
Cytochalasin B-Induced Membrane Vesicles from Human Mesenchymal Stem Cells Overexpressing IL2 Are Able to Stimulate CD8+ T-Killers to Kill Human Triple Negative Breast Cancer Cells

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
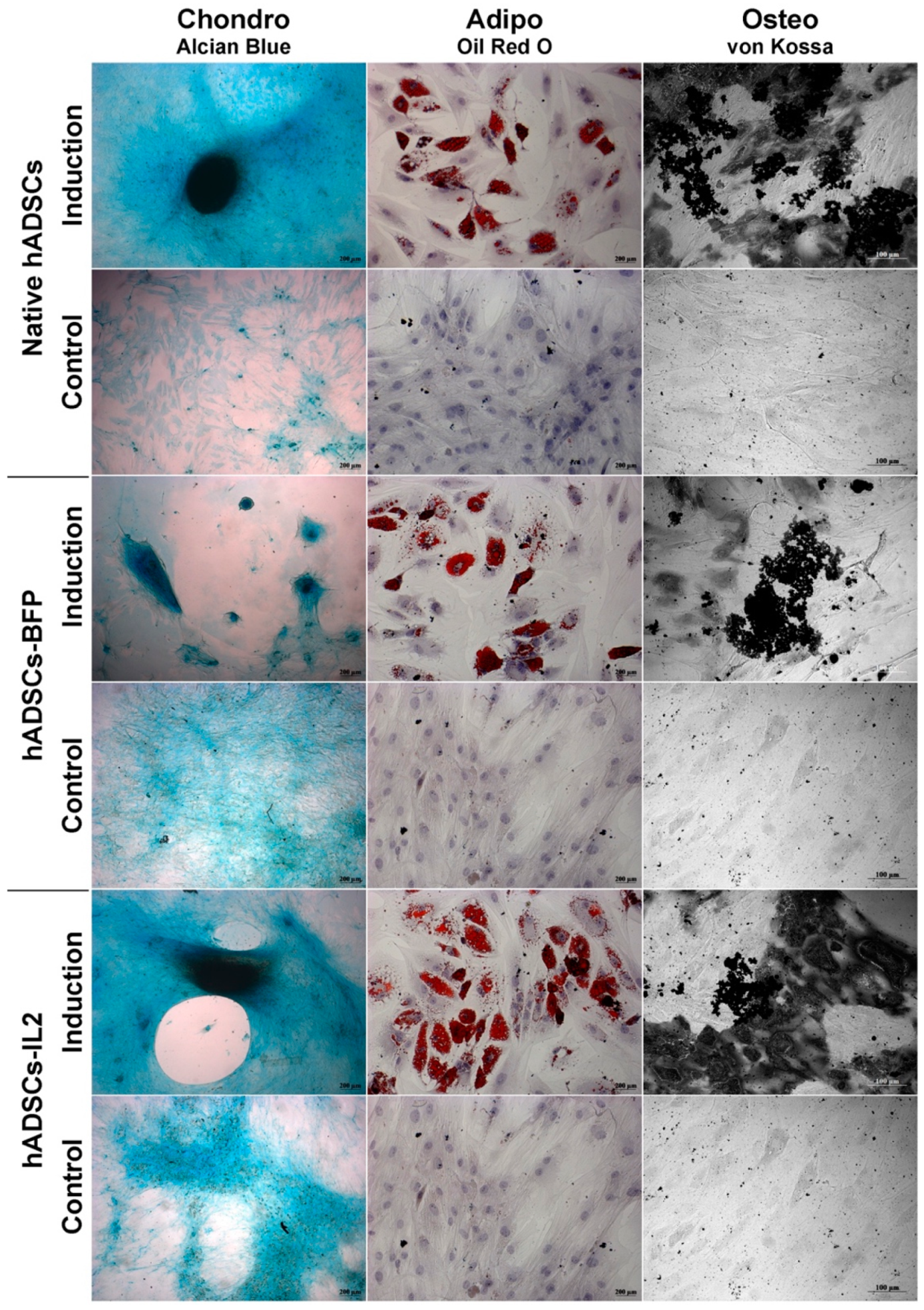
2.2. Differentiation of hADSCs into Adipocytes, Chondrocytes and Osteoblasts
2.3. Lentivirus Production
2.4. Genetic Modification and Selection
2.5. Isolation of Cytochalasin B-Induced Membrane Vesicles
2.6. Quantitative Polymerase Chain Reaction (qPCR)
2.7. Western Blot Analysis
2.8. Immunophenotyping
2.9. Apoptosis and Necrosis Detection
2.10. MTS Cell Proliferation Assays
2.11. Determination of CIMV Size
2.12. Assessment of the Nuclear and Mitochondrial Components in CIMVs
2.13. Cytokine Multiplex Analysis
2.14. IL2 ELISA
2.15. Immunofluorescence Analysis of CIMVs
2.16. Immune Cell Activation
2.17. T-Cell Proliferation Assay
2.18. Analysis of Activated PBMC Cytotoxicity on Human Triple Negative Breast Cancer Cells
2.19. Animals
2.20. Isolation of Murine Peripheral Blood Mononuclear Cells
2.21. Analysis of Murine PBMC Populations
2.22. Multiplex Analysis of Mouse Cytokines
2.23. Statistical Analysis
3. Results
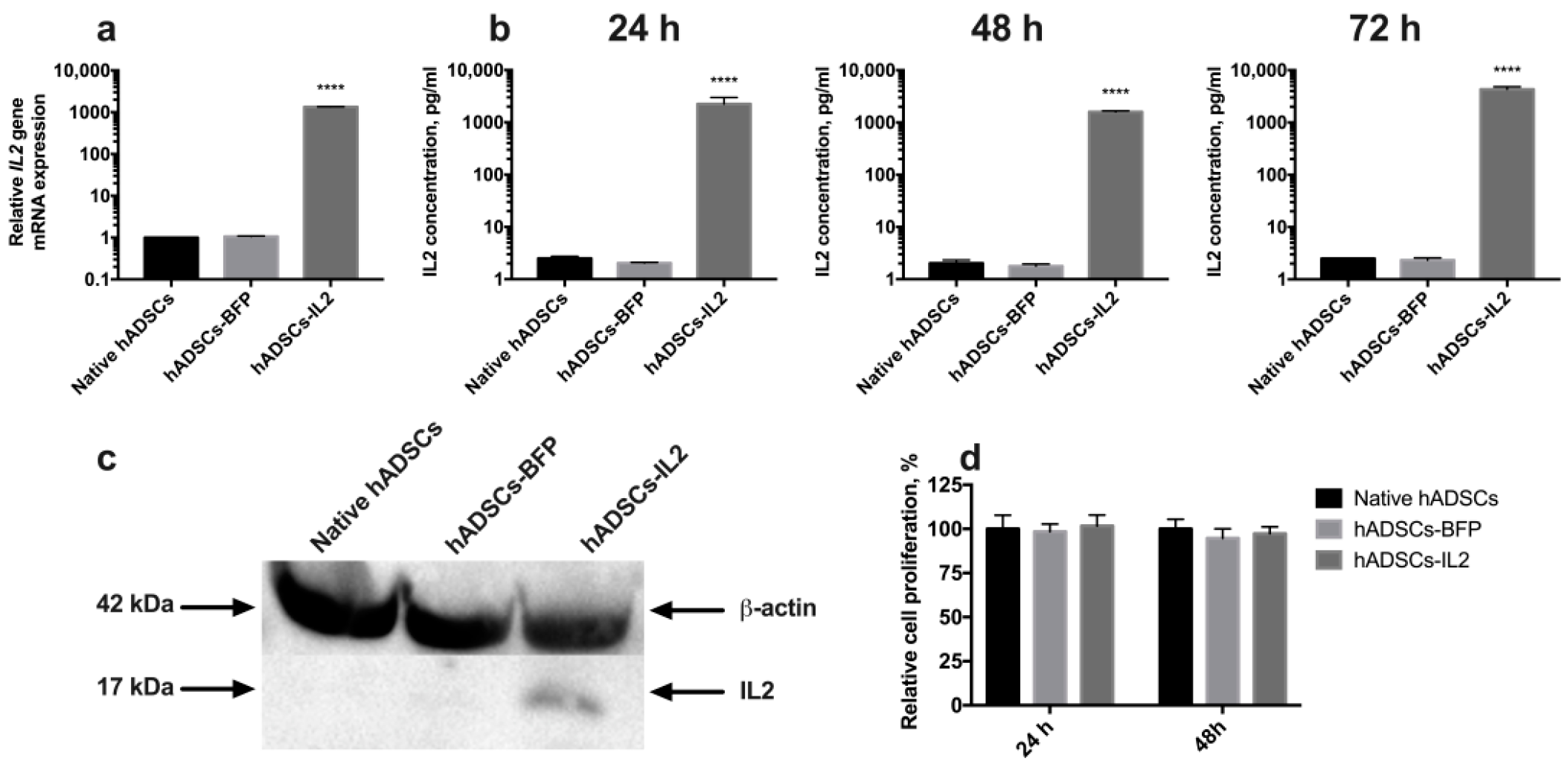
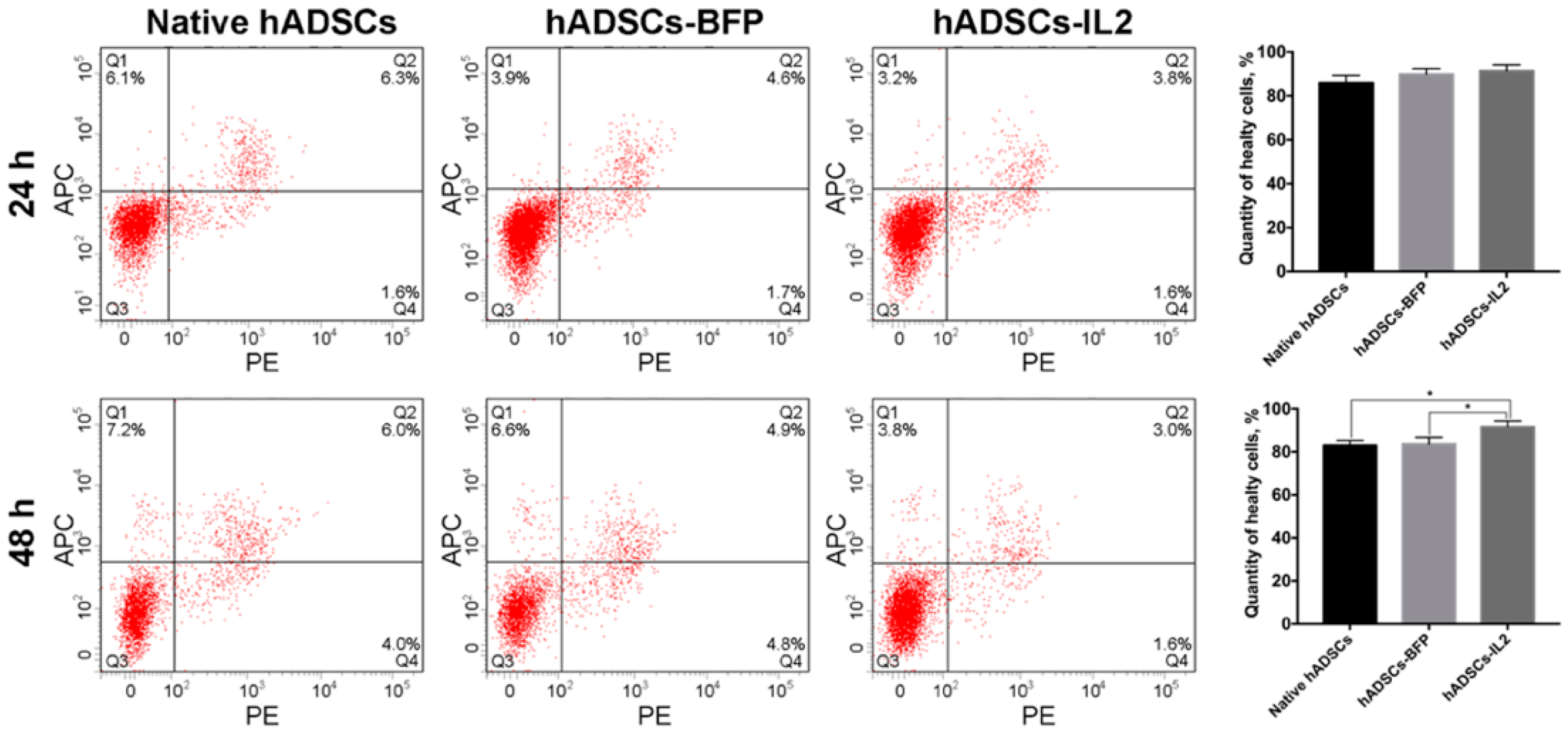
3.1. IL2 Expression Failed to Affect the Viability, Proliferative Activity and Ability to Direct Differentiation of hADSCs
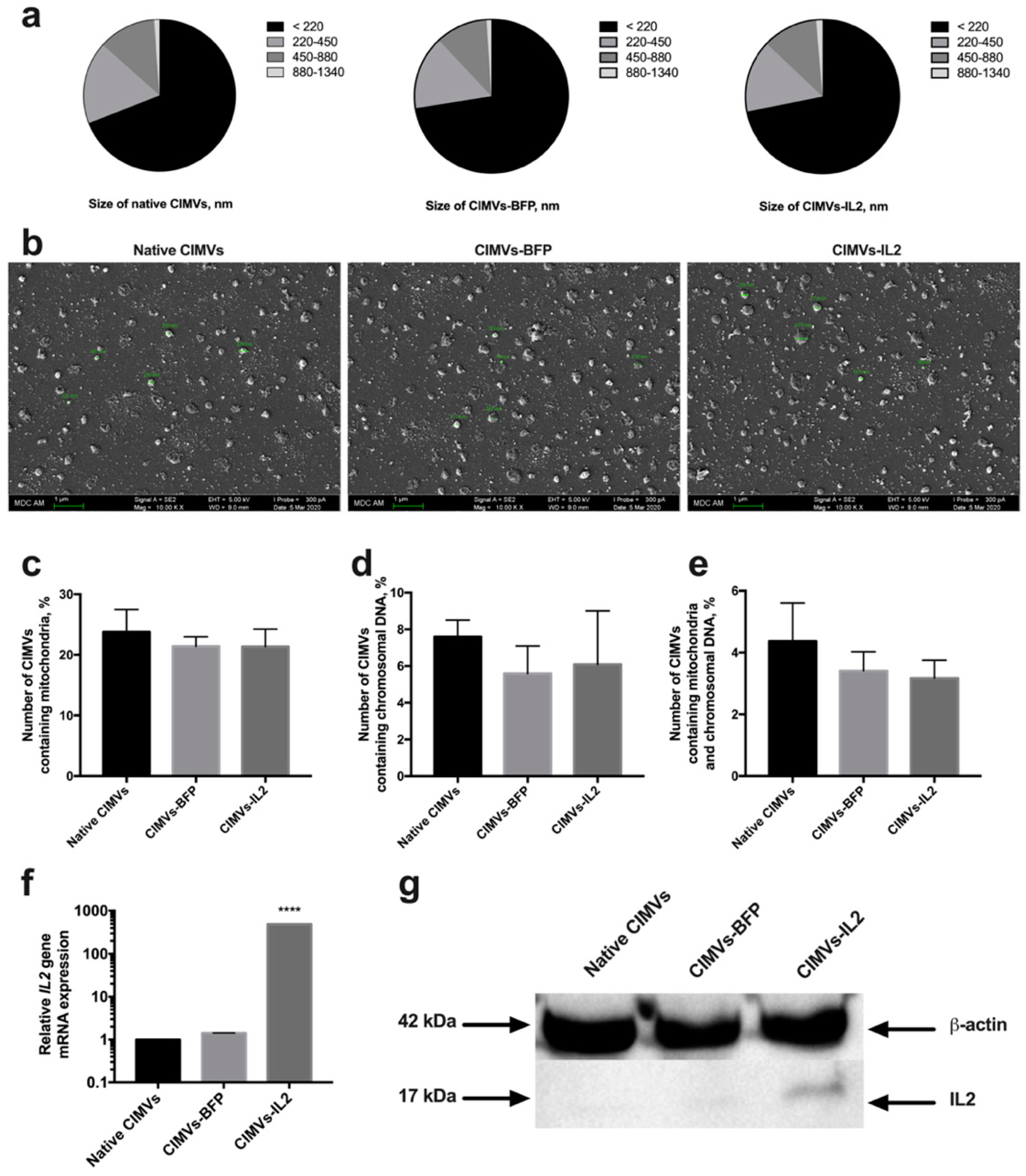
3.2. CIMVs Isolated from hADSCs-IL2 Carry IL2 mRNA and Protein
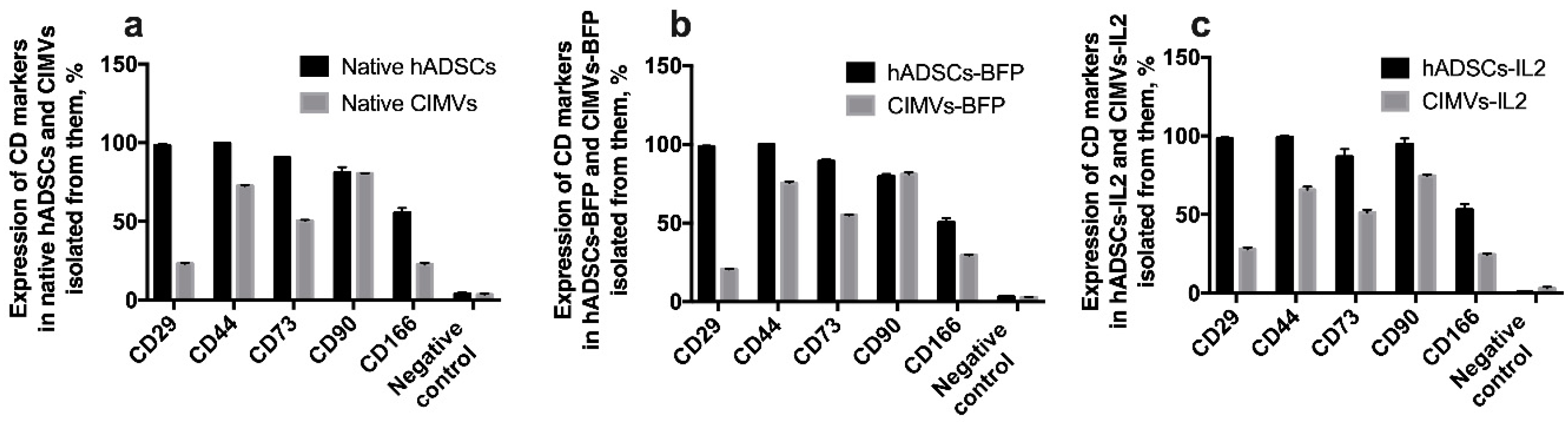
3.3. CIMVs Carry a Reduced Number of Parental Membrane CD Markers
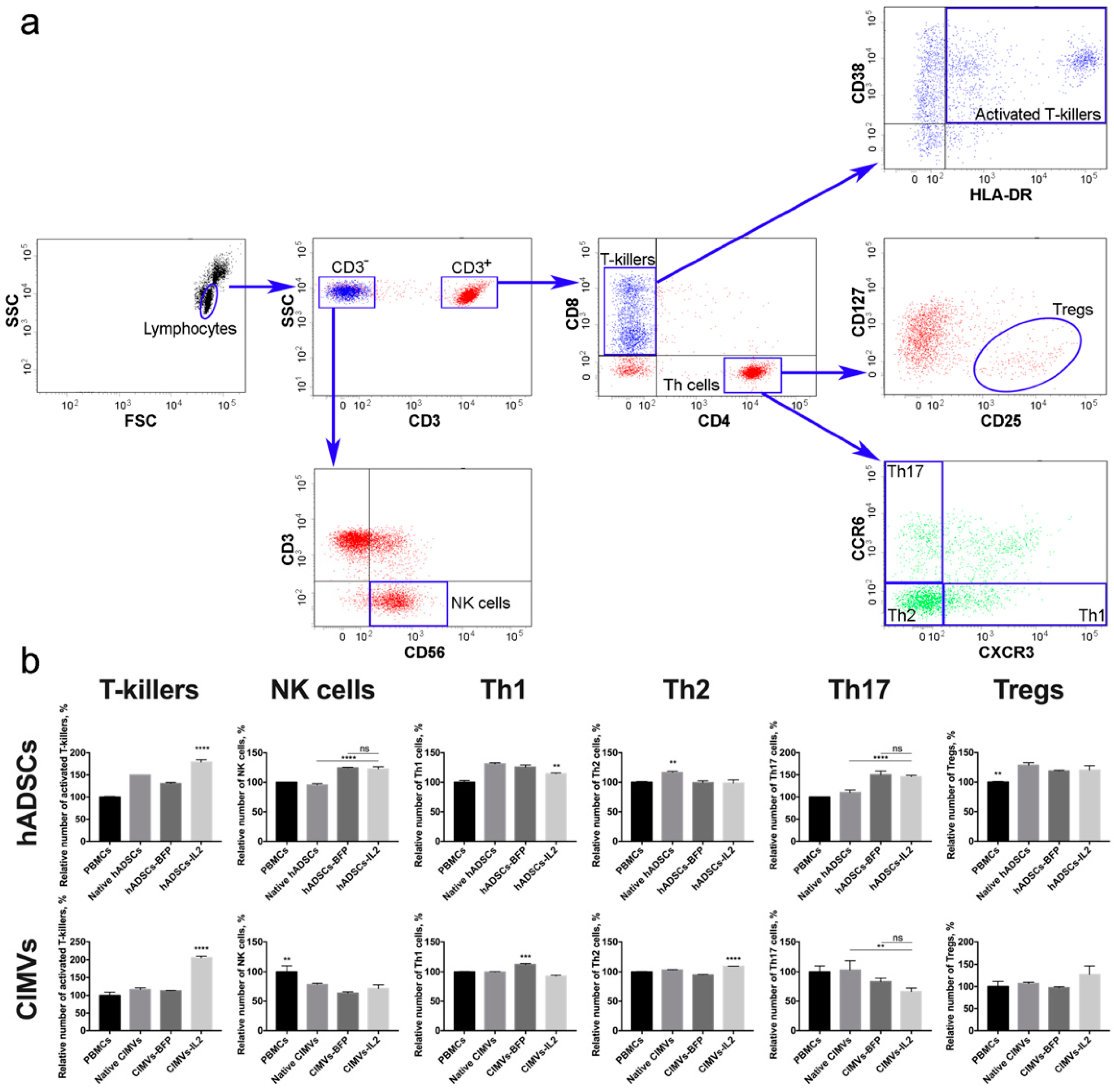
3.4. Both hADSCs-IL2 and CIMVs-IL2 Can Activate Human PBMCs
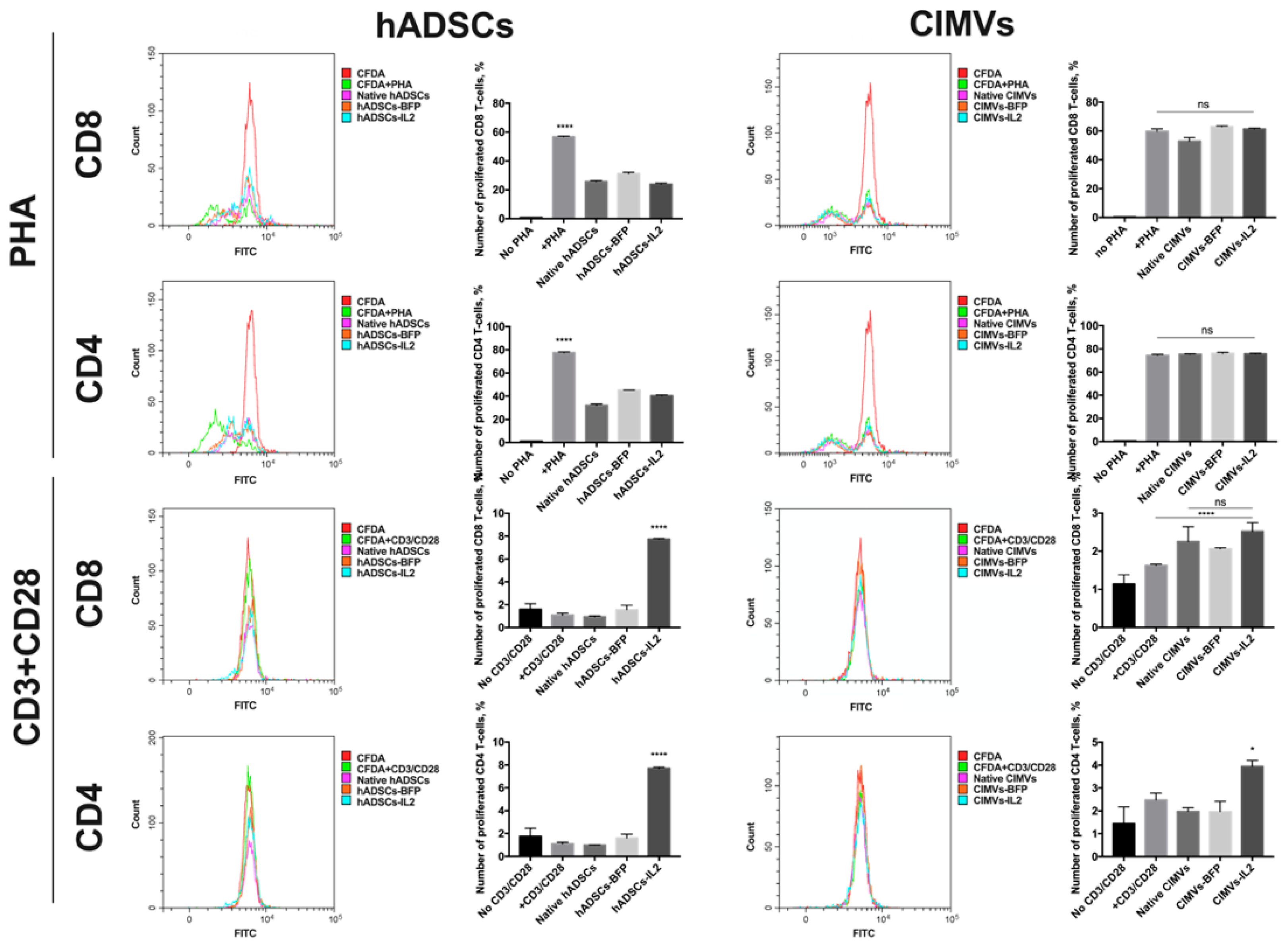
3.5. CIMVs Had Failed to Influence T-Cell Proliferation
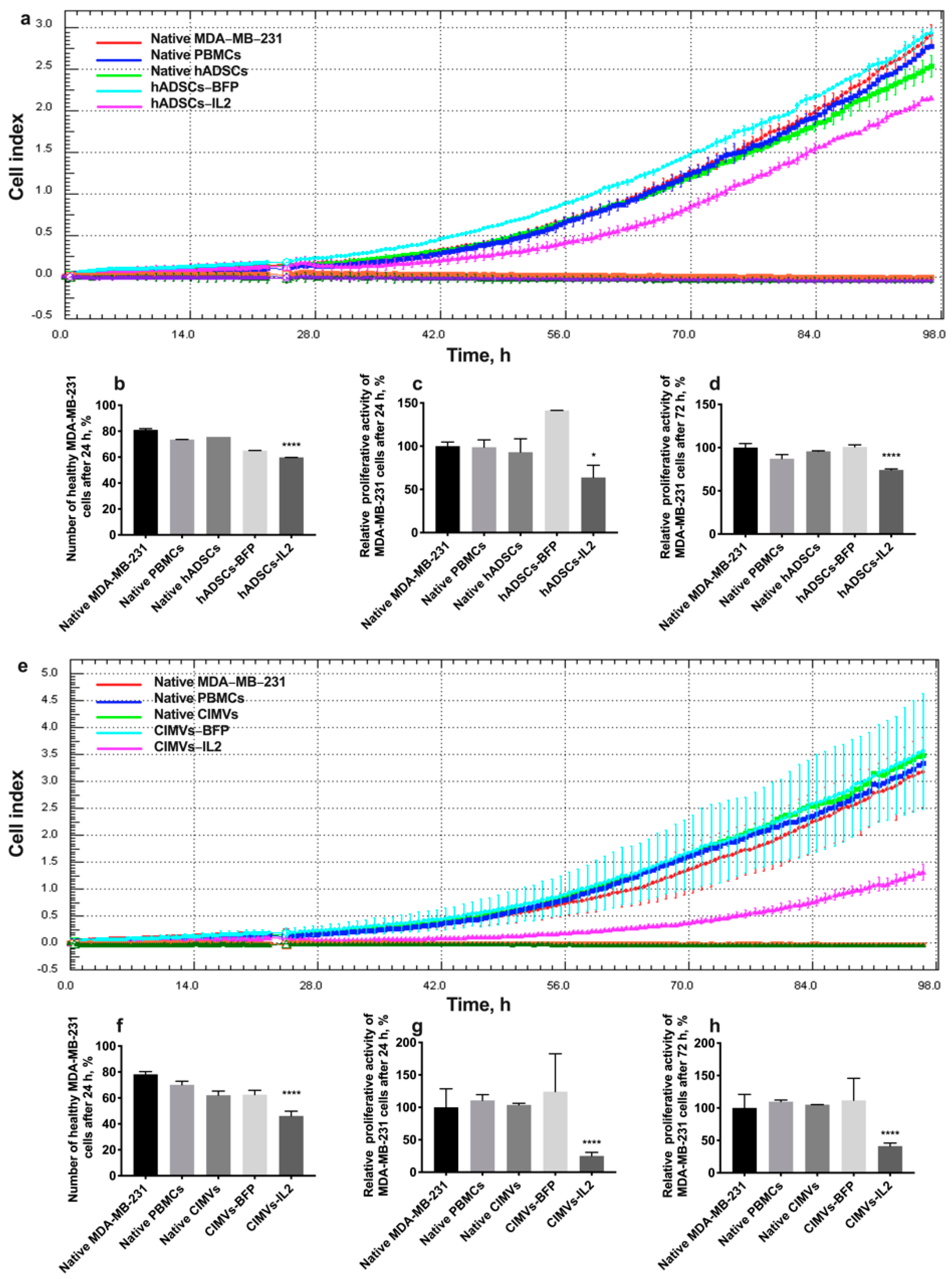
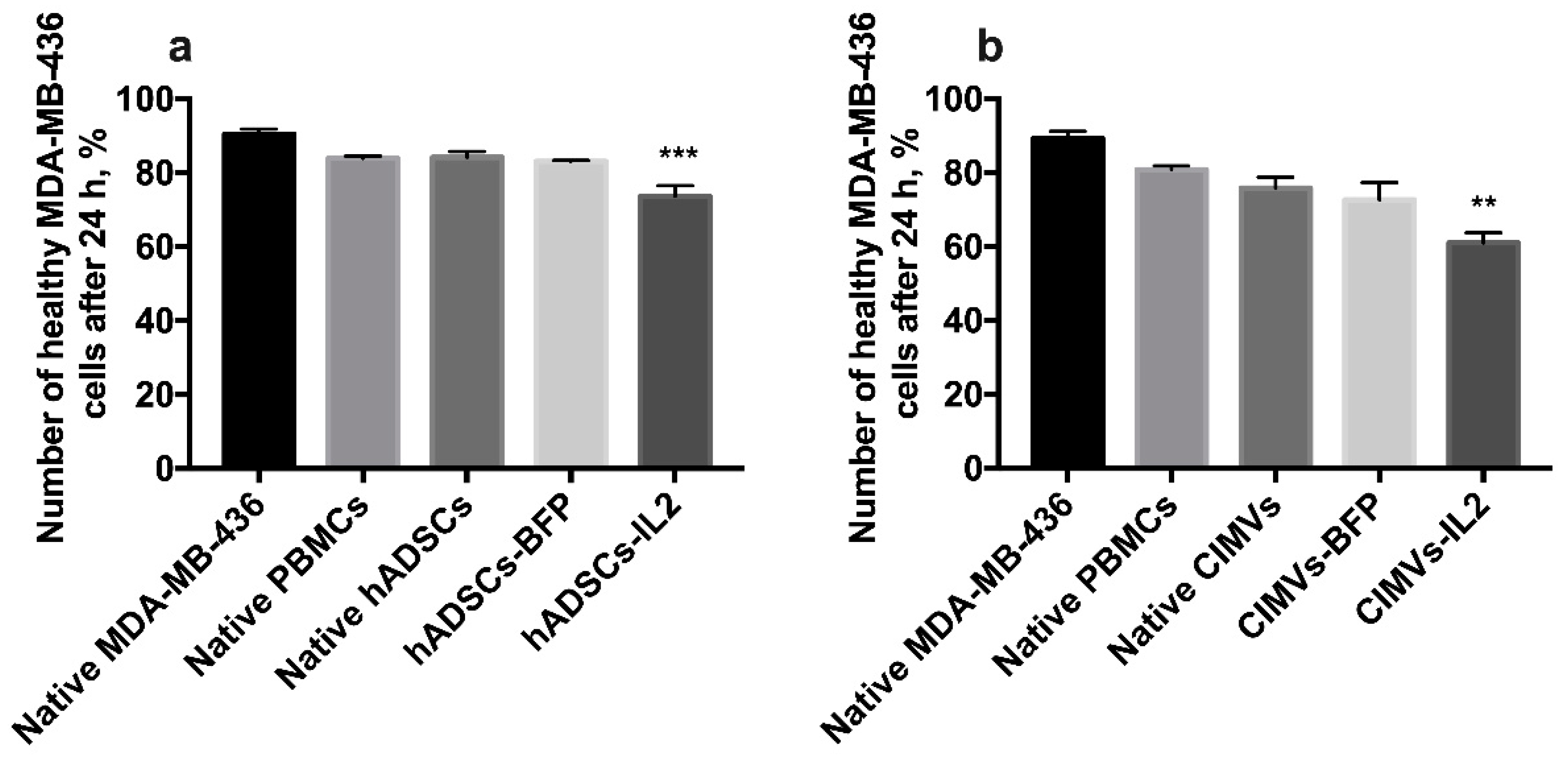
3.6. CIMV-IL2-Activated T-Cells Can Kill Human Triple Negative Breast Cancer Cells
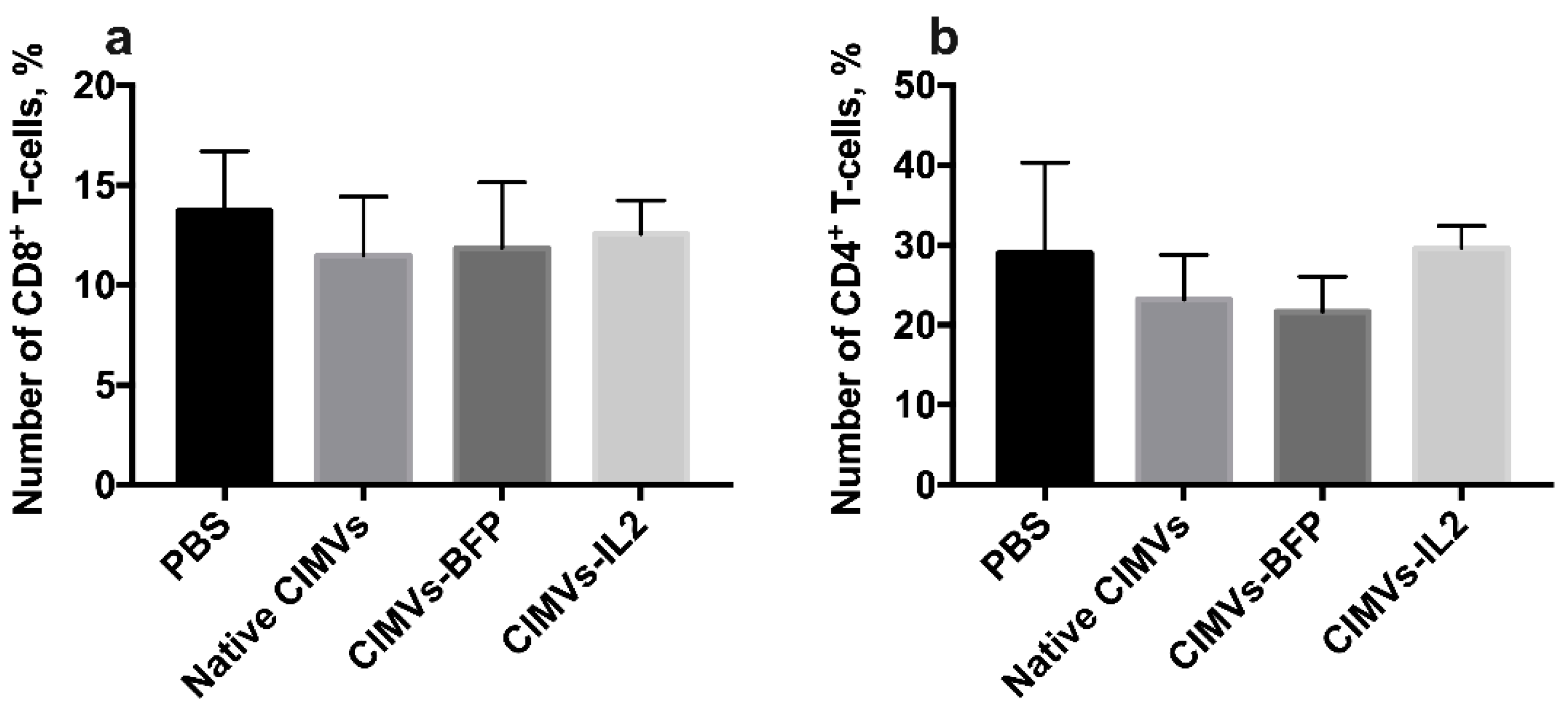
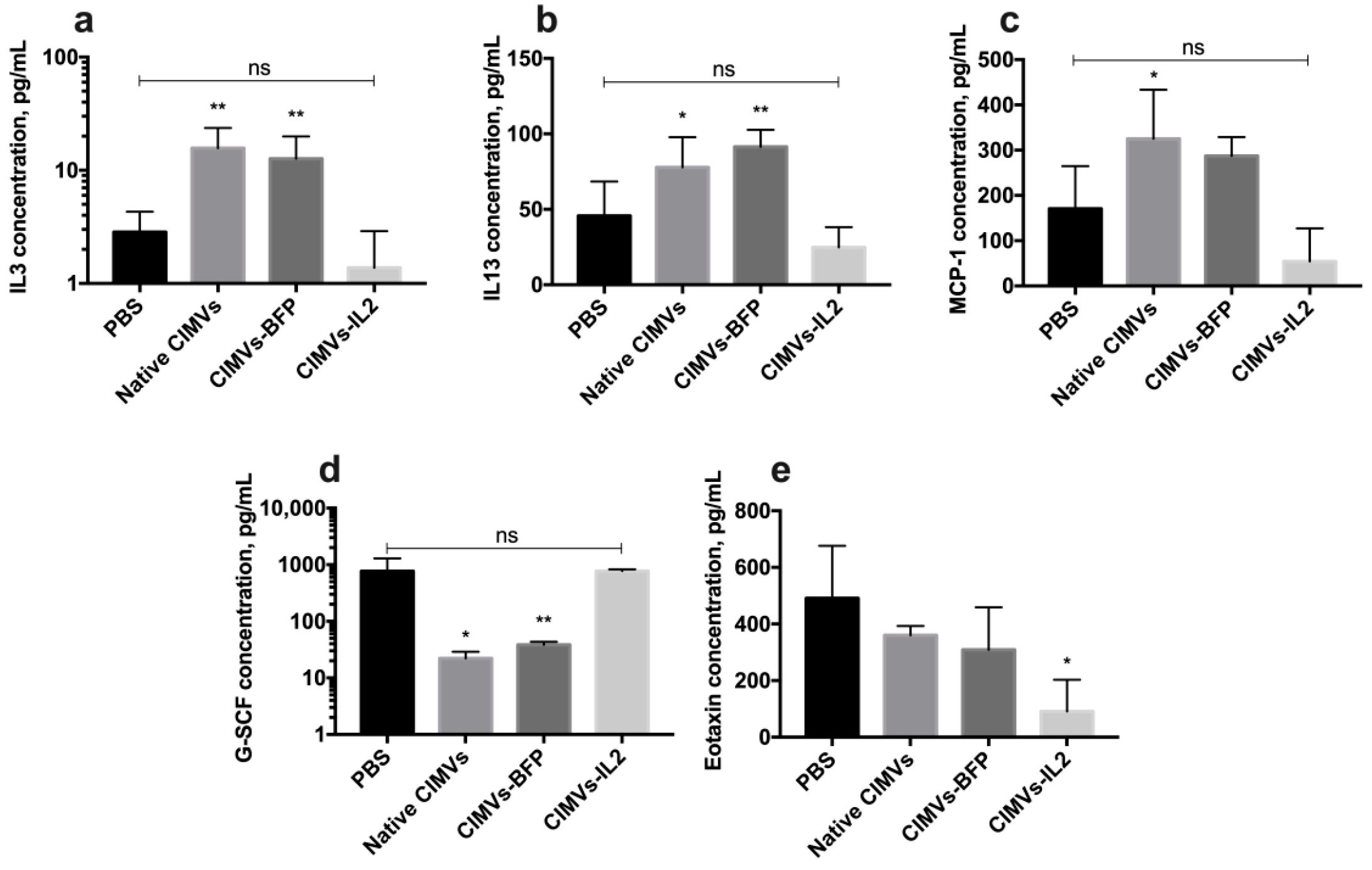
3.7. CIMVs-IL2 Failed to Affect Murine CD8+ T-Cells but Affected Cytokine Profile
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the treatment of cancer. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef] [PubMed]
- West, W.H. Continuous infusion recombinant interleukin-2 (ril-2) in adoptive cellular therapy of renal carcinoma and other malignancies. Cancer Treat. Rev. 1989, 16 (Suppl. A), 83–89. [Google Scholar] [CrossRef]
- Stoter, G.; Fossa, S.D.; Rugarli, C.; Symann, M.; Jasmin, C.; Israel, L.; Bijman, J.T.; Palmer, P.; Franks, C.R.; Philip, T. Metastatic renal cell cancer treated with low-dose interleukin-2. A phase-ii multicentre study. Cancer Treat. Rev. 1989, 16 (Suppl. A), 111–113. [Google Scholar] [CrossRef]
- McDermott, D.F.; Cheng, S.C.; Signoretti, S.; Margolin, K.A.; Clark, J.I.; Sosman, J.A.; Dutcher, J.P.; Logan, T.F.; Curti, B.D.; Ernstoff, M.S.; et al. The high-dose aldesleukin “select” trial: A trial to prospectively validate predictive models of response to treatment in patients with metastatic renal cell carcinoma. Clin. Cancer Res. 2015, 21, 561–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.I.; Wong, M.K.K.; Kaufman, H.L.; Daniels, G.A.; Morse, M.A.; McDermott, D.F.; Agarwala, S.S.; Lewis, L.D.; Stewart, J.H.; Vaishampayan, U.; et al. Impact of sequencing targeted therapies with high-dose interleukin-2 immunotherapy: An analysis of outcome and survival of patients with metastatic renal cell carcinoma from an on-going observational il-2 clinical trial: Proclaim(sm). Clin. Genitourin. Cancer 2017, 15, 31–41.e34. [Google Scholar] [CrossRef]
- Davar, D.; Ding, F.; Saul, M.; Sander, C.; Tarhini, A.A.; Kirkwood, J.M.; Tawbi, H.A. High-dose interleukin-2 (hd il-2) for advanced melanoma: A single center experience from the university of pittsburgh cancer institute. J. Immunother. Cancer 2017, 5, 74. [Google Scholar] [CrossRef]
- Choudhry, H.; Helmi, N.; Abdulaal, W.H.; Zeyadi, M.; Zamzami, M.A.; Wu, W.; Mahmoud, M.M.; Warsi, M.K.; Rasool, M.; Jamal, M.S. Prospects of il-2 in cancer immunotherapy. BioMed. Res. Int. 2018, 2018, 9056173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yui, M.A.; Sharp, L.L.; Havran, W.L.; Rothenberg, E.V. Preferential activation of an il-2 regulatory sequence transgene in tcr gamma delta and nkt cells: Subset-specific differences in il-2 regulation. J. Immunol. 2004, 172, 4691–4699. [Google Scholar] [CrossRef] [Green Version]
- Paliard, X.; de Waal Malefijt, R.; Yssel, H.; Blanchard, D.; Chretien, I.; Abrams, J.; de Vries, J.; Spits, H. Simultaneous production of il-2, il-4, and ifn-gamma by activated human cd4+ and cd8+ t cell clones. J. Immunol. 1988, 141, 849–855. [Google Scholar]
- Hershko, A.Y.; Suzuki, R.; Charles, N.; Alvarez-Errico, D.; Sargent, J.L.; Laurence, A.; Rivera, J. Mast cell interleukin-2 production contributes to suppression of chronic allergic dermatitis. Immunity 2011, 35, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granucci, F.; Vizzardelli, C.; Pavelka, N.; Feau, S.; Persico, M.; Virzi, E.; Rescigno, M.; Moro, G.; Ricciardi-Castagnoli, P. Inducible il-2 production by dendritic cells revealed by global gene expression analysis. Nat. Immunol. 2001, 2, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, C.; Ren, S. Role of il-2 in cancer immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, M.; Li, Y. Anti-colorectal cancer effect of interleukin-2 and interferon-beta fusion gene driven by carcinoembryonic antigen promoter. Onco Targets Ther. 2016, 9, 3259–3267. [Google Scholar] [PubMed] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-cd19 chimeric-antigen-receptor-transduced t cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Donohue, J.H.; Rosenberg, S.A. The fate of interleukin-2 after in vivo administration. J. Immunol. 1983, 130, 2203–2208. [Google Scholar]
- McDermott, D.F.; Atkins, M.B. Application of il-2 and other cytokines in renal cancer. Expert Opin. Biol. Ther. 2004, 4, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Kovar, M.; Rubinstein, M.P.; Surh, C.D.; Sprent, J. Selective stimulation of t cell subsets with antibody-cytokine immune complexes. Science 2006, 311, 1924–1927. [Google Scholar] [CrossRef] [Green Version]
- West, E.E.; Jin, H.T.; Rasheed, A.U.; Penaloza-Macmaster, P.; Ha, S.J.; Tan, W.G.; Youngblood, B.; Freeman, G.J.; Smith, K.A.; Ahmed, R. Pd-l1 blockade synergizes with il-2 therapy in reinvigorating exhausted t cells. J. Clin. Investig. 2013, 123, 2604–2615. [Google Scholar] [CrossRef]
- Prochazkova, J.; Pokorna, K.; Holan, V. Il-12 inhibits the tgf-beta-dependent t cell developmental programs and skews the tgf-beta-induced differentiation into a th1-like direction. Immunobiology 2012, 217, 74–82. [Google Scholar] [CrossRef]
- Attridge, K.; Wang, C.J.; Wardzinski, L.; Kenefeck, R.; Chamberlain, J.L.; Manzotti, C.; Kopf, M.; Walker, L.S. Il-21 inhibits t cell il-2 production and impairs treg homeostasis. Blood 2012, 119, 4656–4664. [Google Scholar] [CrossRef] [Green Version]
- Lissoni, P. Therapy implications of the role of interleukin-2 in cancer. Expert Rev. Clin. Immunol. 2017, 13, 491–498. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Tazetdinova, L.G.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Application of mesenchymal stem cells for therapeutic agent delivery in anti-tumor treatment. Front. Pharm. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic prospects of extracellular vesicles in cancer treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakowski, M.; Buller, B.; Zheng, X.; Lu, Y.; Rogers, T.; Osobamiro, O.; Shu, W.; Jiang, F.; Chopp, M. Exosomes from marrow stromal cells expressing mir-146b inhibit glioma growth. Cancer Lett. 2013, 335, 201–204. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Kolluri, K.K.; Gowers, K.H.; Janes, S.M. Trail delivery by msc-derived extracellular vesicles is an effective anticancer therapy. J. Extracell. Vesicles 2017, 6, 1265291. [Google Scholar] [CrossRef]
- Phan, J.; Kumar, P.; Hao, D.; Gao, K.; Farmer, D.; Wang, A. Engineering mesenchymal stem cells to improve their exosome efficacy and yield for cell-free therapy. J. Extracell. Vesicles 2018, 7, 1522236. [Google Scholar] [CrossRef]
- Oshchepkova, A.; Neumestova, A.; Matveeva, V.; Artemyeva, L.; Morozova, K.; Kiseleva, E.; Zenkova, M.; Vlassov, V. Cytochalasin-b-inducible nanovesicle mimics of natural extracellular vesicles that are capable of nucleic acid transfer. Micromachines 2019, 10, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solovyeva, V.V.; Salafutdinov, I.I.; Tazetdinova, L.G.; Masgutov, R.F.; Khaiboullina, S.F.; Rizvanov, A.A. Genetic modification of adipose derived stem cells with recombinant plasmid DNA pbud-vegf-fgf2 results in increased of il-8 and mcp-1 secretion. J. Pure Appl. Microbiol. 2014, 8, 523–528. [Google Scholar]
- Islamov, R.R.; Rizvanov, A.A.; Mukhamedyarov, M.A.; Salafutdinov, I.I.; Garanina, E.E.; Fedotova, V.Y.; Solovyeva, V.V.; Mukhamedshina, Y.O.; Safiullov, Z.Z.; Izmailov, A.A.; et al. Symptomatic improvement, increased life-span and sustained cell homing in amyotrophic lateral sclerosis after transplantation of human umbilical cord blood cells genetically modified with adeno-viral vectors expressing a neuro-protective factor and a neural cell adhesion molecule. Curr. Gene Ther. 2015, 15, 266–276. [Google Scholar] [PubMed]
- Kolobynina, K.; Solovyeva, V.; Gomzikova, M.; Tazetdinova, L.; Rizvanov, A. Generation of human adipose-derived stem cell lines with expression of tesc gene. BioNanoScience 2017, 7, 92–96. [Google Scholar] [CrossRef]
- Kitaeva, K.V.; Prudnikov, T.S.; Gomzikova, M.O.; Kletukhina, S.K.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Analysis of the interaction and proliferative activity of adenocarcinoma, peripheral blood mononuclear and mesenchymal stromal cells after co-cultivation in vitro. BioNanoScience 2019, 9, 502–509. [Google Scholar] [CrossRef]
- Killer, M.C.; Nold, P.; Henkenius, K.; Fritz, L.; Riedlinger, T.; Barckhausen, C.; Frech, M.; Hackstein, H.; Neubauer, A.; Brendel, C. Immunosuppressive capacity of mesenchymal stem cells correlates with metabolic activity and can be enhanced by valproic acid. Stem Cell Res. Ther. 2017, 8, 100. [Google Scholar] [CrossRef] [PubMed]
- Gomzikova, M.O.; Kletukhina, S.K.; Kurbangaleeva, S.V.; Neustroeva, O.A.; Vasileva, O.S.; Garanina, E.E.; Khaiboullina, S.F.; Rizvanov, A.A. Mesenchymal stem cell derived biocompatible membrane vesicles demonstrate immunomodulatory activity inhibiting activation and proliferation of human mononuclear cells. Pharmaceutics 2020, 12, 577. [Google Scholar] [CrossRef] [PubMed]
- Niu, P.; Smagul, A.; Wang, L.; Sadvakas, A.; Sha, Y.; Perez, L.M.; Nussupbekova, A.; Amirbekov, A.; Akanov, A.A.; Galvez, B.G.; et al. Transcriptional profiling of interleukin-2-primed human adipose derived mesenchymal stem cells revealed dramatic changes in stem cells response imposed by replicative senescence. Oncotarget 2015, 6, 17938–17957. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Pick, H.; Schmid, E.L.; Tairi, A.P.; Ilegems, E.; Hovius, R.; Vogel, H. Investigating cellular signaling reactions in single attoliter vesicles. J. Am. Chem. Soc. 2005, 127, 2908–2912. [Google Scholar] [CrossRef]
- Gomzikova, M.O.; Rizvanov, A.A. Current trends in regenerative medicine: From cell to cell-free therapy. Bionanoscience 2017, 7, 240–245. [Google Scholar] [CrossRef]
- Di Trapani, M.; Bassi, G.; Midolo, M.; Gatti, A.; Kamga, P.T.; Cassaro, A.; Carusone, R.; Adamo, A.; Krampera, M. Differential and transferable modulatory effects of mesenchymal stromal cell-derived extracellular vesicles on t, b and nk cell functions. Sci. Rep. 2016, 6, 24120. [Google Scholar] [CrossRef] [PubMed]
- Khatri, M.; Richardson, L.A.; Meulia, T. Mesenchymal stem cell-derived extracellular vesicles attenuate influenza virus-induced acute lung injury in a pig model. Stem Cell Res. Ther. 2018, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hubal, M.J.; Kraus, V.B. Immune cell extracellular vesicles and their mitochondrial content decline with ageing. Immun. Ageing 2020, 17, 1. [Google Scholar] [CrossRef]
- Phinney, D.G.; Di Giuseppe, M.; Njah, J.; Sala, E.; Shiva, S.; St Croix, C.M.; Stolz, D.B.; Watkins, S.C.; Di, Y.P.; Leikauf, G.D.; et al. Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle micrornas. Nat. Commun. 2015, 6, 8472. [Google Scholar] [CrossRef]
- Hough, K.P.; Trevor, J.L.; Strenkowski, J.G.; Wang, Y.; Chacko, B.K.; Tousif, S.; Chanda, D.; Steele, C.; Antony, V.B.; Dokland, T.; et al. Exosomal transfer of mitochondria from airway myeloid-derived regulatory cells to t cells. Redox Biol. 2018, 18, 54–64. [Google Scholar] [CrossRef]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal stromal cells modulate macrophages in clinically relevant lung injury models by extracellular vesicle mitochondrial transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef]
- Choi, D.S.; Kim, D.K.; Kim, Y.K.; Gho, Y.S. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013, 13, 1554–1571. [Google Scholar] [CrossRef]
- Malkin, E.Z.; Bratman, S.V. Bioactive DNA from extracellular vesicles and particles. Cell Death Dis. 2020, 11, 584. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Ren, H.; Chen, C.; He, D.; Zhou, L.; Eisner, G.M.; Asico, L.D.; Jose, P.A.; Zeng, C. Extracellular vesicle-mediated transfer of donor genomic DNA to recipient cells is a novel mechanism for genetic influence between cells. J. Mol. Cell Biol. 2013, 5, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Gomzikova, M.; Kletukhina, S.; Kurbangaleeva, S.; Rizvanov, A. Evaluation of cytochalasin b-induced membrane vesicles fusion specificity with target cells. BioMed. Res. Int. 2018, 2018, 7053623. [Google Scholar] [CrossRef] [Green Version]
- Nery, A.A.; Nascimento, I.C.; Glaser, T.; Bassaneze, V.; Krieger, J.E.; Ulrich, H. Human mesenchymal stem cells: From immunophenotyping by flow cytometry to clinical applications. Cytom. Part A J. Int. Soc. Anal. Cytol. 2013, 83, 48–61. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Patterson, A.; Johnson, C.B.; Neitzke, D.J.; Mehrotra, S.; Denlinger, C.E.; Paulos, C.M.; Li, Z.; Cole, D.J.; Rubinstein, M.P. Il-2 and beyond in cancer immunotherapy. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2018, 38, 45–68. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Solovyeva, V.V.; James, V.; Arkhipova, S.S.; Gomzikova, M.O.; Garanina, E.E.; Akhmetzyanova, E.R.; Tazetdinova, L.G.; Khaiboullina, S.F.; Rizvanov, A.A. Human mesenchymal stem cells overexpressing interleukin 2 can suppress proliferation of neuroblastoma cells in co-culture and activate mononuclear cells in vitro. Bioengineering 2020, 7, 59. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. Cd8(+) cytotoxic t lymphocytes in cancer immunotherapy: A review. J. Cell Physiol 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular aspects and future perspectives of cytokine-based anti-cancer immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, hyperproliferation, activation of natural killer cells and cd8 t cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef]
- Hof-Nahor, I.; Leshansky, L.; Shivtiel, S.; Eldor, L.; Aberdam, D.; Itskovitz-Eldor, J.; Berrih-Aknin, S. Human mesenchymal stem cells shift cd8+ t cells towards a suppressive phenotype by inducing tolerogenic monocytes. J. Cell Sci. 2012, 125, 4640–4650. [Google Scholar] [CrossRef] [Green Version]
- Brodbeck, T.; Nehmann, N.; Bethge, A.; Wedemann, G.; Schumacher, U. Perforin-dependent direct cytotoxicity in natural killer cells induces considerable knockdown of spontaneous lung metastases and computer modelling-proven tumor cell dormancy in a ht29 human colon cancer xenograft mouse model. Mol. Cancer 2014, 13, 244. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, M.; Matos, T.; Liu, H.Y.; Koreth, J.; Kim, H.T.; Paul, N.E.; Murase, K.; Whangbo, J.; Alho, A.C.; Nikiforow, S.; et al. Low-dose il-2 selectively activates subsets of cd4(+) tregs and nk cells. JCI Insight 2016, 1, e89278. [Google Scholar] [CrossRef] [Green Version]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. Il-2 regulates foxp3 expression in human cd4(+)cd25(+) regulatory t cells through a stat-dependent mechanism and induces the expansion of these cells in vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gore, A.V.; Bible, L.E.; Song, K.; Livingston, D.H.; Mohr, A.M.; Sifri, Z.C. Mesenchymal stem cells increase t-regulatory cells and improve healing following trauma and hemorrhagic shock. J. Trauma Acute Care Surg. 2015, 79, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, R.I.; Minskaia, E.; Fernandes-Platzgummer, A.; Vieira, A.I.S.; da Silva, C.L.; Cabral, J.M.S.; Lacerda, J.F. Mesenchymal stromal cells induce regulatory t cells via epigenetic conversion of human conventional cd4 t cells in vitro. Stem Cells 2020, 38, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Blankenstein, T. Cd4+ t cell--mediated tumor rejection involves inhibition of angiogenesis that is dependent on ifn gamma receptor expression by nonhematopoietic cells. Immunity 2000, 12, 677–686. [Google Scholar] [CrossRef] [Green Version]
- Chraa, D.; Naim, A.; Olive, D.; Badou, A. T lymphocyte subsets in cancer immunity: Friends or foes. J. Leukoc. Biol. 2019, 105, 243–255. [Google Scholar] [CrossRef]
- Steinbrink, K.; Graulich, E.; Kubsch, S.; Knop, J.; Enk, A.H. Cd4(+) and cd8(+) anergic t cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood 2002, 99, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of th17 cell functions in tumor immunity. Cell Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Duffy, M.M.; Pindjakova, J.; Hanley, S.A.; McCarthy, C.; Weidhofer, G.A.; Sweeney, E.M.; English, K.; Shaw, G.; Murphy, J.M.; Barry, F.P.; et al. Mesenchymal stem cell inhibition of t-helper 17 cell-differentiation is triggered by cell-cell contact and mediated by prostaglandin e2 via the ep4 receptor. Eur. J. Immunol. 2011, 41, 2840–2851. [Google Scholar] [CrossRef]
- Xie, M.; Xiong, W.; She, Z.; Wen, Z.; Abdirahman, A.S.; Wan, W.; Wen, C. Immunoregulatory effects of stem cell-derived extracellular vesicles on immune cells. Front. Immunol. 2020, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Hyvarinen, K.; Holopainen, M.; Skirdenko, V.; Ruhanen, H.; Lehenkari, P.; Korhonen, M.; Kakela, R.; Laitinen, S.; Kerkela, E. Mesenchymal stromal cells and their extracellular vesicles enhance the anti-inflammatory phenotype of regulatory macrophages by downregulating the production of interleukin (il)-23 and il-22. Front. Immunol. 2018, 9, 771. [Google Scholar] [CrossRef]
- Noack, M.; Miossec, P. Th17 and regulatory t cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Luciano, R.; Pascucci, L.; Goffredo, B.M.; Giorda, E.; Scapaticci, M.; Fierabracci, A.; Muraca, M. Immunoregulatory effects of mesenchymal stem cell-derived extracellular vesicles on t lymphocytes. Cell Transplant. 2015, 24, 2615–2627. [Google Scholar] [CrossRef] [Green Version]
- Movafagh, A.; Heydary, H.; Mortazavi-Tabatabaei, S.A.; Azargashb, E. The significance application of indigenous phytohemagglutinin (pha) mitogen on metaphase and cell culture procedure. Iran. J. Pharm. Res. IJPR 2011, 10, 895–903. [Google Scholar]
- Cheng, L.E.; Ohlen, C.; Nelson, B.H.; Greenberg, P.D. Enhanced signaling through the il-2 receptor in cd8+ t cells regulated by antigen recognition results in preferential proliferation and expansion of responding cd8+ t cells rather than promotion of cell death. Proc. Natl. Acad. Sci. USA 2002, 99, 3001–3006. [Google Scholar] [CrossRef] [Green Version]
- Azamjah, N.; Soltan-Zadeh, Y.; Zayeri, F. Global trend of breast cancer mortality rate: A 25-year study. Asian Pac. J. Cancer Prev. 2019, 20, 2015–2020. [Google Scholar] [CrossRef] [PubMed]
- Tong, C.W.S.; Wu, M.; Cho, W.C.S.; To, K.K.W. Recent advances in the treatment of breast cancer. Front. Oncol. 2018, 8, 227. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Kleef, R.; Moss, R.; Szasz, A.M.; Bohdjalian, A.; Bojar, H.; Bakacs, T. Complete clinical remission of stage iv triple-negative breast cancer lung metastasis administering low-dose immune checkpoint blockade in combination with hyperthermia and interleukin-2. Integr. Cancer Ther. 2018, 17, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Roberti, M.P.; Barrio, M.M.; Bravo, A.I.; Rocca, Y.S.; Arriaga, J.M.; Bianchini, M.; Mordoh, J.; Levy, E.M. Il-15 and il-2 increase cetuximab-mediated cellular cytotoxicity against triple negative breast cancer cell lines expressing egfr. Breast Cancer Res. Treat. 2011, 130, 465–475. [Google Scholar] [CrossRef]
- Sparano, J.A.; Fisher, R.I.; Weiss, G.R.; Margolin, K.; Aronson, F.R.; Hawkins, M.J.; Atkins, M.B.; Dutcher, J.P.; Gaynor, E.R.; Boldt, D.H.; et al. Phase ii trials of high-dose interleukin-2 and lymphokine-activated killer cells in advanced breast carcinoma and carcinoma of the lung, ovary, and pancreas and other tumors. J. Immunother. Emphas. Tumor Immunol. Off. J. Soc. Biol. Ther. 1994, 16, 216–223. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Kuznetsova, M.; Lopatnikova, J.; Shevchenko, J.; Silkov, A.; Maksyutov, A.; Sennikov, S. Cytotoxic activity and memory t cell subset distribution of in vitro-stimulated cd8(+) t cells specific for her2/neu epitopes. Front. Immunol. 2019, 10, 1017. [Google Scholar] [CrossRef]
- Perez Horta, Z.; Saseedhar, S.; Rakhmilevich, A.L.; Carmichael, L.; Hank, J.A.; Boyden, M.; Gillies, S.D.; Sondel, P.M. Human and murine il2 receptors differentially respond to the human-il2 component of immunocytokines. Oncoimmunology 2019, 8, e1238538. [Google Scholar] [CrossRef] [Green Version]
- Arenas-Ramirez, N.; Woytschak, J.; Boyman, O. Interleukin-2: Biology, design and application. Trends Immunol. 2015, 36, 763–777. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.K. Species specificity of interleukin 2 binding to individual receptor components. Eur. J. Immunol. 1989, 19, 1517–1520. [Google Scholar] [CrossRef]
- Borriello, F.; Galdiero, M.R.; Varricchi, G.; Loffredo, S.; Spadaro, G.; Marone, G. Innate immune modulation by gm-csf and il-3 in health and disease. Int. J. Mol. Sci. 2019, 20, 834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapmana, A.M.; Malkin, D.J.; Camacho, J.; Schiestl, R.H. Il-13 overexpression in mouse lungs triggers systemic genotoxicity in peripheral blood. Mutat. Res. 2014, 769, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, P.; Caprioli, F.; Facciotti, F.; Di Sabatino, A. The role of interleukin-13 in chronic inflammatory intestinal disorders. Autoimmun. Rev. 2019, 18, 549–555. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (mcp-1): An overview. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Fang, H.; Hua, C.; Weiss, S.; Liu, A.; Cheng, W.; Claus, R.; Rodel, J.; Dirsch, O.; Dahmen, U. Modulation of innate immunity by g-csf and inflammatory response by lbpk95a improves the outcome of sepsis in a rat model. J. Immunol. Res. 2018, 2018, 6085095. [Google Scholar] [CrossRef]
- Rothenberg, M.E.; Ownbey, R.; Mehlhop, P.D.; Loiselle, P.M.; van de Rijn, M.; Bonventre, J.V.; Oettgen, H.C.; Leder, P.; Luster, A.D. Eotaxin triggers eosinophil-selective chemotaxis and calcium flux via a distinct receptor and induces pulmonary eosinophilia in the presence of interleukin 5 in mice. Mol. Med. 1996, 2, 334–348. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) | TaqMan Probe (5′–3′) |
|---|---|---|---|
| 18S rRNA | GCCGCTAGAGGTGAAATTCTTG | CATTCTTGGCAAATGCTTTCG | [HEX] ACCGGCGCAAGACGGACCAG [BH2] |
| IL2 | CACCAGGATGCTCACATTTAAG | GTCCCTGGGTCTTAAGTGAAAG | [FAM] CCCAAGAAGGCCACAGAACTGAAACA [BH1] |
| Protein | Protein Concentration in Mouse Serum, pg/mL | |||
|---|---|---|---|---|
| PBS (n = 5) | Native CIMVs (n = 5) | CIMVs-BFP (n = 5) | CIMVs-IL2 (n = 5) | |
| GM-CSF | 30.4 ± 6.9 | 69.5 ± 16.7 | 57.8 ± 31.8 | 31.8 ± 17.4 |
| IFN-γ | 27.1 ± 9.1 | 51.2 ± 19.4 | 43.9 ± 27.0 | 16.0 ± 1.0 |
| IL10 | 40.1 ± 52.7 | 21.5 ± 6.9 | 24.1 ± 8.8 | 17.4 ± 13.4 |
| IL2 | 10.6 ± 2.5 | 21.9 ± 12.8 | 20.2 ± 9.7 | 6.4 ± 0.5 |
| IL4 | 13.5 ± 3.9 | 0.1 ± 0.1 | 14.0 ± 12.4 | 9.8 ± 2.7 |
| IL5 | 0.2 ± 0.3 | 17.2 ± 8.9 | 5.9 ± 9.1 | 0.2 ± 0.3 |
| IL6 | 7.5 ± 3.4 | 2.8 ± 1.1 | 8.9 ± 4.9 | 6.3 ± 4.0 |
| IL12p40 | 37.2 ± 6.9 | 142.1 ± 59.5 | 87.9 ± 70.2 | 11.1 ± 2.2 |
| IL12p70 | 182.4 ± 49.1 | 39.7 ± 11.1 | 112.1 ± 92.6 | 91.4 ± 71.5 |
| KC | 75.3 ± 31.0 | 106.7 ± 20.8 | 120.7 ± 14.0 | 48.9 ± 18.9 |
| MIP-1α | 5.8 ± 1.9 | 9.2 ± 3.4 | 9.2 ± 2.0 | 5.5 ± 0.6 |
| MIP-1δ | 12.4 ± 8.0 | 32.0 ± 15.6 | 28.8 ± 23.7 | 8.2 ± 1.9 |
| RANTES | 55.0 ± 19.7 | 42.5 ± 10.6 | 28.1 ± 19.3 | 38.2 ± 26.5 |
| TNF-α | 61.9 ± 14.8 | 88.3 ± 48.7 | 99.0 ± 51.4 | 68.0 ± 41.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chulpanova, D.S.; Gilazieva, Z.E.; Kletukhina, S.K.; Aimaletdinov, A.M.; Garanina, E.E.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Cytochalasin B-Induced Membrane Vesicles from Human Mesenchymal Stem Cells Overexpressing IL2 Are Able to Stimulate CD8+ T-Killers to Kill Human Triple Negative Breast Cancer Cells. Biology 2021, 10, 141. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10020141
Chulpanova DS, Gilazieva ZE, Kletukhina SK, Aimaletdinov AM, Garanina EE, James V, Rizvanov AA, Solovyeva VV. Cytochalasin B-Induced Membrane Vesicles from Human Mesenchymal Stem Cells Overexpressing IL2 Are Able to Stimulate CD8+ T-Killers to Kill Human Triple Negative Breast Cancer Cells. Biology. 2021; 10(2):141. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10020141
Chicago/Turabian StyleChulpanova, Daria S., Zarema E. Gilazieva, Sevindzh K. Kletukhina, Aleksandr M. Aimaletdinov, Ekaterina E. Garanina, Victoria James, Albert A. Rizvanov, and Valeriya V. Solovyeva. 2021. "Cytochalasin B-Induced Membrane Vesicles from Human Mesenchymal Stem Cells Overexpressing IL2 Are Able to Stimulate CD8+ T-Killers to Kill Human Triple Negative Breast Cancer Cells" Biology 10, no. 2: 141. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10020141