
Identification of Five Hub Genes as Key Prognostic Biomarkers in Liver Cancer via Integrated Bioinformatics Analysis

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Collection
2.2. Datasets Preprocessing and Differential Gene Expression Analysis
2.3. Weighted Gene Co-Expression Network Construction
2.4. Module–Trait Relationship Analysis of Liver Cancer
2.5. Function Enrichment Analysis
2.6. Gene Regulatory Network
2.7. Protein–Protein Interaction Network Construction
2.8. Methylation Analysis
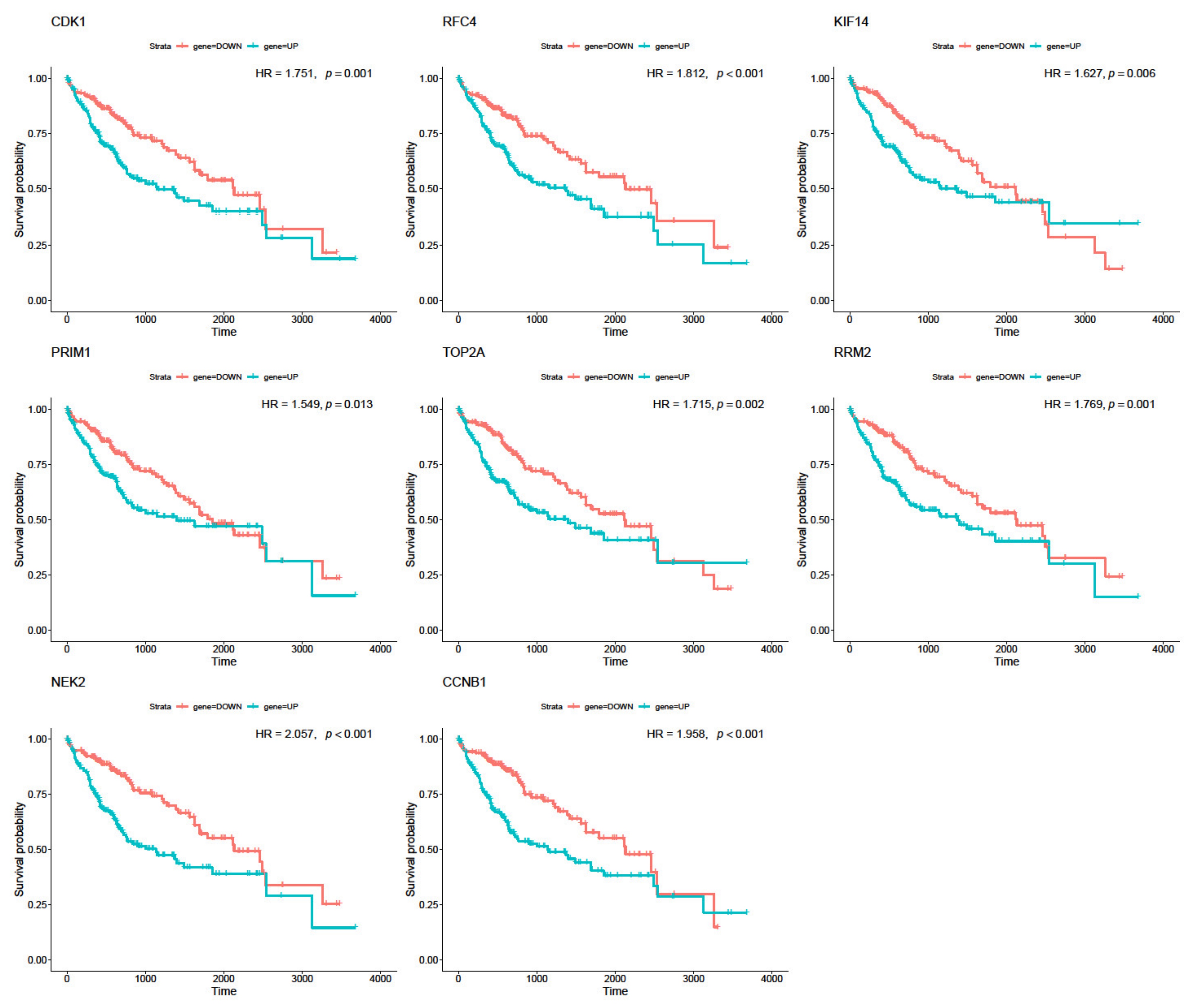
2.9. Survival Analysis
2.10. The Protein Expressions of the Prognostic Hub Genes
2.11. Gene–Drug Interaction Analysis
3. Results
3.1. Key Modules Identification by Weighted Gene Co-Expression Network
3.2. Identification of Hub Genes through Gene Regulatory Networks
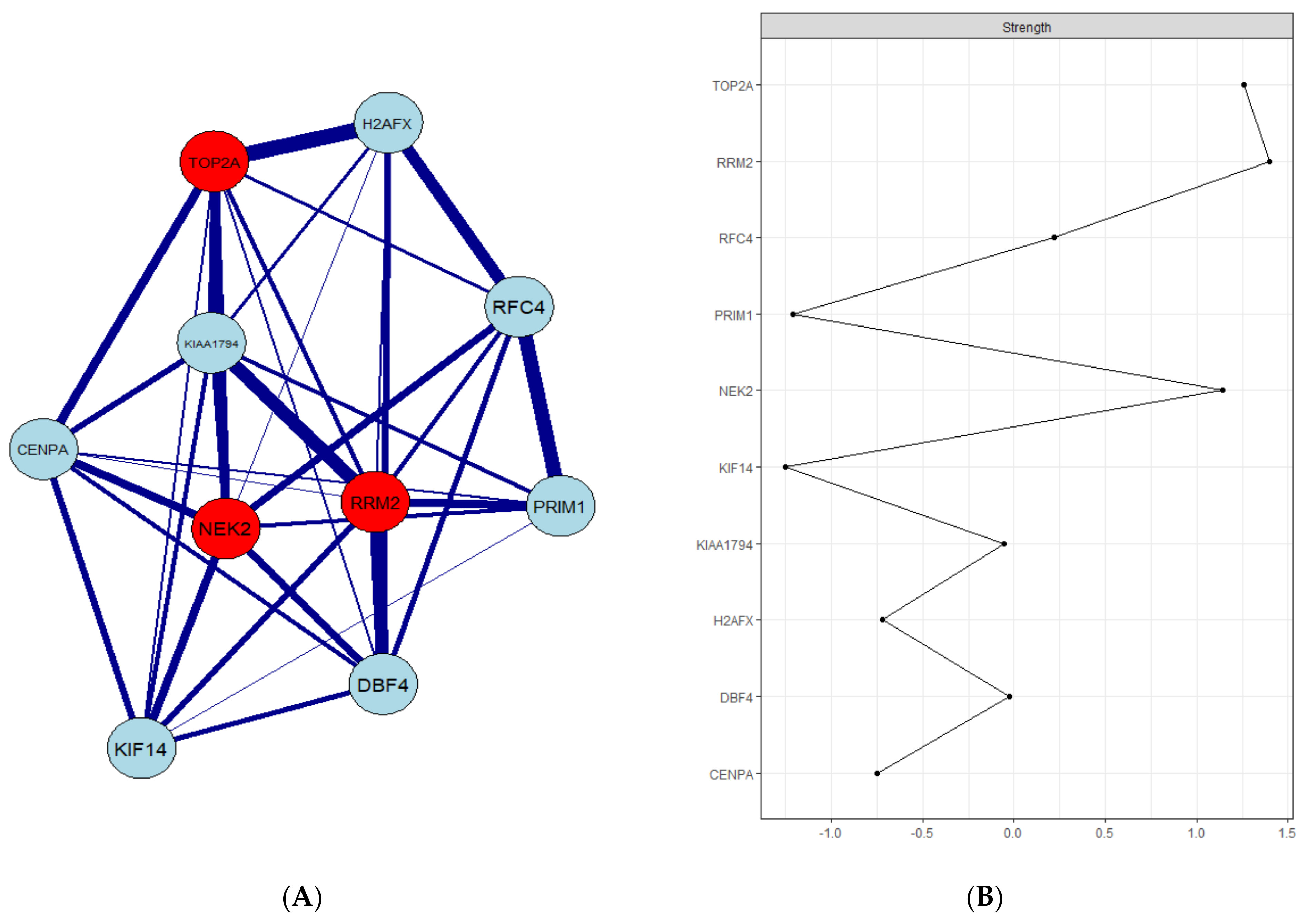
3.3. PPI Network Construction and Hub Gene Validation
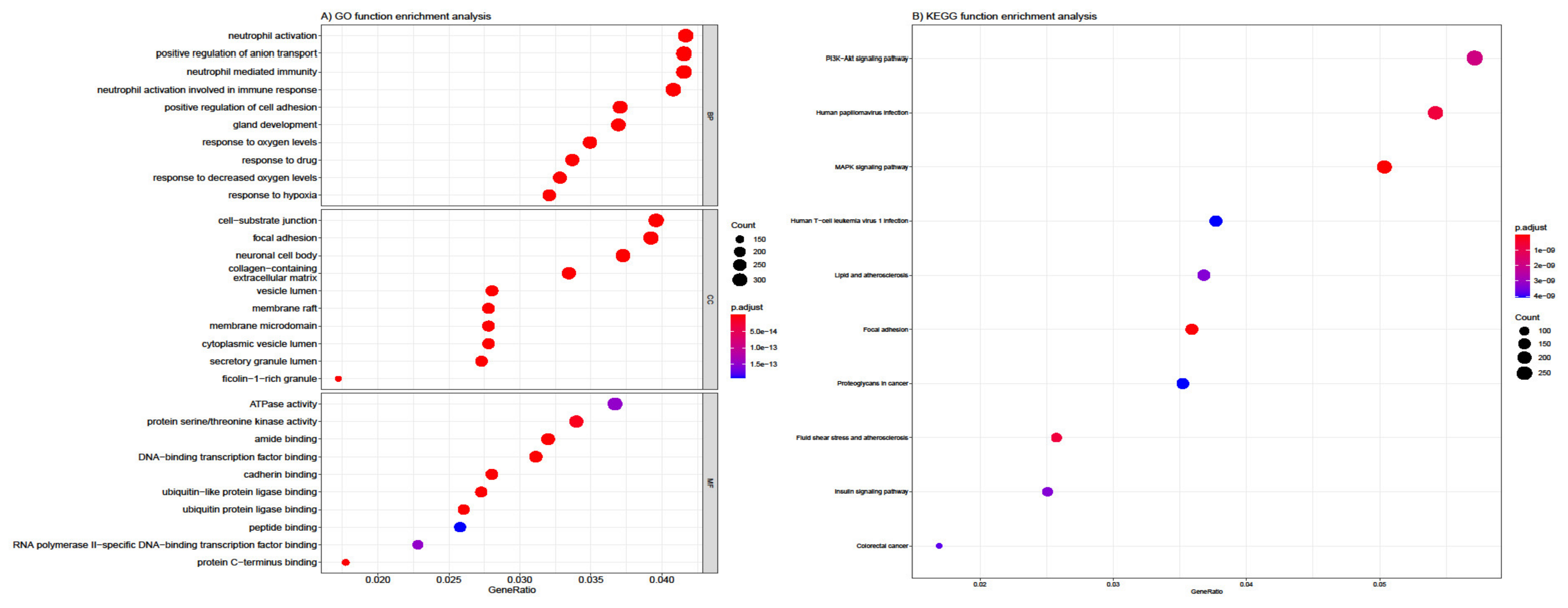
3.4. Functional and Pathway Enrichment Analysis
3.5. Real Hub Genes Identification and Validation
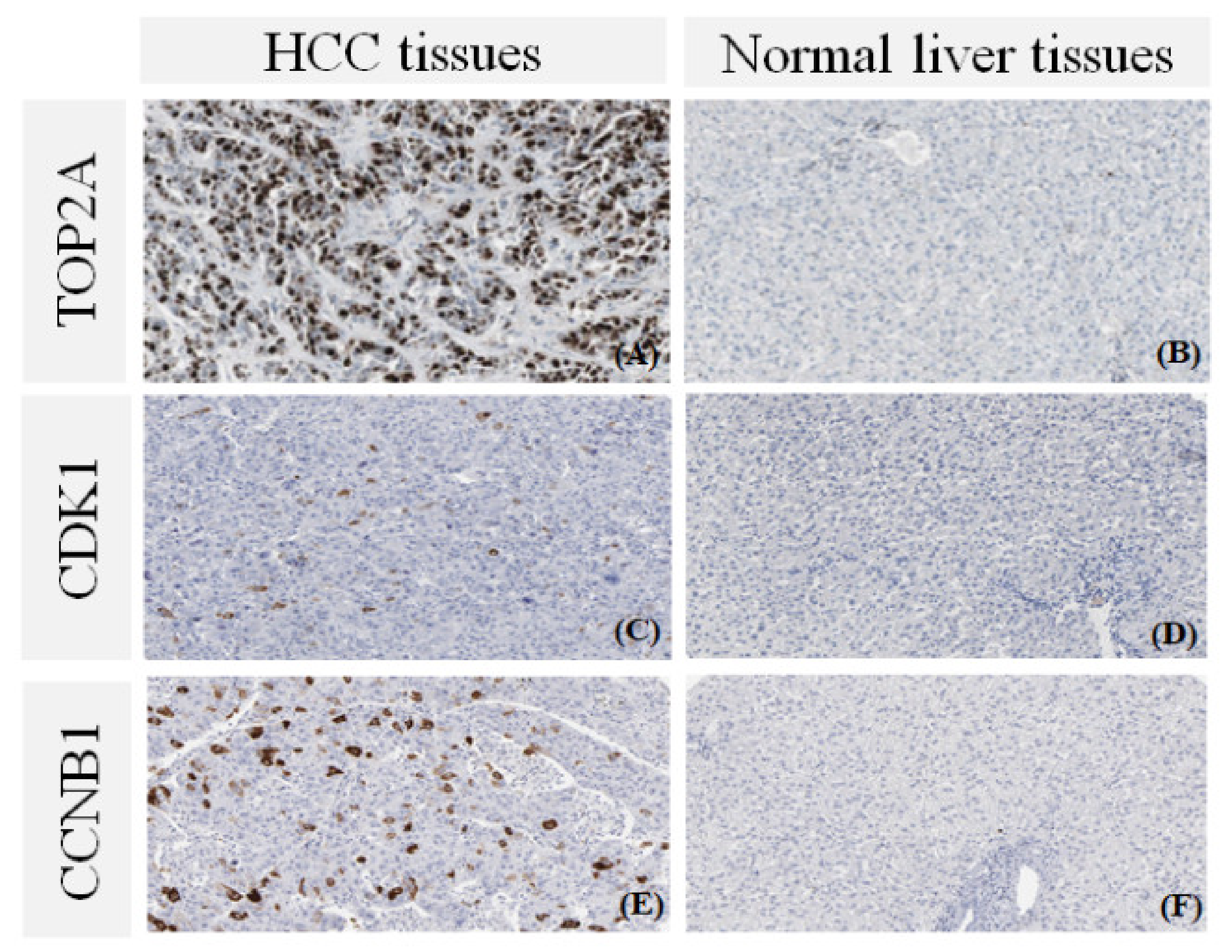
3.6. The Protein Expression of Hub Genes
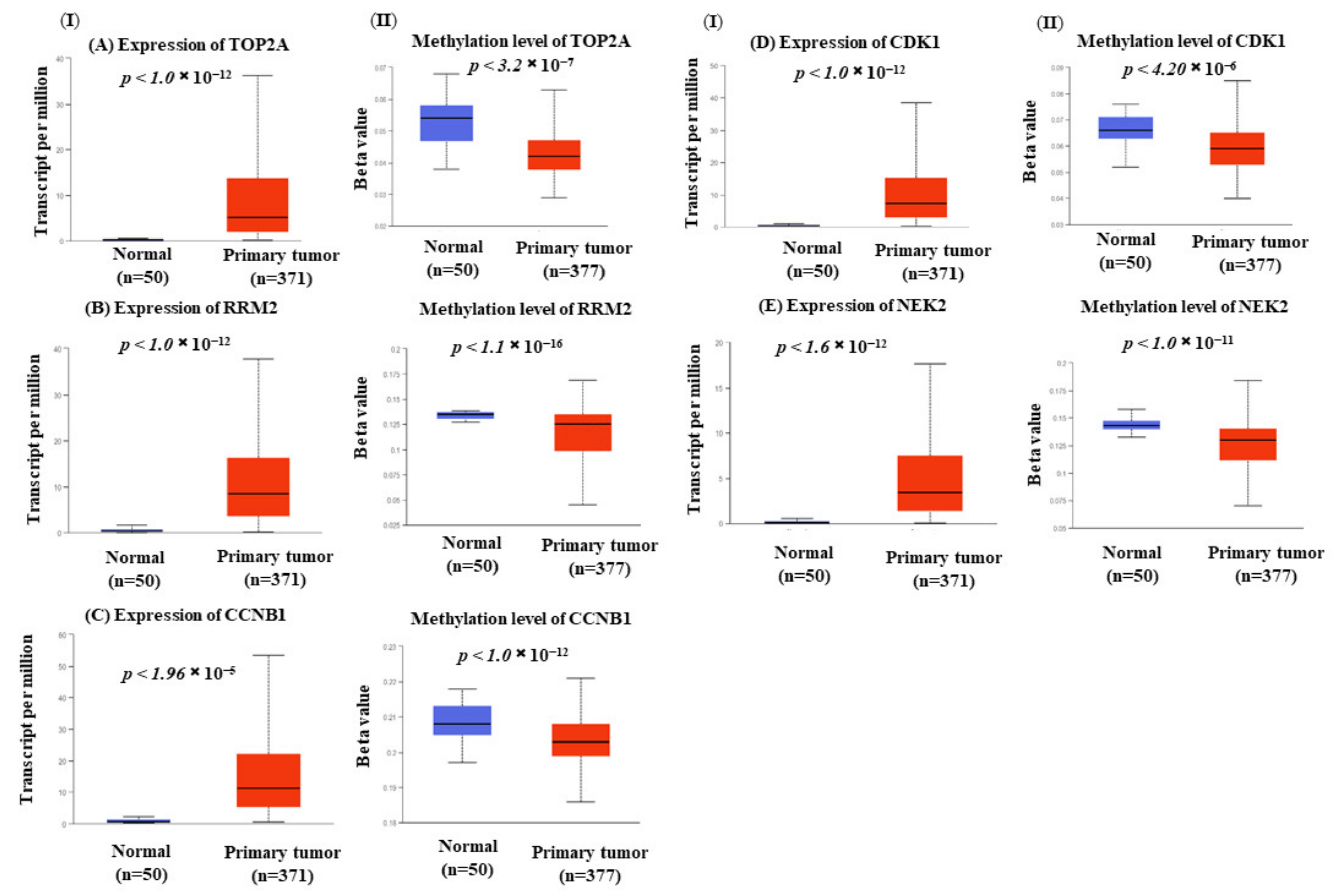
3.7. Hub Genes Expression Is Correlated with Methylation
3.8. Gene–Drug Interaction Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.; Ji, Z.; Quan, Y.; Zhan, M.; Wang, H.; Li, W.; Hua, S.; Ji, Z.; Quan, Y.; Zhan, M.; et al. Identification of hub genes in hepatocellular carcinoma using integrated bioinformatic analysis. Aging (Albany. NY) 2020, 12, 5439–5468. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Long, J.; Liu, Z.; Lin, J.; Huang, H.; Wang, D.; Yang, X.; Miao, F.; Mao, Y.; Sang, X.; et al. Comprehensive analysis of a ceRNA network reveals potential prognostic cytoplasmic lncRNAs involved in HCC progression. J. Cell Physiol. 2019, 234, 18837–18848. [Google Scholar] [CrossRef] [Green Version]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]
- Adler, A.S.; Chang, H.Y. From description to causality: Mechanisms of gene expression signatures in cancer. Cell Cycle 2006, 5, 1148–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Ljungberg, B.; Grankvist, K.; Rasmuson, T.; Tibshirani, R.; Brooks, J.D. Gene expression profiling predicts survival in conventional renal cell carcinoma. PLoS Med. 2005, 3, e13. [Google Scholar] [CrossRef]
- Spentzos, D.; Levine, D.A.; Ramoni, M.F.; Joseph, M.; Gu, X.; Boyd, J.; Libermann, T.A.; Cannistra, S.A. Gene expression signature with independent prognostic significance in epithelial ovarian cancer. J. Clin. Oncol. 2004, 22, 4700–4710. [Google Scholar] [CrossRef]
- Bullinger, L.; Döhner, K.; Bair, E.; Fröhling, S.; Schlenk, R.F.; Tibshirani, R.; Döhner, H.; Pollack, J.R. Use of gene-expression profiling to identify prognostic subclasses in adult acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1605–1616. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Xiao, H.; Guo, S.; Dong, L.; Chen, J. Identification of breast cancer mechanism based on weighted gene coexpression network analysis. Cancer Gene Ther. 2017, 24, 333–341. [Google Scholar] [CrossRef]
- Roessler, S.; Jia, H.-L.; Budhu, A.; Forgues, M.; Ye, Q.-H.; Lee, J.-S.; Thorgeirsson, S.S.; Sun, Z.; Tang, Z.-Y.; Qin, L.-X.; et al. A unique metastasis gene signature enables prediction of tumor relapse in early-stage hepatocellular carcinoma patients. Cancer Res. 2010, 70, 10202–10212. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Li, K.; Zhang, Y. Identification of modules and novel prognostic biomarkers in liver cancer through integrated bioinformatics analysis. FEBS Open Bio 2020, 10, 2388–2403. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Churchill, G.A. Statistical tests for differential expression in cDNA microarray experiments. Genome Biol. 2003, 4, 210. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma. powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- Ai, D.; Wang, Y.; Li, X.; Pan, H. Colorectal Cancer Prediction Based on Weighted Gene Co-Expression Network Analysis and Variational Auto-Encoder. Biomolecules 2020, 10, 1207. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining clusters from a hierarchical cluster tree: The dynamic tree cut package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Pan, J.; Xin, Y.; Mi, X.; Wang, J.; Gao, Q.; Luo, H. Gene expression analysis reveals novel gene signatures between young and old adults in human prefrontal cortex. Front. Aging Neurosci. 2018, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Tian, G.-Y.; Song, Y.; Liu, Y.-L.; Chen, Y.-D.; Shi, J.-P.; Yang, J. Characterization of transcriptional modules related to fibrosing-NAFLD progression. Sci. Rep. 2017, 7, 4748. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, J.; Liu, S.; Li, W. Identification of crucial genes associated with immune cell infiltration in hepatocellular carcinoma by weighted gene co-expression network analysis. Front. Genet. 2020, 11, 342. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Epskamp, S.; Cramer, A.O.; Waldorp, L.J.; Schmittmann, V.D.; Borsboom, D. qgraph: Network visualizations of relationships in psychometric data. J. Stat. Softw. 2012, 48, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Geng, R.; Li, Z.; Yu, S.; Yuan, C.; Hong, W.; Wang, Z.; Wang, Q.; Yi, Z.; Fang, Y. Weighted gene co-expression network analysis identifies specific modules and hub genes related to subsyndromal symptomatic depression. World. J. Biol. Psychiatry 2020, 21, 102–110. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, G.; Hossain, A.; Danilova, I.G.; Bolkov, M.A.; Liu, G.; Tuzankina, I.A.; Tan, W. A co-expression network for differentially expressed genes in bladder cancer and a risk score model for predicting survival. Hereditas 2019, 156, 24. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978, 1, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Zhou, B.; Chen, J.; Hu, W.; Bai, R.; Ye, C.; Weng, X.; Zheng, S. Significant prognostic values of differentially expressed-aberrantly methylated hub genes in breast cancer. J. Cancer 2019, 10, 6618–6634. [Google Scholar] [CrossRef]
- Foygel, R.; Drton, M. Extended Bayesian information criteria for Gaussian graphical models. Adv. Neural Inform. Process Syst. 2010, 23, 2020–2028. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Sparse inverse covariance estimation with the graphical lasso. Biostatistics 2008, 9, 432–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrat, A.; Barthelemy, M.; Pastor-Satorras, R.; Vespignani, A. The architecture of complex weighted networks. PNAS 2004, 101, 3747–3752. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Kartsonaki, C. Survival analysis. Diagn. Histopathol. 2016, 22, 263–270. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Wang, Q.; Zhang, G.; Sun, F.; Zhang, L.; Li, H.; Li, Y.; Peng, Y.; Zhu, W.; Ji, W.; et al. OSlihc: An online prognostic biomarker analysis tool for hepatocellular carcinoma. Front. Pharmacol. 2020, 11, 875. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Sun, J.; Chen, X.; Liu, L.; Wu, D. Nek2 augments sorafenib resistance by regulating the ubiquitination and localization of β-catenin in hepatocellular carcinoma. J. Exp. Clin. Cancer. Res. 2019, 38, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, G.; Rong, X.; Zhao, J.; Yang, X.; Li, H.; Jiang, H.; Zhou, Q.; Ji, T.; Huang, S.; Zhang, J.; et al. MicroRNA-105 suppresses cell proliferation and inhibits PI3K/AKT signaling in human hepatocellular carcinoma. Carcinogenesis 2014, 35, 2748–2755. [Google Scholar] [CrossRef] [Green Version]
- Moukharskaya, J.; Verschraegen, C. Topoisomerase 1 inhibitors and cancer therapy. Hematol. Oncol. Clin. N. Am. 2012, 26, 507–525. [Google Scholar] [CrossRef]
- Song, H.; Ding, N.; Li, S.; Liao, J.; Xie, A.; Yu, Y.; Zhang, C.; Ni, C. Identification of Hub Genes Associated With Hepatocellular Carcinoma Using Robust Rank Aggregation Combined With Weighted Gene Co-expression Network Analysis. Front. Genet. 2020, 11, 895. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.-M.; Chan, N.-L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef]
- Liu, L.-M.; Xiong, D.-D.; Lin, P.; Yang, H.; Dang, Y.-W.; Chen, G. DNA topoisomerase 1 and 2A function as oncogenes in liver cancer and may be direct targets of nitidine chloride. Int. J. Oncol. 2018, 53, 1897–1912. [Google Scholar] [CrossRef]
- Russo, P.; Del Bufalo, A.; Cesario, A. Flavonoids acting on DNA topoisomerases: Recent advances and future perspectives in cancer therapy. Curr. Med. Chem. 2012, 19, 5287–5293. [Google Scholar] [CrossRef]
- Cowell, I.G.; Okorokov, A.L.; Cutts, S.A.; Padget, K.; Bell, M.; Milner, J.; Austin, C.A. Human topoisomerase IIα and IIβ interact with the C-terminal region of p53. Exp. Cell. Res. 2000, 255, 86–94. [Google Scholar] [CrossRef]
- Wang, Y.; Zhi, Q.; Ye, Q.; Zhou, C.; Zhang, L.; Yan, W.; Wu, Q.; Zhang, D.; Li, P.; Huo, K. SCYL1-BP1 affects cell cycle arrest in human hepatocellular carcinoma cells via cyclin F and RRM2. Anticancer Agents Med. Chem. 2016, 16, 440–446. [Google Scholar] [CrossRef] [PubMed]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.-T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fry, A.M.; O’Regan, L.; Sabir, S.R.; Bayliss, R. Cell cycle regulation by the NEK family of protein kinases. J. Cell Sci. 2012, 125, 4423–4433. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhang, X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle 2016, 15, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Feo, F.; Frau, M.; Pascale, R.M. Interaction of major genes predisposing to hepatocellular carcinoma with genes encoding signal transduction pathways influences tumor phenotype and prognosis. World J. Gastroenterol. 2008, 14, 6601–6615. [Google Scholar] [CrossRef] [PubMed]
- Diril, M.K.; Ratnacaram, C.K.; Padmakumar, V.; Du, T.; Wasser, M.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. PNAS 2012, 109, 3826–3831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qinfeng, H.; Junhong, L.; Ailing, W. Identification of potential therapeutic targets in hepatocellular carcinoma using an integrated bioinformatics approach. Transl. Cancer Res. 2018, 7, 849–858. [Google Scholar]
- Manjang, K.; Tripathi, S.; Yli-Harja, O.; Dehmer, M.; Glazko, G.; Emmert-Streib, F. Prognostic gene expression signatures of breast cancer are lacking a sensible biological meaning. Sci. Rep. 2021, 11, 1–18. [Google Scholar]
- Povedano, E.; Ruiz-Valdepeñas, M.V.; Gamella, M.; Pedrero, M.; Barderas, R.; Peláez-García, A.; Mendiola, M.; Hardisson, D.; Feliú, J.; Yáñez-Sedeño, P.; et al. Amperometric bioplatforms to detect regional DNA methylation with single-base sensitivity. Anal. Chem. 2020, 92, 5604–5612. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, B.; Meng, D.; Ge, C. Comprehensive analysis of DNA methylation and gene expression profiles in cholangiocarcinoma. Cancer Cell. Int. 2019, 19, 352. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Correlation | p-Value | Number of Genes |
|---|---|---|---|
| Black | 0.872 | <0.001 | 656 |
| Blue | −0.104 | 0.028 | 1677 |
| Brown | −0.663 | <0.001 | 2396 |
| Cyan | 0.57 | <0.001 | 220 |
| Dark green | −0.042 | 0.382 | 65 |
| Dark grey | 0.677 | <0.001 | 7105 |
| Dark orange | 0.503 | <0.001 | 58 |
| Dark red | 0.506 | <0.001 | 74 |
| Dark turquoise | 0.303 | <0.001 | 59 |
| Green yellow | 0.4 | <0.001 | 684 |
| Grey | 0.114 | 0.016 | 290 |
| Grey60 | −0.305 | <0.001 | 487 |
| Light cyan | −0.031 | 0.515 | 176 |
| Light green | −0.711 | <0.001 | 153 |
| Light yellow | −0.374 | <0.001 | 143 |
| Magenta | −0.332 | <0.001 | 455 |
| Midnight blue | −0.112 | 0.018 | 189 |
| Orange | −0.406 | <0.001 | 58 |
| Pale turquoise | −0.463 | <0.001 | 33 |
| Pink | −0.169 | <0.001 | 563 |
| Royal blue | 0.393 | <0.001 | 119 |
| Saddle brown | −0.485 | <0.001 | 40 |
| Salmon | −0.017 | 0.713 | 232 |
| Sky blue | 0.407 | <0.001 | 53 |
| Steel blue | −0.447 | <0.001 | 35 |
| White | −0.109 | 0.021 | 54 |
| Genes | FC | Ave. Expr. | t | p-Value | Adj. p-Value | MM. Black | GS | Kin |
|---|---|---|---|---|---|---|---|---|
| CENPA | 1.313 | 2.053 | −20.644 | 4.89 × 10−67 | 2.46 × 10−65 | 0.881 | 0.699 | 182.908 |
| DBF4 | 1.293 | 2.248 | −23.814 | 1.30 × 10−81 | 1.61 × 10−79 | 0.883 | 0.748 | 188.790 |
| H2AFX | 1.291 | 2.683 | −26.595 | 3.18 × 10−94 | 8.84 × 10−92 | 0.868 | 0.783 | 183.809 |
| KIAA1794 | 1.294 | 2.135 | −21.209 | 1.22 × 10−69 | 7.04 × 10−68 | 0.891 | 0.709 | 181.842 |
| KIF14 | 1.249 | 2.063 | −20.100 | 1.57 × 10−67 | 6.81 × 10−63 | 0.852 | 0.689 | 185.458 |
| NEK2 | 1.468 | 2.136 | −26.636 | 2.09 × 10−94 | 5.96 × 10−92 | 0.926 | 0.783 | 184.615 |
| PRIM1 | 1.341 | 2.307 | −24.049 | 1.10 × 10−82 | 1.47 × 10−80 | 0.853 | 0.751 | 186.033 |
| RFC4 | 1.443 | 2.535 | −29.438 | 8.27 × 10−107 | 5.17 × 10−104 | 0.910 | 0.812 | 181.994 |
| RRM2 | 1.785 | 2.360 | −31.691 | 1.62 × 10−116 | 1.81 × 10−103 | 0.929 | 0.832 | 184.521 |
| TOP2A | 1.686 | 2.292 | −31.427 | 2.15 × 10−115 | 2.04 × 10−112 | 0.932 | 0.824 | 185.060 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.B.; Do, D.N.; Nguyen-Thanh, T.; Tatipamula, V.B.; Nguyen, H.T. Identification of Five Hub Genes as Key Prognostic Biomarkers in Liver Cancer via Integrated Bioinformatics Analysis. Biology 2021, 10, 957. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10100957
Nguyen TB, Do DN, Nguyen-Thanh T, Tatipamula VB, Nguyen HT. Identification of Five Hub Genes as Key Prognostic Biomarkers in Liver Cancer via Integrated Bioinformatics Analysis. Biology. 2021; 10(10):957. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10100957
Chicago/Turabian StyleNguyen, Thong Ba, Duy Ngoc Do, Tung Nguyen-Thanh, Vinay Bharadwaj Tatipamula, and Ha Thi Nguyen. 2021. "Identification of Five Hub Genes as Key Prognostic Biomarkers in Liver Cancer via Integrated Bioinformatics Analysis" Biology 10, no. 10: 957. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10100957