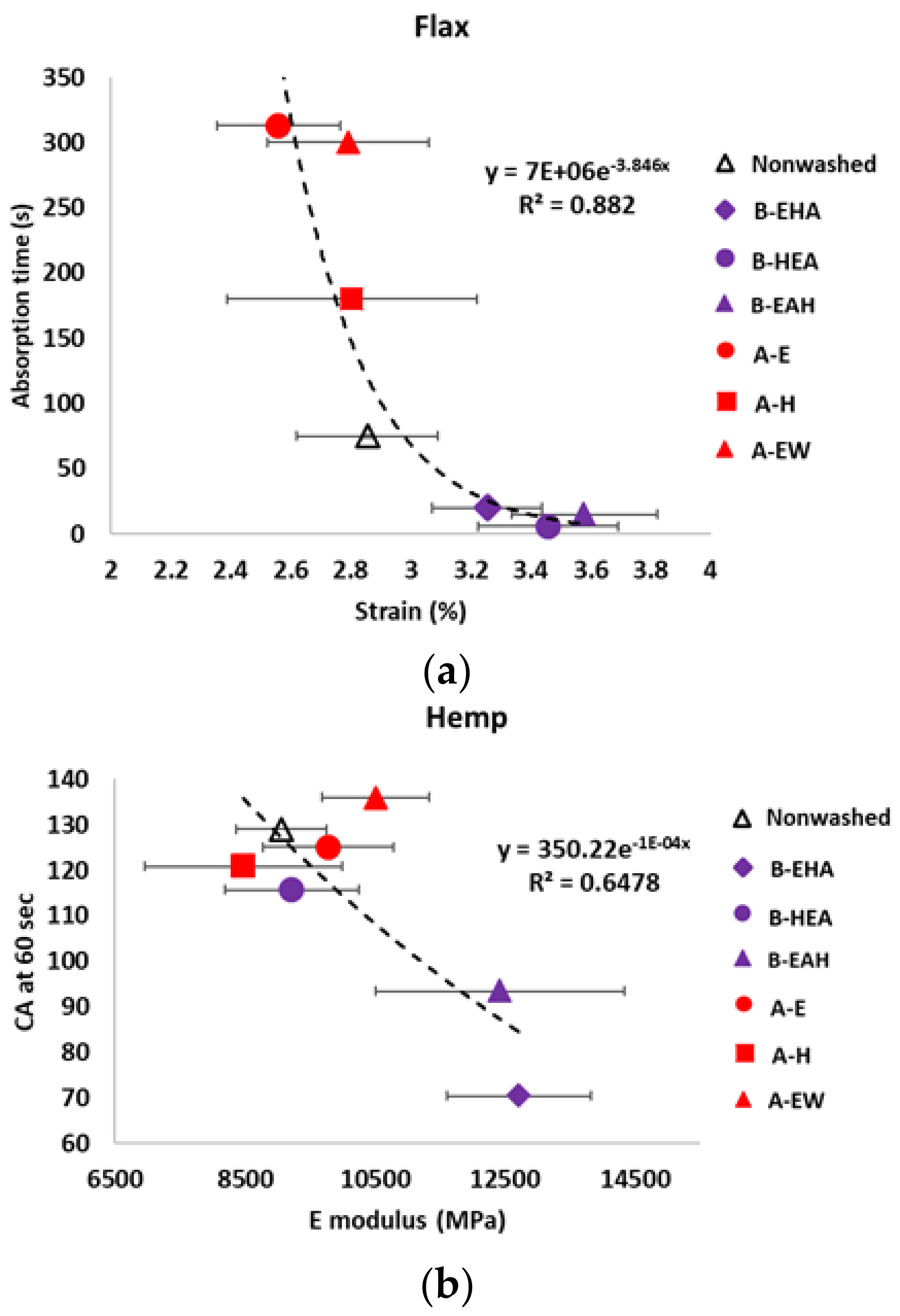
3.3. Wettability Characterization
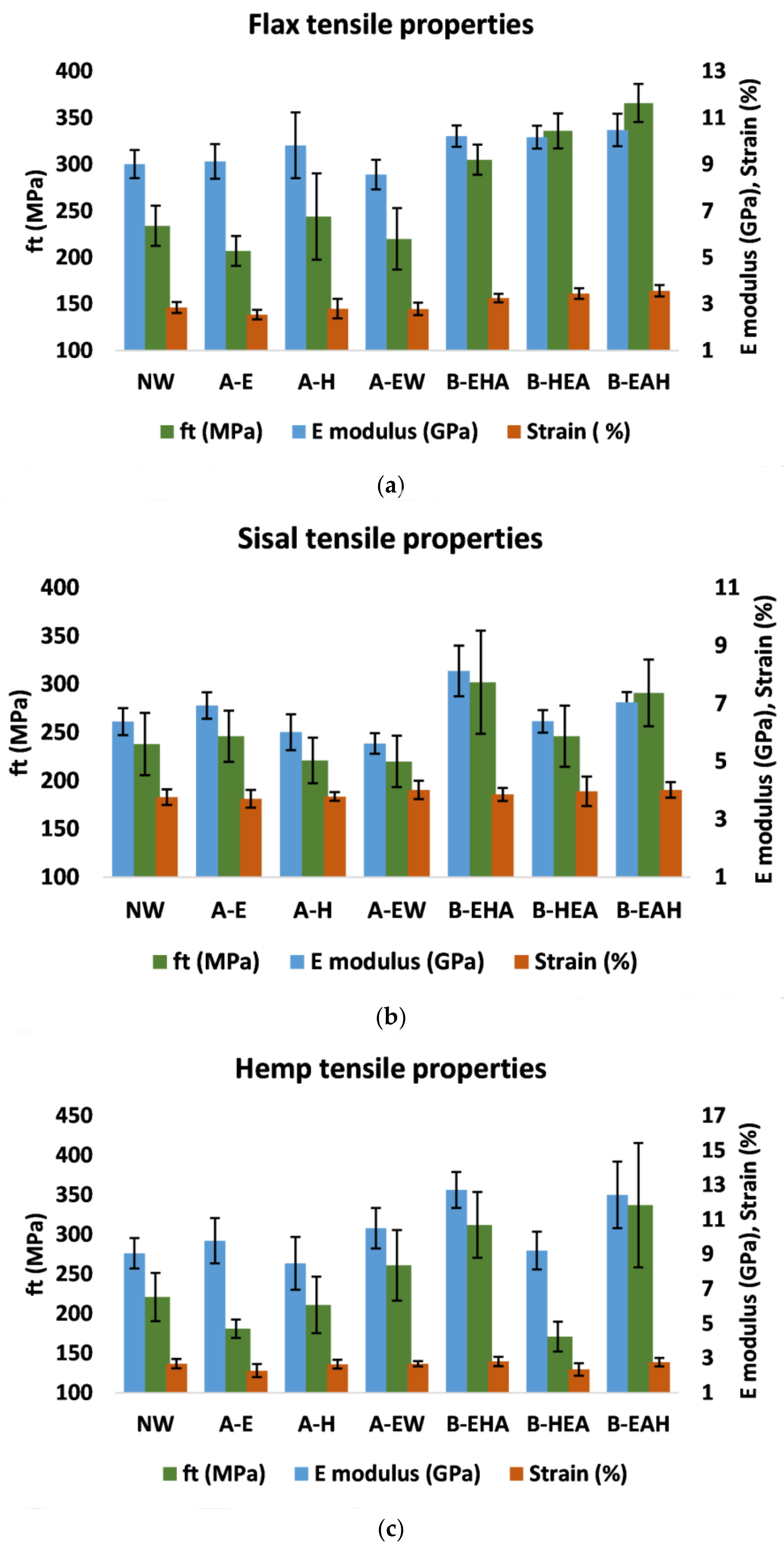
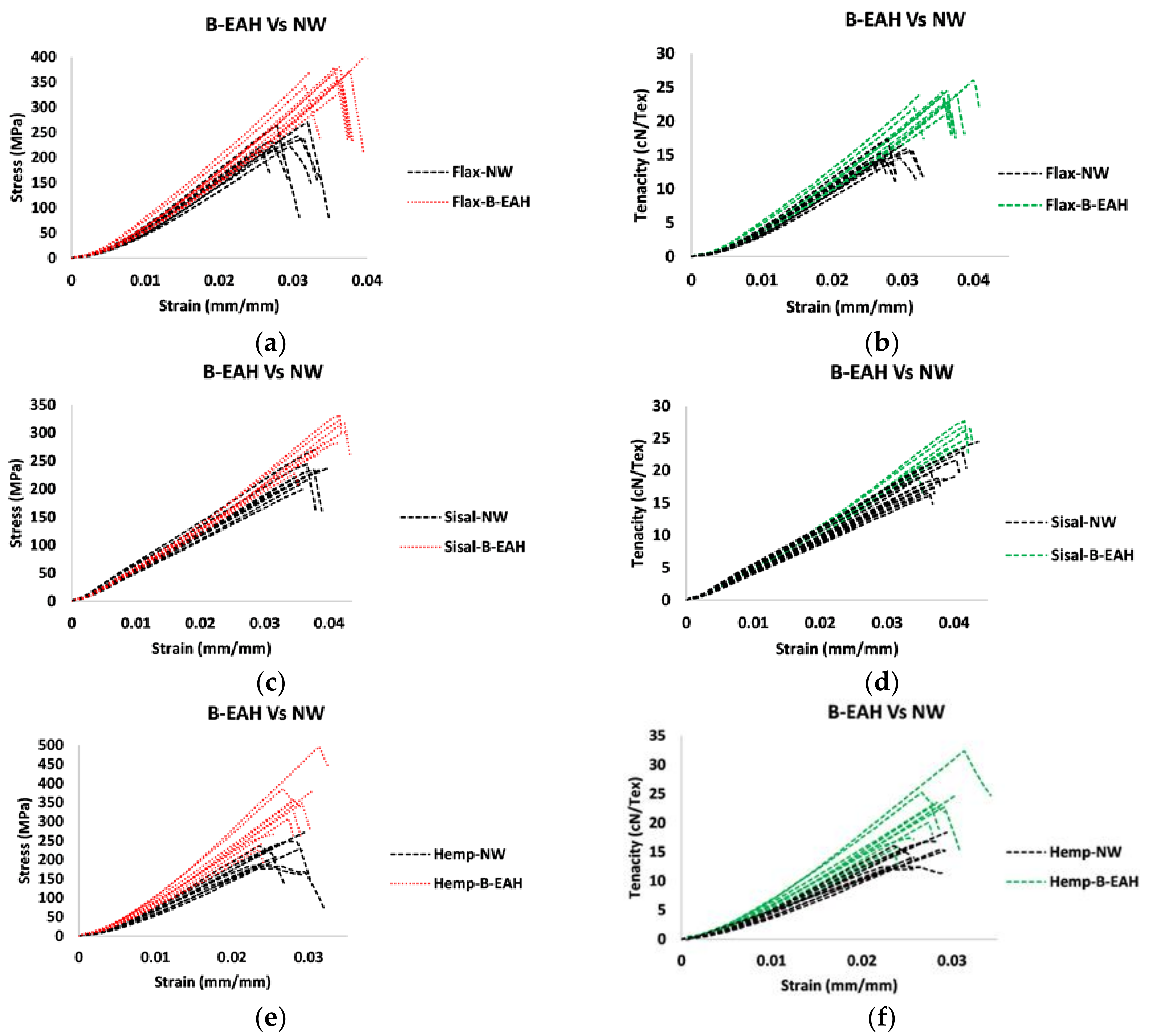
3.4. Tensile Properties Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pickering, K.L.; Efendy, M.G.A.; Le, T.M. A review of recent developments in natural fibre composites and their mechanical performance. Compos. Part A Appl. Sci. Manuf. 2016, 83, 98–112. [Google Scholar] [CrossRef] [Green Version]
- Abbass, A.; Lourenço, P.B.; Oliveira, D.V. The use of natural fibers in repairing and strengthening of cultural heritage buildings. Mater. Today Proc. 2020, 31, S321–S328. [Google Scholar] [CrossRef]
- Wei, J.; Meyer, C. Degradation mechanisms of natural fiber in the matrix of cement composites. Cem. Concr. Res. 2015, 73, 1–16. [Google Scholar] [CrossRef]
- De Almeida, J.; Filho, M.; De Andrade, F.; Dias, R.; Filho, T. Degradation kinetics and aging mechanisms on sisal fiber cement composite systems. Cem. Concr. Compos. 2013, 40, 30–39. [Google Scholar] [CrossRef]
- Claramunt, J.; Ardanuy, M.; García-Hortal, J.A.; Filho, R.D.T. The hornification of vegetable fibers to improve the durability of cement mortar composites. Cem. Concr. Compos. 2011, 33, 586–595. [Google Scholar] [CrossRef]
- de Carvalho Bello, C.B.; Cecchi, A. Experiments on natural fibers: Durability and mechanical properties. Adv. Mater. Process. Technol. 2017, 3, 632–639. [Google Scholar] [CrossRef]
- Wei, J. Degradation behavior and kinetics of sisal fiber in pore solutions of sustainable cementitious composite containing metakaolin. Polym. Degrad. Stab. 2018, 150, 1–12. [Google Scholar] [CrossRef]
- Filho, R.D.T.; Scrivener, K.; England, G.L.; Ghavami, K. Durability of alkali-sensitive sisal and coconut ® bres in cement mortar composites. Cem. Concr. Compos. 2000, 22, 127–143. [Google Scholar] [CrossRef]
- Grilo, J.; Silva, A.S.; Faria, P.; Gameiro, A.; Veiga, R.; Velosa, A. Mechanical and mineralogical properties of natural hydraulic lime-metakaolin mortars in different curing conditions. Constr. Build. Mater. 2014, 51, 287–294. [Google Scholar] [CrossRef]
- Arizzi, A.; Cultrone, G. Aerial lime-based mortars blended with a pozzolanic additive and different admixtures: A mineralogical, textural and physical-mechanical study. Constr. Build. Mater. 2012, 31, 135–143. [Google Scholar] [CrossRef]
- Sawpan, M.A.; Pickering, K.L.; Fernyhough, A. Effect of various chemical treatments on the fibre structure and tensile properties of industrial hemp fibres. Compos. Part A Appl. Sci. Manuf. 2011, 42, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Lv, N.; Wang, X.; Peng, S.; Zhang, H.; Luo, L. Study of the Kinetics and Equilibrium of the Adsorption of Oils onto Hydrophobic Jute Fiber Modified via the Sol-Gel Method. Int. J. Environ. Res. Public Health 2018, 15, 969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, A.; Fan, X.; Wang, Q.; Yu, Y.; Cavaco-Paulo, A. Hydrophobic surface functionalization of lignocellulosic jute fabrics by enzymatic grafting of octadecylamine. Int. J. Biol. Macromol. 2015, 79, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Dong, A.; Fan, X.; Yu, Y.; Yuan, J.; Wang, P.; Wang, Q.; Cavaco-Paulo, A. Enzymatic Hydrophobic Modification of Jute Fibers via Grafting to Reinforce Composites. Appl. Biochem. Biotechnol. 2016, 178, 1612–1629. [Google Scholar] [CrossRef] [Green Version]
- Kudanga, T.; Prasetyo, E.N.; Sipilä, J.; Guebitz, G.M.; Nyanhongo, G.S. Reactivity of long chain alkylamines to lignin moieties: Implications on hydrophobicity of lignocellulose materials. J. Biotechnol. 2010, 149, 81–87. [Google Scholar] [CrossRef]
- Wang, K.; Dong, Y.; Zhang, W.; Zhang, S.; Li, J. Preparation of Stable Superhydrophobic Coatings on Wood Substrate Surfaces via Mussel-Inspired. Polymers 2017, 9, 218. [Google Scholar] [CrossRef] [Green Version]
- Dong, A.; Yu, Y.; Yuan, J.; Wang, Q.; Fan, X. Hydrophobic modification of jute fiber used for composite reinforcement via laccase-mediated grafting. Appl. Surf. Sci. 2014, 301, 418–427. [Google Scholar] [CrossRef]
- Amiri, A.; Triplett, Z.; Moreira, A.; Brezinka, N.; Alcock, M.; Ulven, C.A. Standard density measurement method development for flax fiber. Ind. Crop. Prod. 2017, 96, 196–202. [Google Scholar] [CrossRef]
- Kandemir, A.; Pozegic, T.R.; Hamerton, I.; Eichhorn, S.J.; Longana, M.L. Characterisation of Natural Fibres for Sustainable Discontinuous Fibre Composite Materials. Materials 2020, 13, 2129. [Google Scholar] [CrossRef] [PubMed]
- Truong, M.; Zhong, W.; Boyko, S.; Alcock, M. A comparative study on natural fibre density measurement. J. Text. Inst. 2009, 100, 525–529. [Google Scholar] [CrossRef]
- Le Gall, M.; Davies, P.; Martin, N.; Baley, C. Recommended flax fibre density values for composite property predictions. Ind. Crop. Prod. 2018, 114, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.; Mouret, N.; Davies, P.; Baley, C. Influence of the degree of retting of flax fibers on the tensile properties of single fibers and short fiber/polypropylene composites. Ind. Crop. Prod. 2013, 49, 755–767. [Google Scholar] [CrossRef] [Green Version]
- B.S.I. Part 5: Determination of Linear Density of Yarn Removed from Fabric; 07211-8-1984 (2002) ISO; 1984. Available online: http://www.bsi-global.com (accessed on 25 May 2021).
- American National Standard. Dealing with Outlying Observations, E 178–02. In Annual Book of ASTM Standards; ASTM International: West Conshohocken, PA, USA, 2002. [Google Scholar]
- Advisory Committee on Technical Recommendations for Construction. Guide for the Design and Construction of Externally Bonded FRP Systems for Strengthening Existing Structures; CNR: Rome, Italy, 2014. [Google Scholar]
- Codispoti, R.; Oliveira, D.V.; Olivito, R.S.; Lourenço, P.B.; Fangueiro, R. Mechanical performance of natural fiber-reinforced composites for the strengthening of masonry. Compos. Part B Eng. 2015, 77, 74–83. [Google Scholar] [CrossRef]
- Traoré, M.; Kaal, J.; Cortizas, A.M. Application of FTIR spectroscopy to the characterization of archeological wood. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 153, 63–70. [Google Scholar] [CrossRef] [PubMed]
- TCI/24 Technical Committee. Determination of Single-End Breaking Force and Elongation at Break. EN ISO 2062:2009. Available online: https://www.iso.org/standard/45642.html (accessed on 25 May 2021).
- Koutsianitis, D.; Mitani, C.; Giagli, K.; Tsalagkas, D.; Halász, K.; Kolonics, O.; Gallis, C.; Csóka, L. Properties of ultrasound extracted bicomponent lignocellulose thin films. Ultrason. Sonochemistry 2015, 23, 148–155. [Google Scholar] [CrossRef]
- Ferreira, S.R.; Silva, F.D.A.; Lima, P.R.L.; Filho, R.D.T. Effect of fiber treatments on the sisal fiber properties and fiber-matrix bond in cement based systems. Constr. Build. Mater. 2015, 101, 730–740. [Google Scholar] [CrossRef]
- Pucci, M.F.; Liotier, P.J.; Drapier, S. Capillary wicking in flax fabrics—Effects of swelling in water. Colloids Surf. A Physicochem. Eng. Asp. 2016, 498, 176–184. [Google Scholar] [CrossRef]
- Baley, C. Analysis of the flax fibres tensile behaviour and analysis of the tensile stiffness increase. Compos. Part A Appl. Sci. Manuf. 2002, 33, 939–948. [Google Scholar] [CrossRef]
- Ferreira, S.R.; de Andrade Silva, F.; Lima, P.R.L.; Filho, R.D.T. Effect of hornification on the structure, tensile behavior and fiber matrix bond of sisal, jute and curauá fiber cement based composite systems. Constr. Build. Mater. 2017, 139, 551–561. [Google Scholar] [CrossRef]
- Schwanninger, M.; Rodrigues, J.C.; Pereira, H.; Hinterstoisser, B. Effects of short-time vibratory ball milling on the shape of FT-IR spectra of wood and cellulose. Vib. Spectrosc. 2004, 36, 23–40. [Google Scholar] [CrossRef]
- Poletto, M.; Zattera, A.J.; Santana, R.M.C. Structural Differences Between Wood Species: Evidence from Chemical Composition, FTIR Spectroscopy, and Thermogravimetric Analysis. J. Appl. Polym. Sci. 2012, 126, E336–E343. [Google Scholar] [CrossRef]
- Popescu, M.; Popescu, C.; Lisa, G.; Sakata, Y. Evaluation of morphological and chemical aspects of different wood species by spectroscopy and thermal methods. J. Mol. Struct. 2011, 988, 65–72. [Google Scholar] [CrossRef]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Shen, T.; Gnanakaran, S. The stability of cellulose: A statistical perspective from a coarse-grained model of hydrogen-bond networks. Biophys. J. 2009, 96, 3032–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, K.N.; Pang, S.; Staiger, M.P. Ionic Liquids and Their Interaction with Cellulose. Am. Chem. Soc. 2009, 109, 6712–6728. [Google Scholar] [CrossRef]
- El Oudiani, A.; Msahli, S.; Sakli, F. In-depth study of agave fiber structure using Fourier transform infrared spectroscopy. Carbohydr. Polym. 2017, 164, 242–248. [Google Scholar] [CrossRef]
- HS, M.; RK, M. Effect of Microorganism on Flax and Linen. J. Text. Sci. Eng. 2015, 6, 229. [Google Scholar] [CrossRef]
- Gonzalez-lopez, L.; Claramunt, J.; Hsieh, Y.; Ventura, H. Surface modification of flax nonwovens for the development of sustainable, high performance, and durable calcium aluminate cement composites. Compos. Part B 2020, 191, 107955. [Google Scholar] [CrossRef]
- Ornaghi, H.L., Jr.; Poletto, M.; Jose, A. Correlation of the thermal stability and the decomposition kinetics of six different vegetal fibers. Cellulose 2014, 21, 177–188. [Google Scholar] [CrossRef]
- Poletto, M.; Pistor, V.; Marlene, R.; Santana, C.; José, A. Materials Produced From Plant Biomass: Part II: Evaluation of Crystallinity and Degradation Kinetics of Cellulose. Mater. Res. 2012, 15, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Feng, Y.; He, L. Effect of thermal treatment on properties of ramie fibers. Polym. Degrad. Stab. 2016, 133, 303–311. [Google Scholar] [CrossRef]
- Cichosz, S.; Masek, A. IR study on cellulose with the varied moisture contents: Insight into the supramolecular structure. Materials 2020, 13, 4573. [Google Scholar] [CrossRef]
- Kim, J.T.; Netravali, A.N. Mercerization of sisal fibers: Effect of tension on mechanical properties of sisal fiber and fiber-reinforced composites. Compos. Part A 2010, 41, 1245–1252. [Google Scholar] [CrossRef]
- Zhang, H.; Ming, R.; Yang, G.; Li, Y.; Li, Q.; Shao, H. Influence of Alkali Treatment on Flax Fiber for Use as Reinforcements in Polylactide Stereocomplex Composites. Polym. Eng. Sci. 2015, 55, 2553–2558. [Google Scholar] [CrossRef]
- Lionetto, F.; Del Sole, R.; Cannoletta, D.; Vasapollo, G.; Maffezzoli, A. Monitoring Wood Degradation during Weathering by Cellulose Crystallinity. Materials 2012, 5, 1910–1922. [Google Scholar] [CrossRef] [Green Version]
- Morán, J.I.; Alvarez, V.A.; Cyras, V.P.; Vázquez, A. Extraction of cellulose and preparation of nanocellulose from sisal fibers. Cellulose 2008, 15, 149–159. [Google Scholar] [CrossRef]
- Zhuang, J.; Li, M.; Pu, Y.; Ragauskas, A.J.; Yoo, C.G. Observation of Potential Contaminants in Processed Biomass Using Fourier Transform Infrared Spectroscopy. Appl. Sci 2020, 10, 4345. [Google Scholar] [CrossRef]
- Makarem, M.; Lee, C.M.; Kafle, K.; Huang, S.; Chae, I.; Yang, H.; Kubicki, J.D.; Kim, S.H. Probing cellulose structures with vibrational spectroscopy. Cellulose 2019, 26, 35–79. [Google Scholar] [CrossRef]
- Popescu, C.; Singurel, G.; Popescu, M.; Vasile, C.; Dimitris, S.; Willför, S. Vibrational spectroscopy and X-ray diffraction methods to establish the differences between hardwood and softwood. Carbohydr. Polym. 2009, 77, 851–857. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D., II; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, I.; Pogorzelec-glaser, K.; Tritt-goc, J. Cellulose microfibers surface treated with imidazole as new proton conductors. Mater. Chem. Phys. 2020, 239, 122056. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, I.; Shin, Y.; Kim, C.; Kim, Y. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef] [PubMed]
- Gominho, J.; Costa, R.; Lourenço, A.; Neiva, D.M.; Pereira, H. The effect of different pre-treatments to improve delignification of eucalypt stumps in a biorefinery context. Bioresour. Technol. Reports J. 2019, 6, 89–95. [Google Scholar] [CrossRef]
- Alix, S.; Philippe, E.; Bessadok, A.; Lebrun, L.; Morvan, C.; Marais, S. Effect of chemical treatments on water sorption and mechanical properties of flax fibres. Bioresour. Technol. 2009, 100, 4742–4749. [Google Scholar] [CrossRef]
- Sinha, E.; Rout, S.K. Influence of fibre-surface treatment on structural, thermal and mechanical properties of jute. J. Mater. Sci. 2008, 43, 2590–2601. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Široký, J.; Blackburn, R.S.; Bechtold, T.; Taylor, J.; White, P. Attenuated total reflectance Fourier-transform Infrared spectroscopy analysis of crystallinity changes in lyocell following continuous treatment with sodium hydroxide. Cellulose 2010, 17, 103–115. [Google Scholar] [CrossRef]
- Spiridon, I.; Teacă, C.-A.; Bodîrlău, R. Structural changes evidenced by FTIR spectroscopy in cellulosic materials after pre-treatment with ionic liquid and enzymatic hydrolysis. BioResources 2011, 6, 400–413. [Google Scholar] [CrossRef]
- Hospodarova, V.; Singovszka, E.; Stevulova, N. Characterization of Cellulosic Fibers by FTIR Spectroscopy for Their Further Implementation to Building Materials. Am. J. Anal. Chem. 2018, 9, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Nada, A.M.A.; Kamel, S.; El-sakhawy, M. Thermal behaviour and infrared spectroscopy of cellulose carbamates. Polym. Degrad. Stab. 2000, 70, 347–355. [Google Scholar] [CrossRef]
- Lazić, B.D.; Pejić, B.M.; Kramar, A.D.; Vukčević, M.M.; Mihajlovski, K.R.; Rusmirović, J.D.; Kostić, M.M. Influence of hemicelluloses and lignin content on structure and sorption properties of flax fibers (Linum usitatissimum L.). Cellulose 2018, 25, 697–709. [Google Scholar] [CrossRef]
- George, M.; Mussone, P.G.; Abboud, Z.; Bressler, D.C. Characterization of chemically and enzymatically treated hemp fibres using atomic force microscopy and spectroscopy. Appl. Surf. Sci. 2014, 314, 1019–1025. [Google Scholar] [CrossRef]
- Lazic, B.D.; Janjic, S.D.; Rijavec, T.; Kostic, M.M. Effect of chemical treatments on the chemical composition and properties of flax fibers. J. Serbian Chem. Soc. 2017, 82, 83–97. [Google Scholar] [CrossRef]
- Corbin, A.C.; Soulat, D.; Ferreira, M.; Labanieh, A.R.; Gabrion, X.; Placet, V. Multi-scale analysis of flax fibres woven fabrics for composite applications Multi-scale analysis of flax fibres woven fabrics for composite applications. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Milan, Italy, 17–19 September 2018; Volume 406, p. 012016. Available online: https://0-iopscience-iop-org.brum.beds.ac.uk/article/10.1088/1757-899X/406/1/012016 (accessed on 25 May 2021). [CrossRef] [Green Version]
- Bledzki, A.K.; Mamun, A.A.; Lucka-Gabor, M.; Gutowski, V.S. The effects of acetylation on properties of flax fibre and its polypropylene composites. Express Polym. Lett. 2008, 2, 413–422. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Protocol ID | Time-Solvent-Stir |
|---|---|---|
| A | A-E | 1 h-ethanol + ultrapure water rinsing-no stir |
| A | A-H | 45 min-hexane + ultrapure water rinsing-no stir |
| A | A-EW | 1 h-ethanol/water: 1:2 v/v + ultrapure water rinsing-no stir |
| B | B-EHA | 1 h (20 min each)-ethanol/hexane/acetone-ultrasonication |
| B | B-HEA | 1 h (20 min each)-hexane/ethanol/acetone-ultrasonication |
| B | B-EAH | 1 h (20 min each)-ethanol/acetone/hexane-ultrasonication |
| Fiber | ID | Density (g/cm3) | Density (SD) | Area (mm2) | Area (SD) |
|---|---|---|---|---|---|
| Flax | NW | 1.523 | 0.076 | 0.985 | 0.049 |
| A-E | 1.508 | 0.030 | 0.942 | 0.019 | |
| A-H | 1.535 | 0.090 | 0.885 | 0.051 | |
| A-EW | 1.467 | 0.025 | 0.941 | 0.017 | |
| B-EHA | 1.463 | 0.029 | 1.085 | 0.022 | |
| B-HEA | 1.469 | 0.020 | 1.039 | 0.014 | |
| B-EAH | 1.558 | 0.128 | 0.971 | 0.080 | |
| Sisal | NW | 1.248 | 0.032 | 3.928 | 0.101 |
| A-E | 1.249 | 0.019 | 2.755 | 0.041 | |
| A-H | 1.168 | 0.003 | 3.152 | 0.008 | |
| A-EW | 1.275 | 0.061 | 3.358 | 0.159 | |
| B-EHA | 1.197 | 0.015 | 2.929 | 0.036 | |
| B-HEA | 1.198 | 0.033 | 2.952 | 0.08 | |
| B-EAH | 1.197 | 0.031 | 3.530 | 0.0919 | |
| Hemp | NW | 1.473 | 0.033 | 0.752 | 0.004 |
| A-E | 1.501 | 0.017 | 0.599 | 0.004 | |
| A-H | 1.474 | 0.015 | 0.755 | 0.004 | |
| A-EW | 1.684 | 0.173 | 0.556 | 0.007 | |
| B-EHA | 1.480 | 0.009 | 0.629 | 0.008 | |
| B-HEA | 1.471 | 0.009 | 0.639 | 0.021 | |
| B-EAH | 1.530 | 0.055 | 0.586 | 0.055 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbass, A.; Paiva, M.C.; Oliveira, D.V.; Lourenço, P.B.; Fangueiro, R. Insight into the Effects of Solvent Treatment of Natural Fibers Prior to Structural Composite Casting: Chemical, Physical and Mechanical Evaluation. Fibers 2021, 9, 54. https://0-doi-org.brum.beds.ac.uk/10.3390/fib9090054
Abbass A, Paiva MC, Oliveira DV, Lourenço PB, Fangueiro R. Insight into the Effects of Solvent Treatment of Natural Fibers Prior to Structural Composite Casting: Chemical, Physical and Mechanical Evaluation. Fibers. 2021; 9(9):54. https://0-doi-org.brum.beds.ac.uk/10.3390/fib9090054
Chicago/Turabian StyleAbbass, Ali, Maria C. Paiva, Daniel V. Oliveira, Paulo B. Lourenço, and Raul Fangueiro. 2021. "Insight into the Effects of Solvent Treatment of Natural Fibers Prior to Structural Composite Casting: Chemical, Physical and Mechanical Evaluation" Fibers 9, no. 9: 54. https://0-doi-org.brum.beds.ac.uk/10.3390/fib9090054