
Comparative Transcriptome Analysis Revealed Genes Involved in Sexual and Polyploid Growth Dimorphisms in Loach (Misgurnus anguillicaudatus)

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethic Statement
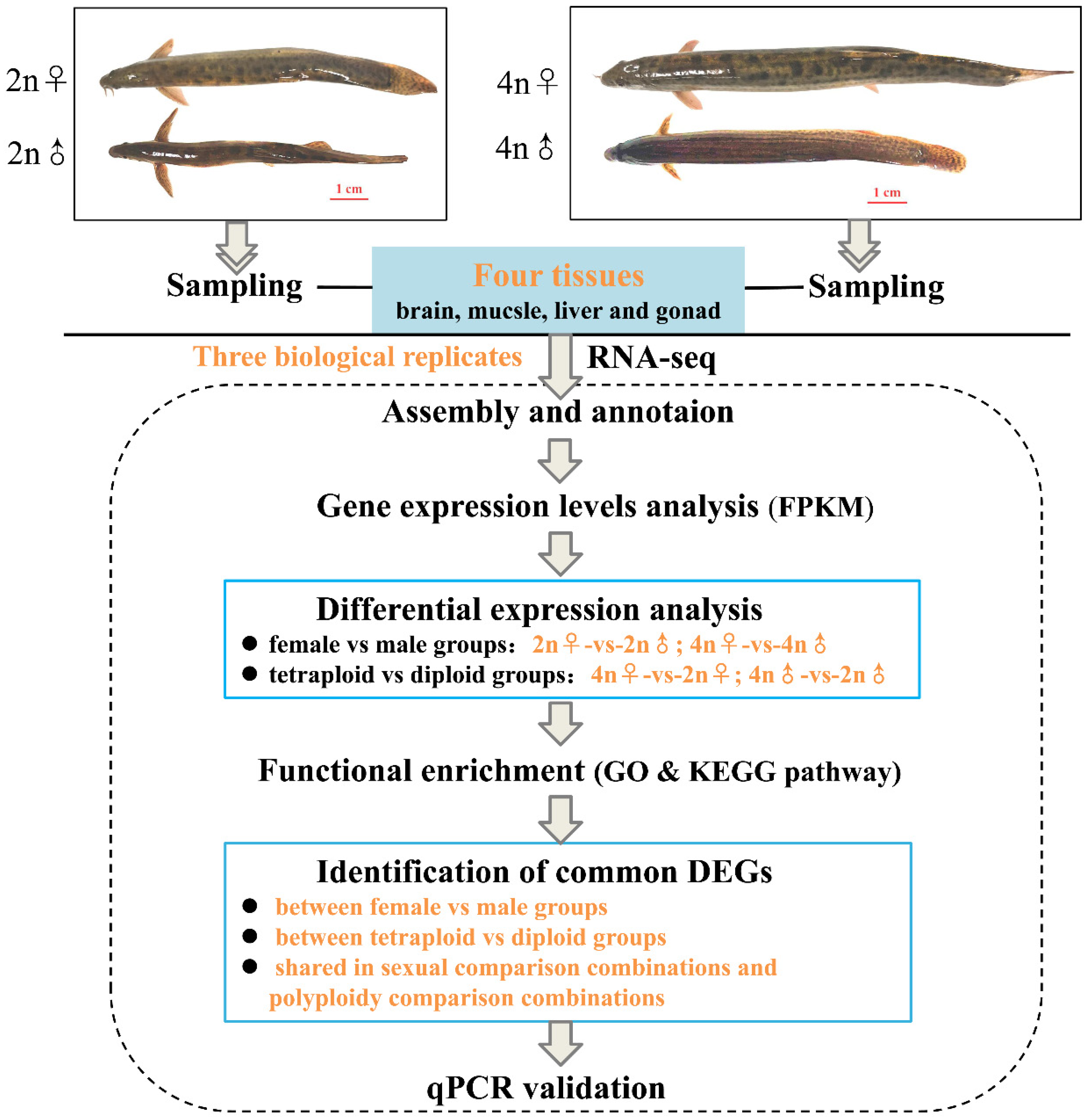
2.2. Sampling and RNA Extraction
2.3. Library Construction and Sequencing
2.4. Preliminary Analysis of the Transcriptome Data
2.5. Screening and Functional Analysis of Differentially Expressed Genes (DEGs)
2.6. Validation of RNA-Seq Data by Quantitative Real-Time PCR
3. Results
3.1. De novo Transcriptome Assembly and Quality Evaluation
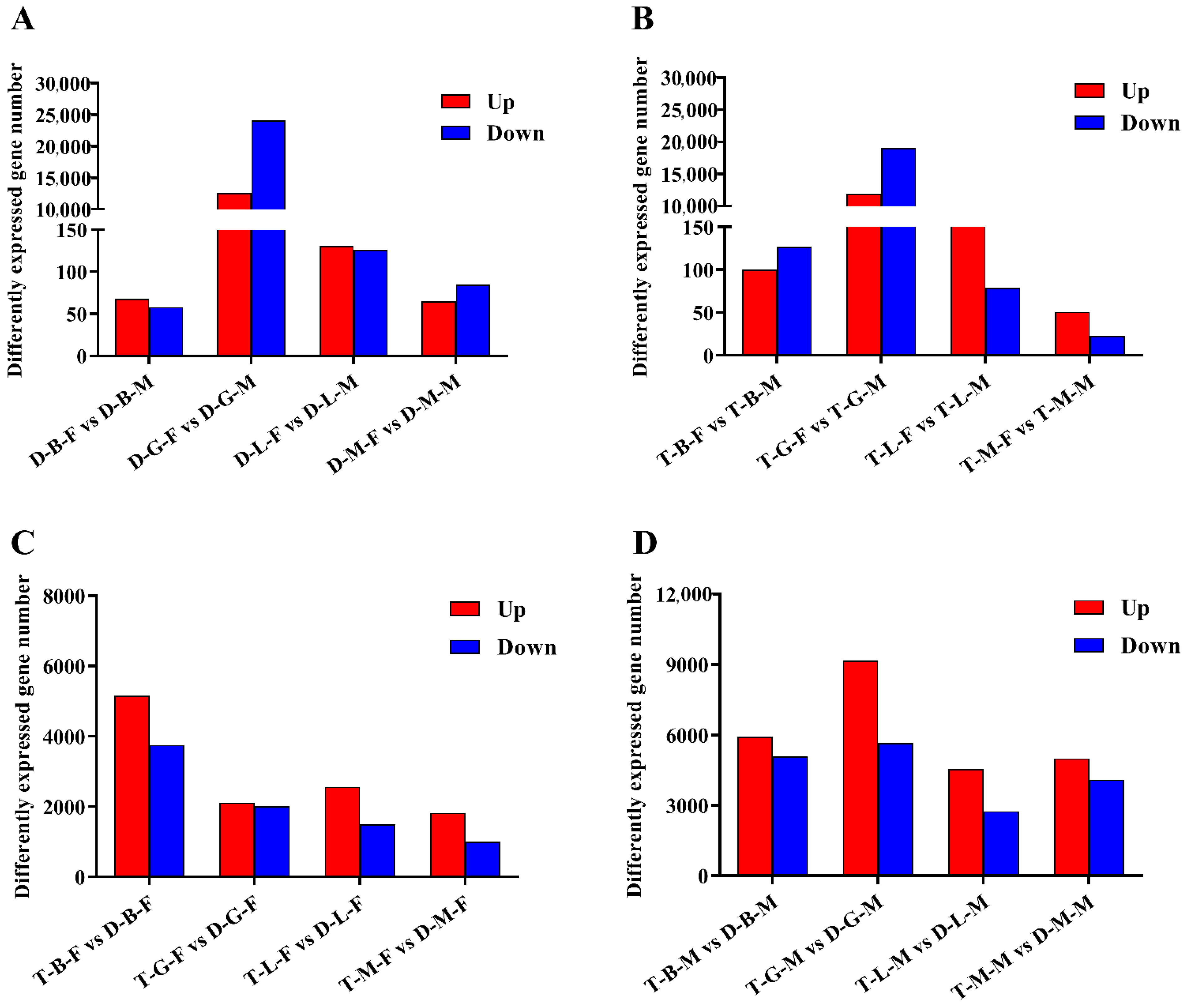
3.2. Analysis of DEGs between Female and Male Loaches
3.3. Analysis of DEGs between Tetraploid and Diploid Loaches
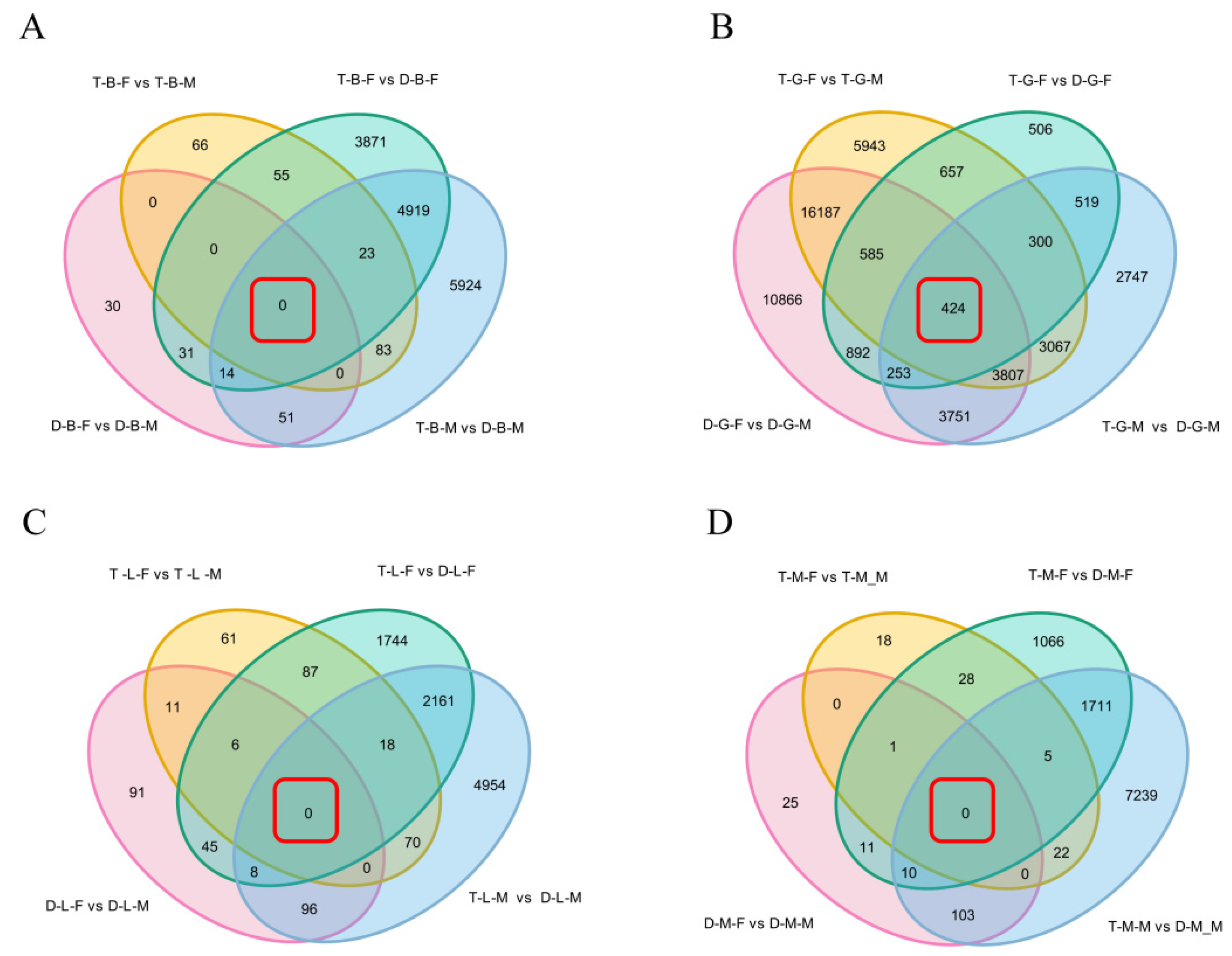
3.4. Identification of Common DEGs Shared in Different Comparison Groups in the Same Tissue
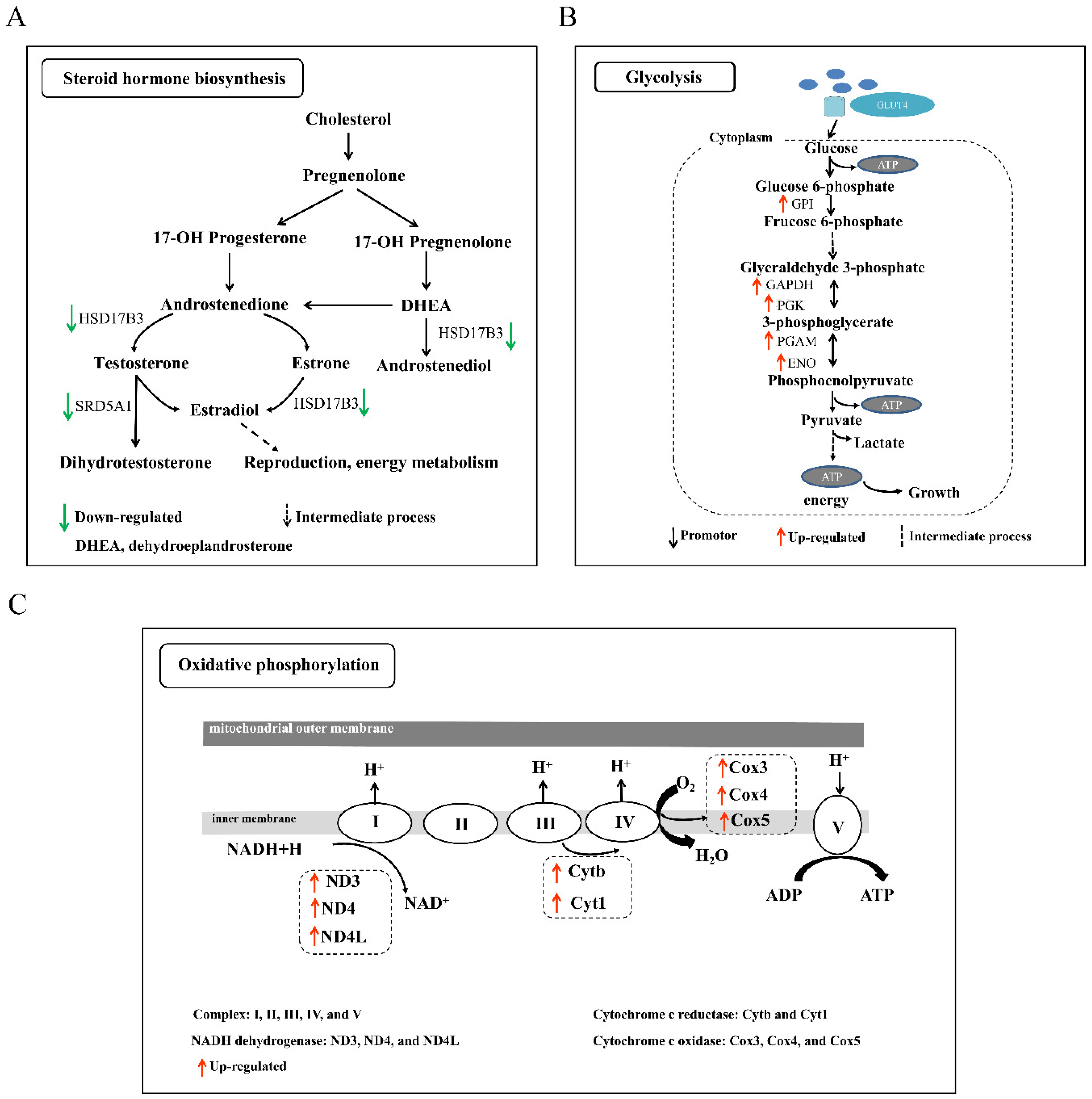
3.5. Analysis of KEGG Pathways Related to Sexual and Polyploid Growth Dimorphisms of Loaches
3.6. Verification of RNA-seq Data by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mei, J.; Gui, J. Genetic basis and biotechnological manipulation of sexual dimorphism and sex determination in fish. Sci. China Life Sci. 2015, 58, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Connallon, T.; Knowles, L. Intergenomic conflict revealed by patterns of sex-biased gene expression. Trends Genet. 2005, 21, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.; Snyder, M. Sexual dimorphism in mammalian gene expression. Trends Genet. 2005, 21, 298–305. [Google Scholar] [CrossRef]
- Ji, X. Artificial Gynogenesis, Genetic Analysis and Differential Expression of Growth-Related Genes in Half-Smooth Tongue Sole; Shandong Agricultural University: Shandong, China, 2009. (In Chinese) [Google Scholar]
- Wu, B. Difference of Growth Performance, Digestive Enzyme Activities and Growth Hormone (GH) Expressions between Male and Female Scatophagus argus Linnaeus; Guangdong Ocean University: Guangdong, China, 2013. [Google Scholar]
- Cutting, A.; Chue, J.; Smith, C. Just how conserved is vertebrate sex determination? Dev. Dyn. 2013, 242, 380–387. [Google Scholar] [CrossRef]
- Wang, B.; Guo, G.; Wang, C.; Lin, Y.; Wang, X.; Zhao, M.; Guo, Y.; He, M.; Zhang, Y.; Pan, L. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010, 38, 5075–5087. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, G.; Tian, J.; Liu, H.; Yang, H.; Yi, Y.; Wang, J.; Shi, X.; Jiang, F.; Yao, B.; et al. Transcriptome analysis of the silkworm (Bombyx mori) by high-throughput RNA sequencing. PLoS ONE 2012, 7, e43713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wang, Y.; Wang, S.; Liu, J.; Warren, W.; Mitreva, M.; Walter, R. Transcriptome analysis of female and male Xiphophorus maculatus Jp 163 A. PLoS ONE 2011, 6, e18379. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Rexroad, C.; Wang, J.; Thorgaard, G.; Yao, J. Characterization of the rainbow trout transcriptome using Sanger and 454-pyrosequencing approaches. BMC Genom. 2010, 11, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, W.; Yuan, J.; Zhou, L.; Sun, L.; Sun, Y.; Yang, S.; Li, M.; Zeng, S.; Huang, B.; Wang, D. Characterization of gonadal transcriptomes from nile tilapia (Oreochromis niloticus) reveals differentially expressed genes. PLoS ONE 2013, 8, e63604. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, C.; Huang, L.; Wu, M.; Zuo, Z. Transcriptome analysis of male and female Sebastiscus marmoratus. PLoS ONE 2012, 7, e50676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Liu, S.; Gao, X.; Jiang, Y.; Perera, D.; Wang, X.; Li, C.; Sun, L.; Zhang, J.; Kaltenboeck, L.; et al. Male-biased genes in catfish as revealed by RNA-seq analysis of the testis transcriptome. PLoS ONE 2013, 8, e68452. [Google Scholar] [CrossRef] [Green Version]
- Ribas, L.; Pardo, B.; Fernandez, C.; Antonio Alvarez-Dios, J.; Gomez-Tato, A.; Isabel Quiroga, M.; Planas, J.; Sitja-Bobadilla, A.; Martinez, P.; Piferrer, F. A combined strategy involving Sanger and 454 pyrosequencing increases genomic resources to aid in the management of reproduction, disease control and genetic selection in the turbot (Scophthalmus maximus). BMC Genom. 2013, 14, 180. [Google Scholar] [CrossRef] [Green Version]
- Krysanov, E.; Golubtsov, A. Karyotypes of four fish species from the Nile and Omo-Turkana basins in Ethiopia. J. Ichthyol. 2014, 54, 889–892. [Google Scholar] [CrossRef]
- Comber, S.; Smith, C. Polyploidy in fishes: Patterns and processes. Biol. J. Linn. Soc. 2004, 82, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Zou, T.; Chen, Y.; Chen, L.; Liu, S.; Tao, M.; Zhang, C.; Zhao, R.; Zhou, Y.; Long, Y.; et al. Coexistence of diploid, triploid and tetraploid crucian carp (Carassius auratus) in natural waters. BMC Genet. 2011, 12, 20. [Google Scholar] [CrossRef] [Green Version]
- Tao, M.; Liu, S.; Long, Y.; Zeng, C.; Liu, J.; Liu, L.; Zhang, C.; Duan, W.; Liu, Y. The cloning of Dmc1 cDNAs and a comparative study of its expression in different ploidy cyprinid fishes. Sci. China Ser. C Life Sci. 2008, 51, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ma, T.Y.; Zhang, R.; Xu, Q.; Shen, F.; Qin, Y.; Xu, W.; Wang, Y.; Li, Y. Analysis of different ploidy and parent-offspring genomic DNA methylation in the loach Misgurnus anguillicaudatus. Int. J. Mol. Sci. 2016, 17, 1299. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Yi, Z.; Liu, S.; Tao, M.; Long, Y.; Liu, Z.; Zhang, C.; Duan, W.; Hu, J.; Song, C.; et al. Elevated expressions of GH/IGF axis genes in triploid crucian carp. Gen Comp. Endocr. 2012, 178, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.; Liu, S.; Zhan, Z.; Chen, J.; Liu, W.; Liu, Y. Molecular cloning and comparative expression patterns of cyp19a1a of gene in different ploidy cyprinid fishes. J. Fish. China 2014, 38, 1201–1210. (In Chinese) [Google Scholar]
- Meirmans, P.; Liu, S.; Van Tienderen, P. The Analysis of Polyploid Genetic Data. J. Hered. 2018, 109, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Odei, D.; Hagen, O.; Peruzzi, S.; Falk-Petersen, I.; Fernandes, J. Transcriptome sequencing and histology reveal dosage compensation in the liver of triploid pre-smolt Atlantic salmon. Sci. Rep. 2020, 10, 16836. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Wu, Y.; Tao, M.; Zhang, C.; Liu, S. MicroRNA profiles reveal female allotetraploid hybrid fertility. BMC Genet. 2015, 16, 119. [Google Scholar] [CrossRef] [Green Version]
- Michaeloudes, C.; Kuo, C.; Haji, G.; Finch, D.; Halayko, A.; Kirkham, P.; Chung, K.; Adcock, I. Metabolic re-patterning in COPD airway smooth muscle cells. Eur. Respir. J. 2017, 50, 1700202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosterman, D.; Meyerhof, W.; Cottrell, G. Proton Transport Chains in Glucose Metabolism: Mind the Proton. Front. Neurosci. 2018, 12, 404. [Google Scholar] [CrossRef]
- Tixier, V.; Bataillé, L.; Etard, C.; Jagla, T.; Weger, M.; Daponte, J.; Strähle, U.; Dickmeis, T.; Jagla, K. Glycolysis supports embryonic muscle growth by promoting myoblast fusion. Proc. Natl. Acad. Sci. USA 2013, 110, 18982–18987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Q.; Miao, J.; Yang, Q.; Fang, L.; Fang, Y.; Ding, H.; Zhou, Y.; Jiang, L.; Dai, C.; Zen, K.; et al. Role of pyruvate kinase M2-mediated metabolic reprogramming during podocyte differentiation. Cell Death Dis. 2020, 11, 355. [Google Scholar] [CrossRef]
- Jiang, D.; LaGory, E.; Kenzelmann Brož, D.; Bieging, K.; Brady, C.; Link, N.; Abrams, J.; Giaccia, A.; Attardi, L. Analysis of p53 transactivation domain mutants reveals Acad11 as a metabolic target important for p53 pro-survival function. Cell Rep. 2015, 10, 1096–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.; Wu, S. Reprogramming with Small Molecules instead of Exogenous Transcription Factors. Stem Cells Int. 2015, 794632. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Yi, S.; Li, R.; Zhou, X. Comparison of age and growth performance of diploid and tetraploid loach Misgurnus anguillicaudatus in the Yangtze River basin, China. Environ. Biol. Fish 2017, 100, 815–828. [Google Scholar] [CrossRef]
- Zhong, J.; Yi, S.; Ma, L.; Wang, W. Evolution and phylogeography analysis of diploid and polyploid Misgurnus anguillicaudatus populations across China. Proc. Biol. Sci. 2019, 286, 20190076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.; Haas, B.; Yassour, M.; Levin, J.; Thompson, D.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.; Salzberg, S.; Wold, B.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.; Schmittgen, T. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Blay, C.; Haffray, P.; Bugeon, J.; D’Ambrosio, J.; Dechamp, N.; Collewet, G.; Enez, F.; Petit, V.; Cousin, X.; Corraze, G.; et al. Genetic parameters and genome-wide association studies of quality traits characterised using imaging technologies in rainbow trout, Oncorhynchus mykiss. Front. Genet. 2021, 12, 639223. [Google Scholar] [CrossRef]
- Wang, X.; Li, C.; Xie, Z.; Fan, W.; Zhang, J. Studies on the growth difference of the male and female Siniperca chuatsi. Freshw. Fish. 2006, 36, 34–37. [Google Scholar]
- Toguyeni, A.; Fauconneau, B.; Fostier, A.; Abucay, J.; Mair, G.; Baroiller, J. Influence of sexual phenotype and genotype, and sex ratio on growth performances in tilapia, Oreochromis niloticus. Aquaculture 2002, 207, 249–261. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, W.; He, Y.; Wu, J.; Dawar, F.; Ren, F.; Zhao, X.; Mei, J. Sex biased expression of ghrelin and GHSR associated with sexual size dimorphism in yellow catfish. Gene 2016, 578, 169–176. [Google Scholar] [CrossRef]
- Chatchaiphan, S.; Srisapoome, P.; Kim, J.; Devlin, R.; Na-Nakorn, U. De novo transcriptome characterization and growth-related gene expression profiling of diploid and triploid bighead catfish (Clarias macrocephalus Gunther, 1864). Mar. Biotechnol. 2017, 19, 36–48. [Google Scholar] [CrossRef]
- Ma, W.; Wu, J.; Zhang, J.; He, Y.; Gui, J.; Mei, J. Sex differences in the expression of GH/IGF axis genes underlie sexual size dimorphism in the yellow catfish (Pelteobagrus fulvidraco). Sci. China-Life Sci. 2016, 59, 431–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaxton, W. The organization and regulation of plant glycolysis. Annu. Rev. Plant Physiol. 1996, 47, 185–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes, L.; Le Roy, N.; Perret, M.; de Buffrenil, V.; Castanet, J.; Cubo, J. Relationships between bone growth rate, body mass and resting metabolic rate in growing amniotes: A phylogenetic approach. Biol. J. Linn. Soc. 2007, 92, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Tymchuk, W.; Sakhrani, D.; Devlin, R. Domestication causes large-scale effects on gene expression in rainbow trout: Analysis of muscle, liver and brain transcriptomes. Gen. Comp. Endocrinol. 2009, 164, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Danzmann, R.; Kocmarek, A.; Norman, J.; Rexroad, C.; Palti, Y. Transcriptome profiling in fast versus slow-growing rainbow trout across seasonal gradients. BMC Genom. 2016, 17, 60. [Google Scholar] [CrossRef] [Green Version]
- Robledo, D.; Rubiolo, J.; Cabaleiro, S.; Martínez, P.; Bouza, C. Differential gene expression and SNP association between fast- and slow-growing turbot (Scophthalmus maximus). Sci. Rep. 2017, 7, 12105. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.; Conaghan, J.; Winston, R.; Handyside, A. Increased number of cells and metabolic activity in male human preimplantation embryos following in vitro fertilization. J. Reprod. Fertil. 1995, 104, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Tiffin, G.; Rieger, D.; Betteridge, K.; Yadav, B.; King, W. Glucose and glutamine metabolism in pre-attachment cattle embryos in relation to sex and stage of development. J. Reprod. Fertil. 1991, 93, 125–132. [Google Scholar] [CrossRef]
- Rieger, D. Relationships between energy metabolism and development of early mammalian embryos. Theriogenology 1992, 37, 75–93. [Google Scholar] [CrossRef]
- Sorushanova, A.; Delgado, L.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; Mullen, A.; Bayon, Y.; Pandit, A.; Raghunath, M.; et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv. Mater. 2019, 31, e1801651. [Google Scholar] [CrossRef] [Green Version]
- Izu, Y.; Ezura, Y.; Koch, M.; Birk, D.; Noda, M. Collagens VI and XII form complexes mediating osteoblast interactions during osteogenesis. Cell Tissue Res. 2016, 364, 623–635. [Google Scholar] [PubMed] [Green Version]
- Wen, Y.; Yang, H.; Wu, J.; Wang, A.; Chen, X.; Hu, S.; Zhang, Y.; Bai, D.; Jin, Z. COL4A2 in the tissue-specific extracellular matrix plays important role on osteogenic differentiation of periodontal ligament stem cells. Theranostics 2019, 9, 4265–4286. [Google Scholar] [CrossRef]
- Redecker, T.; Kisko, T.; Schwarting, R.; Wohr, M. Effects of Cacna1c haploinsufficiency on social interaction behavior and 50-kHz ultrasonic vocalizations in adult female rats. Behav. Brain Res. 2019, 367, 35–52. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cao, M.; Zhang, A.; Shi, M.; Tao, B.; Li, Y.; Wang, Y.; Zhu, Z.; Trudeau, V.L.; Hu, W. Growth Hormone Overexpression Disrupts Reproductive Status Through Actions on Leptin. Front. Endocrinol. 2018, 9, 131. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Wangkahart, E.; Secombes, C.J.; Wang, T. Insights into the Evolution of the Suppressors of Cytokine Signaling (SOCS) Gene Family in Vertebrates. Mol. Biol. Evol. 2019, 36, 393–411. [Google Scholar] [CrossRef] [Green Version]
- Colom, B.; Oliver, J.; Roca, P.; Garcia-Palmer, F. Caloric restriction and gender modulate cardiac muscle mitochondrial H2O2 production and oxidative damage. Cardiovasc. Res. 2007, 74, 456–465. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Ge, H.; Li, H.; Lieber, S.; Natividad, F.; Resuello, R.; Kim, S.; Akeju, S.; Sun, A.; Loo, K.; et al. Gender-specific proteomic alterations in glycolytic and mitochondrial pathways in aging monkey hearts. J. Mol. Cell. Cardiol. 2004, 37, 921–929. [Google Scholar] [CrossRef]
- Brand, S.; Ebner, K.; Mikoteit, T.; Lejri, I.; Gerber, M.; Beck, J.; Holsboer-Trachsler, E.; Eckert, A. Influence of Regular Physical Activity on Mitochondrial Activity and Symptoms of Burnout-An Interventional Pilot Study. J. Clin. Med. 2020, 9, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, D. Oxidative phosphorylation: Unique regulatory mechanism and role in metabolic homeostasis. J. Appl. Physiol. 2017, 122, 611–619. [Google Scholar] [CrossRef]
- Klinge, C. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, B.; Clegg, D. The sexual dimorphism of obesity. Mol. Cell. Endocrinol. 2015, 402, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.; Bartholomew, C.; Craft, J. Differential expression of vitellogenin and oestrogen receptor genes in the liver of zebrafish, Danio Rerio. Anal. Bioanal. Chem. 2010, 396, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Qin, F.; Liu, S.; Wu, T.; Li, M.; Xu, P.; Zhang, X.; Wang, X.; Hu, G.; et al. Molecular characterization of estrogen receptor genes in loach Paramisgurnus dabryanus and their expression upon 17 alpha-ethinylestradiol exposure in juveniles. Gen. Comp. Endocr. 2012, 178, 194–205. [Google Scholar] [CrossRef]
- Speijer, D.; Manjeri, G.; Szklarczyk, R. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130446. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
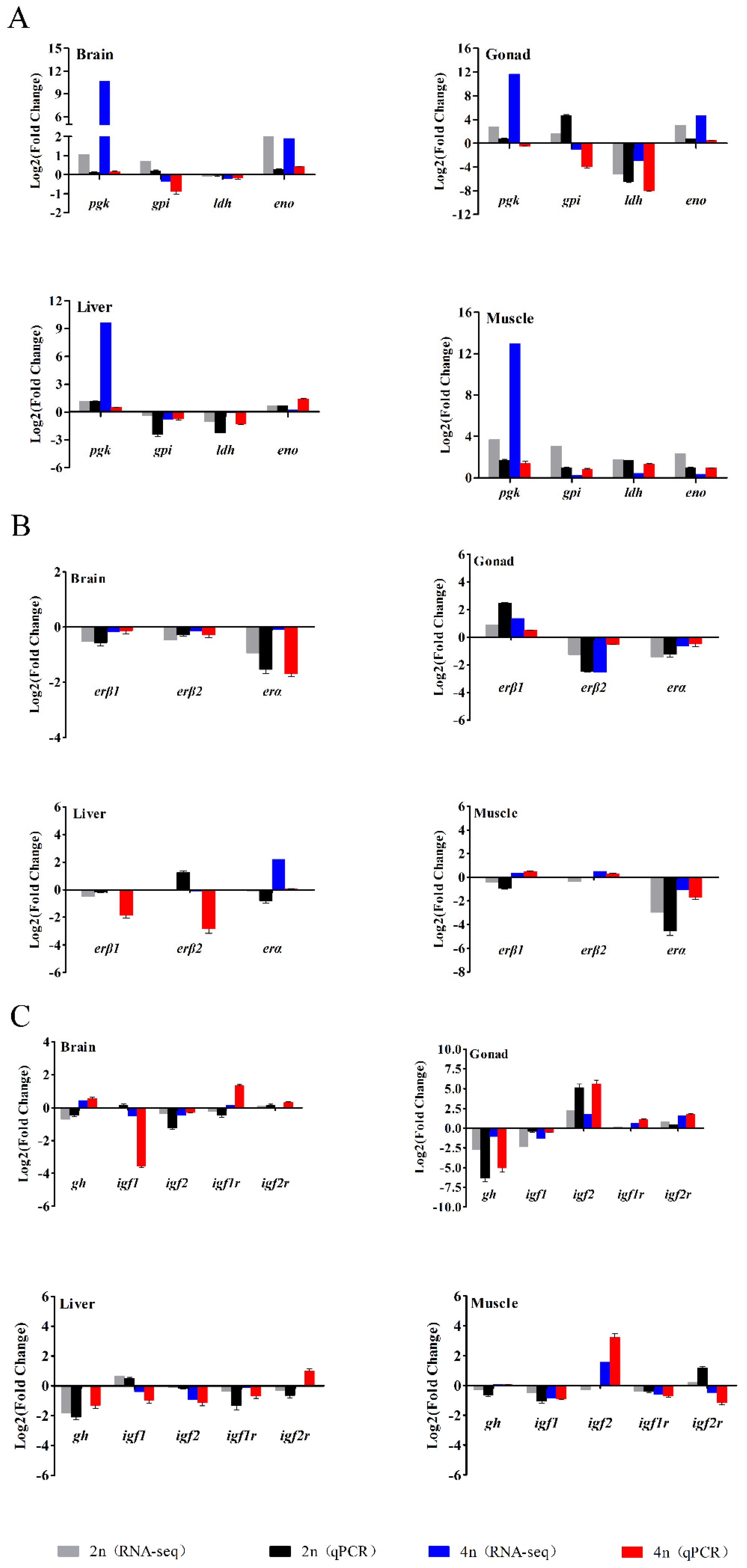{kind=link}
| Group | KEGG Terms | Upregulated Genes | Downregulated Genes | Corrected P-Value |
|---|---|---|---|---|
| D-B-F vs. D-B-M | Porphyrin and chlorophyll metabolism | E2.3.1.37(5-aminolevulinate synthase) | 4.36 × 10−2 | |
| Glycine, serine and threonine metabolism | E2.3.1.37(5-aminolevulinate synthase) | 4.36 × 10−2 | ||
| Arachidonic acid metabolism | alox5 | 4.36 × 10−2 | ||
| Protein digestion and absorption | cola1, cola2 | 8.82 × 10−2 | ||
| Ovarian steroidogenesis | alox5 | 4.36 × 10−2 | ||
| Steroid hormone biosynthesis | hsd17b3, srd5a1 | 4.36 × 10−2 | ||
| D-G-F vs. D-G-M | Cell cycle | cdc45, ccne, apc1, orc1, p53, mcm2 | 4.63 × 10−2 | |
| Spliceosome | cdc5, cype, prp17, prp18, eif4a1 | 7.83 × 10−2 | ||
| DNA replication | pcna, ssb, RNaseHI, mcm2, mcm4 | 8.47 × 10−2 | ||
| D-L-F vs. D-L-M | Taurine and hypotaurine metabolism | csad | 3.39 × 10−1 | |
| Arginine and proline metabolism | prodh2 | aoc1 | 3.39 × 10−1 | |
| D-M-F vs. D-M-M | Glycolysis/gluconeogenesis | gpi, pgk, pgam, ldh | 2.58 × 10−7 | |
| Glucagon signaling pathway | pgam, ldh | 9.64 × 10−3 | ||
| Starch and sucrose metabolism | gpi | 2.30 × 10−2 | ||
| T-B-F vs. T-B-M | Glycolysis/gluconeogenesis | pgk | 4.88 × 10−1 | |
| Protein digestion and absorption | col1a, cola2 | 3.31 × 10−1 | ||
| Drug metabolism-cytochrome P450 | fmo | 3.54 × 10−1 | ||
| Taurine and hypotaurine metabolism | ggt1-5 | 4.88 × 10−1 | ||
| Cardiac muscle contraction | cacna1c, cacna1d | 4.94 × 10−1 | ||
| GnRH signaling pathway | cacna1c, cacna1f | 4.88 × 10−1 | ||
| Insulin secretion | cacna1c | 4.88 × 10−1 | ||
| MAPK signaling pathway | cacna1a | 5.77 × 10−1 | ||
| Calcium signaling pathway | cacna1c | 5.77 × 10-1 | ||
| T-G-F vs. T-G-M | Ribosome biogenesis in eukaryotes | utp22, utp6, imp3, ck2a, emg1 | 2.77 × 10−1 | |
| Protein processing in endoplasmic reticulum | atf6, ire1, eif2ak1, eif2α, ssr1 | 3.17 × 10−1 | ||
| T-L-F vs. T-L-M | Ribosome | lp1, lp2, l11e, l23e | 4.92 × 10−11 | |
| Protein processing in endoplasmic reticulum | ssr1 | hsp70 | 8.01 × 10−2 | |
| Glycolysis/gluconeogenesis | pfka, pgk | 2.93 × 10−1 | ||
| Biosynthesis of unsaturated fatty acids | △9-desaturase | 2.26 × 10−2 | ||
| Steroid hormone biosynthesis | cyp7a1, hsd11b2 | 7.18 × 10−2 | ||
| T-M-F vs. T-M-M | Tight junction | myl6, myl12, myh9 | 3.85 × 10−2 | |
| DNA replication | rfc3/5 | 1.98 × 10−1 | ||
| Linoleic acid metabolism | cyp2j | 1.98 × 10−1 | ||
| Fatty acid degradation | cpt1 | 2.16 × 10−1 | ||
| T-B-F vs. D-B-F | Oxidative phosphorylation | cox3, cox5b, cytb, nd3, nd4 | cox1, atpef0a | 1.04 × 10−4 |
| Cell cycle | pcna, cyca, cycb, apc/c | 4.07 × 10−1 | ||
| PI3K-Akt signaling pathway | creb, akt, pp2a, ras | itga, pkc, bim | 7.58 × 10−1 | |
| PPAR signaling pathway | lpl, acs, scp | 8.70 × 10−1 | ||
| Protein digestion and absorption | slc15a1, mme | 1.20 × 10−1 | ||
| Regulation of actin cytoskeleton | actb-g1, myl2, myl5 | egfr, fgfr2, fgfr3 | 9.99 × 10−1 | |
| Drug metabolism-cytochrome P450 | mao, ugt, gst | 6.32 × 10−1 | ||
| GnRH signaling pathway | slc5a6, rft2 | cubn, abcc1 | 9.99 × 10−1 | |
| Estrogen signaling pathway | gper, mmp, creb1 | 7.58× 10−1 | ||
| Fatty acid degradation | mecr, fada, acaa2 | 9.99 × 10−1 | ||
| Fatty acid elongation | elovl1, mecr, acaa2, ter | 8.70 × 10−1 | ||
| Cardiac muscle contraction | myh6/7, myl2, myl3, atp1a, atp1b | 3.82 × 10−1 | ||
| T-G-F vs. D-G-F | Oxidative phosphorylation | cox3, nd4, nd4l, cytb | cox1, sdhb | 6.35 × 10−1 |
| Arachidonic acid metabolism | prxl2b, cbr1, cbr2, ptgds, cyp4f, alox12 | 6.35 × 10−1 | ||
| p53 signaling pathway | igf, tsp, mdm2, cyclinb, casp8, perp | 6.35 × 10−1 | ||
| Ascorbate and aldarate metabolism | 6.35 × 10−1 | |||
| T-L-F vs. D-L-F | Oxidative phosphorylation | cox3, nd4, nd4l, cytb | cox1 | 1.77 × 10−6 |
| Mineral absorption | zip4, atpase | 1.04 × 10−5 | ||
| Vitamin digestion and absorption | apob-48, lrat, rft2 | 1.64 × 10−4 | ||
| T-M-F vs. D-M-F | Tight junction | actin | myl6, myl1, myh9 | 5.02 × 10−5 |
| Cardiac muscle contraction | actin, tpm1 | 4.64 × 10−13 | ||
| Oxidative phosphorylation | cox3, nd4, nd4l, cytb, qcr2, qcr10 | cox1 | 1.00 × 10−9 | |
| Protein digestion and absorption | slc8a, mme | slc3a2 | 6.82 × 10−5 | |
| T-B-M vs. D-B-M | Oxidative phosphorylation | cox3, cox5, cytb, nd3, nd4 | cox1, atpef0a | 2.30 × 10−2 |
| Glutathione metabolism | gpx, anpep, rrm1 | gpx4, ggct | 1.45 × 10−2 | |
| Steroid hormone biosynthesis | cyp11b1, cyp11b2, srd5a1 | 1.88 × 10−1 | ||
| T-G-M vs. D-G-M | Cell cycle | cdc45, ccne, apc1, orc1, p53, mcm2 | 3.50 × 10−6 | |
| DNA replication | pcna, mcm2, mcm4 | 4.25 × 10−7 | ||
| Regulation of actin cytoskeleton | iqgap, fak, rho, pak1, f-actin, drf3 | 1.04 × 10−1 | ||
| T-L-M vs. D-L-M | Mineral absorption | mt, fpn1 | hmox, znt1 | 1.51 × 10−3 |
| Cardiac muscle contraction | oplah, gpx, ggt | ggct, g6pd | 5.45 × 10−1 | |
| Steroid hormone biosynthesis | cyp2r1, cyp51, dwf, fdft1 | 1.18 × 10−6 | ||
| Fat digestion and absorption | abca1, apoa, dgat | 2.44 × 10−3 | ||
| Oxidative phosphorylation | cox3, cox5, cytb, nd3, nd4 | 1.40 × 10−2 | ||
| Glycolysis/gluconeogenesis | fbp, tpi, eno, adh1, g6pc | 1.93 × 10−1 | ||
| T-M-M vs. D-M-M | Glycolysis/gluconeogenesis | gpi, pgk, pgam, ldh, tpi | 6.20 × 10−7 | |
| Tight junction | actin, lgl1 | claudin, pp2a, myl2 | 1.00 × 10−9 | |
| Cardiac muscle contraction | actin, tpm, atp | tnt, tnc | 7.34 × 10−14 | |
| Oxidative phosphorylation | cox3, nd4, nd4l, cytb, qcr2, qcr10 | cox1 | 7.34 × 10−14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.-F.; Xu, Z.-S.; Elgazzar, E.A.B.E.; Du, H.; Li, D.-Y.; Zhou, X.-Y.; Gao, Z.-X. Comparative Transcriptome Analysis Revealed Genes Involved in Sexual and Polyploid Growth Dimorphisms in Loach (Misgurnus anguillicaudatus). Biology 2021, 10, 935. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090935
Luo L-F, Xu Z-S, Elgazzar EABE, Du H, Li D-Y, Zhou X-Y, Gao Z-X. Comparative Transcriptome Analysis Revealed Genes Involved in Sexual and Polyploid Growth Dimorphisms in Loach (Misgurnus anguillicaudatus). Biology. 2021; 10(9):935. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090935
Chicago/Turabian StyleLuo, Li-Fei, Zi-Sheng Xu, Eman Abdelwareth Baioumy Elsayed Elgazzar, Hang Du, Dan-Yang Li, Xiao-Yun Zhou, and Ze-Xia Gao. 2021. "Comparative Transcriptome Analysis Revealed Genes Involved in Sexual and Polyploid Growth Dimorphisms in Loach (Misgurnus anguillicaudatus)" Biology 10, no. 9: 935. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090935