
Diversity and Evolution of pogo and Tc1/mariner Transposons in the Apoidea Genomes

 , , and
, , and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
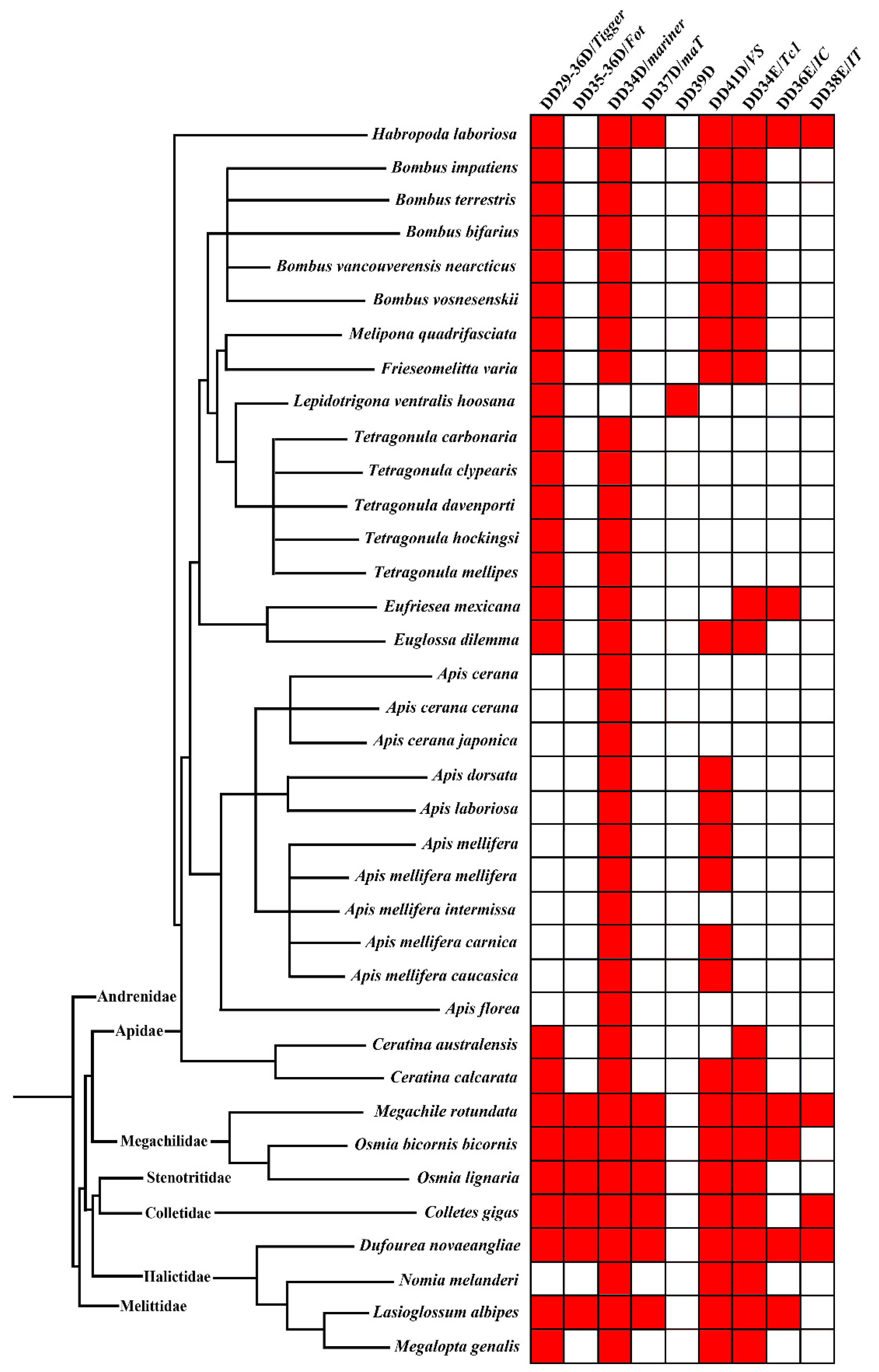
2.1. Distribution of pogo and Tc1/mariner within Apoidea
2.2. Phylogenetic Analysis and Protein Domain Prediction
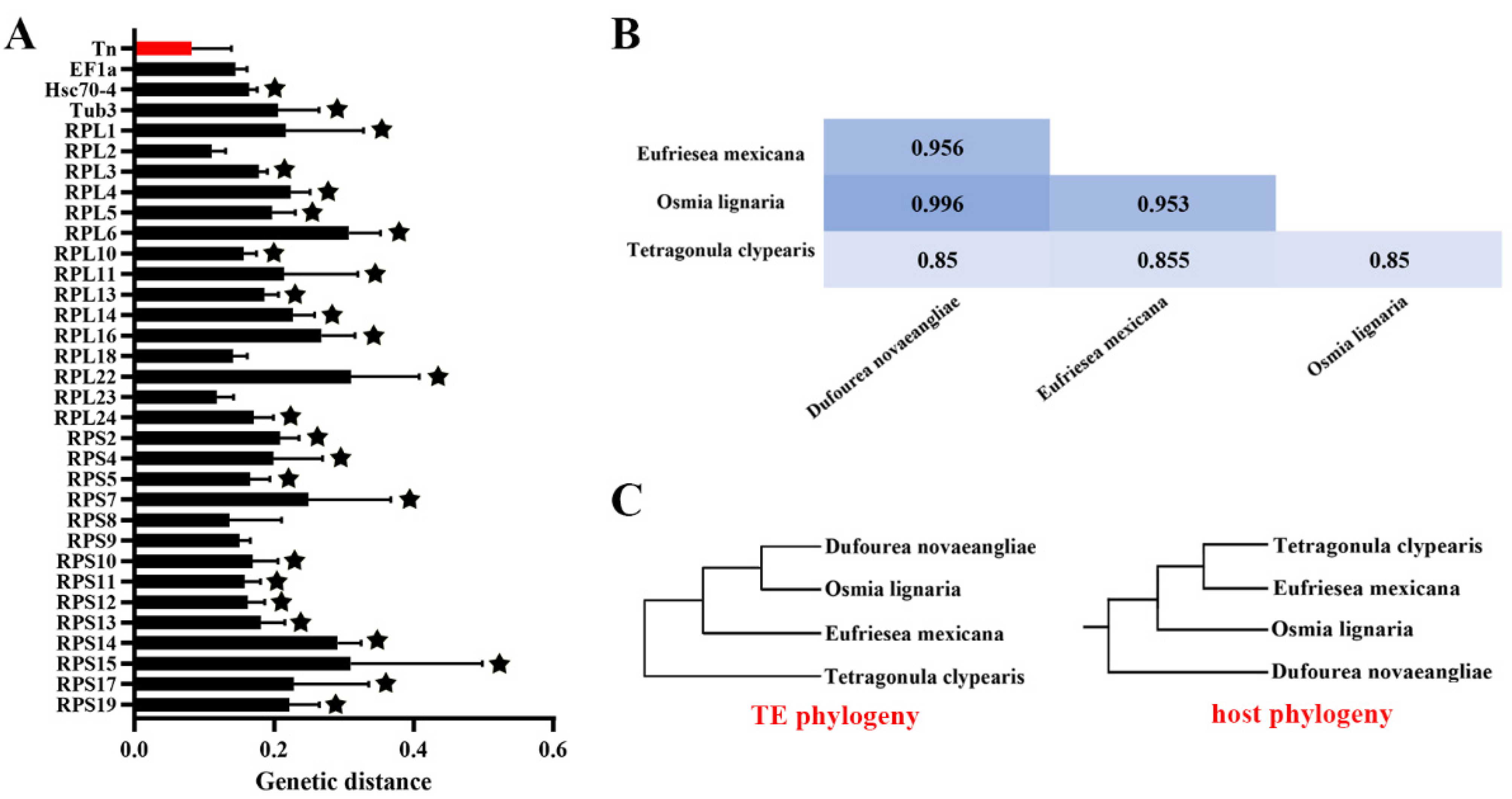
2.3. Horizontal Transfer and Evolutionary Dynamics Analysis
3. Results
3.1. Diversity and Distribution of pogo and Tc1/mariner Elements in the Apoidea Genomes
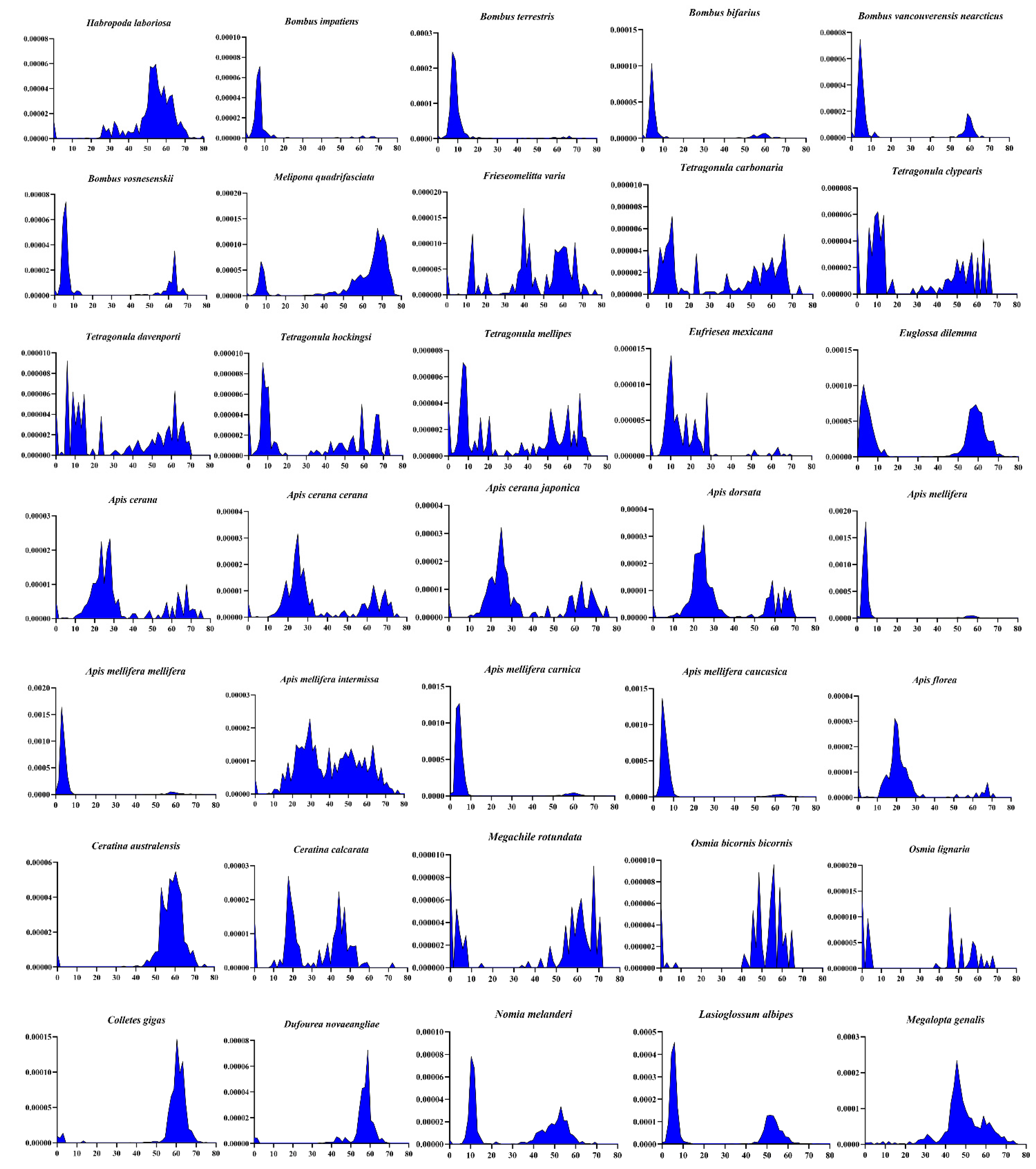
3.2. Invasions of pogo and Tc1/mariner in the Apoidea Genomes
3.3. Evolutionary Patterns of pogo and Tc1/mariner in Apoidea
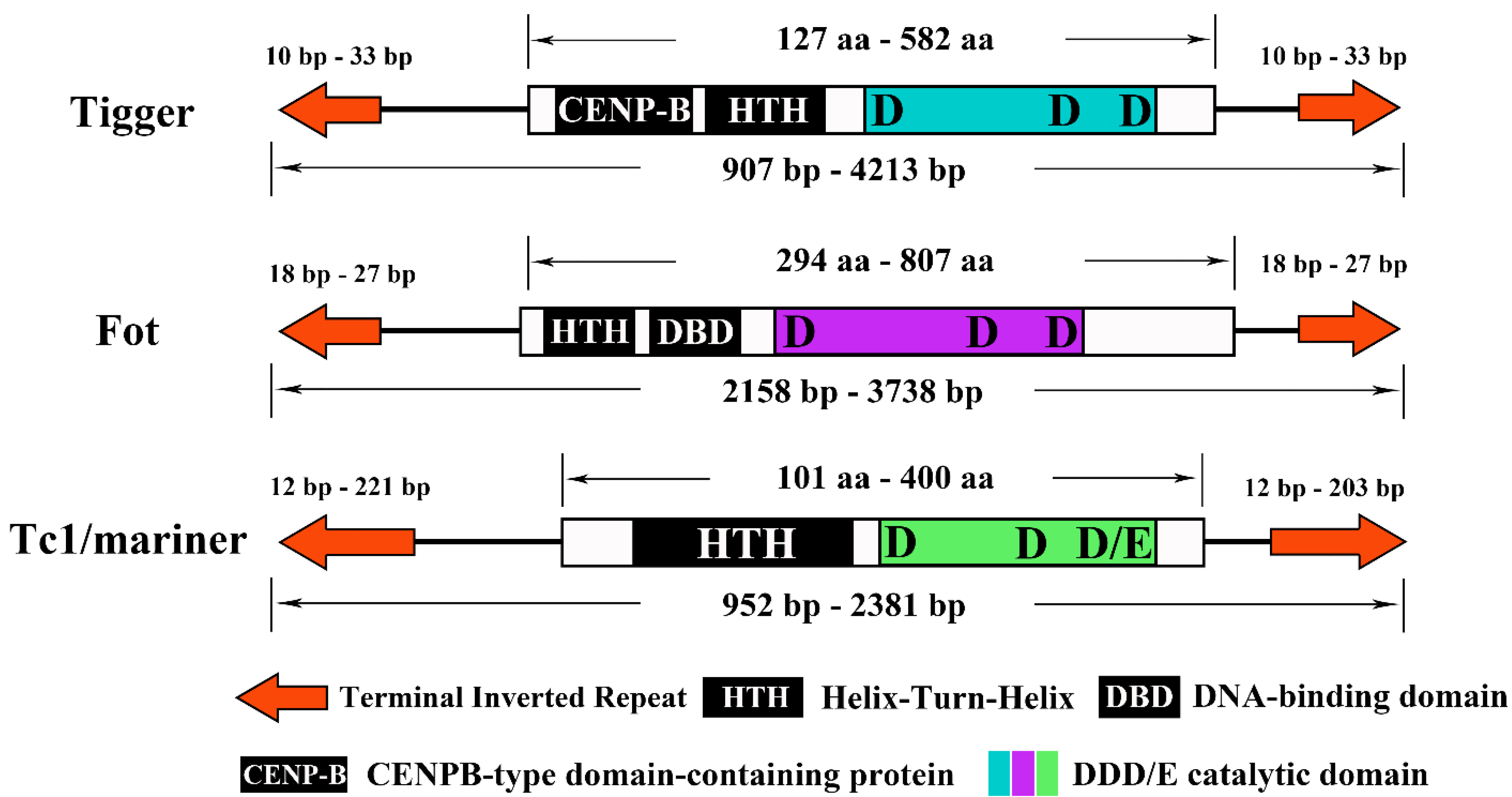
3.4. Structural Organization of the Detected pogo and Tc1/mariner Transposons
4. Discussion
4.1. Distribution, Diversity and Copy Number in Apoidea
4.2. Differential Susceptibility of Apoidea Species to DNA Transposons
4.3. HT Events in Apoidea
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Danforth, B.N.; Sipes, S.; Fang, J.; Brady, S.G. The history of early bee diversification based on five genes plus morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 15118–15123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascher, J.S.; Pickering, J. Discover Life Bee Species Guide and World Checklist (Hymenoptera:Apoidea:Anthophila). Available online: https://bugguide.net/node/view/468583 (accessed on 27 May 2021).
- Danforth, B.N.; Cardinal, S.; Praz, C.; Almeida, E.A.B.; Michez, D. The impact of molecular data on our understanding of bee phylogeny and evolution. Annu. Rev. Entomol. 2013, 58, 57–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potts, S.G.; Imperatriz-Fonseca, V.; Ngo, H.T.; Aizen, M.A.; Biesmeijer, J.C.; Breeze, T.D.; Dicks, L.V.; Garibaldi, L.A.; Hill, R.; Settele, J.; et al. Safeguarding pollinators and their values to human well-being. Nature 2016, 540, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Hepburn, H.R. The Bees of the World. Afr. Zool. 2001. [Google Scholar] [CrossRef]
- Danforth, B.N.; Fang, J.; Sipes, S. Analysis of family-level relationships in bees (Hymenoptera: Apiformes) using 28S and two previously unexplored nuclear genes: CAD and RNA polymerase II. Mol. Phylogenet. Evol. 2006, 39, 358–372. [Google Scholar] [CrossRef]
- Roberts, R.B. Genesis of the Hymenoptera and the Phases of Their Evolution. Bull. Entomol. Soc. Am. 1969, 15. [Google Scholar] [CrossRef] [Green Version]
- Engel, M.S. A monograph of the baltic amber bees and evolution of the apoidea (Hymenoptera). Bull. Am. Mus. Nat. Hist. 2001, 259, 1–192. [Google Scholar] [CrossRef]
- Ilyasov, R.A.; Poskryakov, A.V.; Nikolenko, A.G. Modern methods of assessing the taxonomic affiliation of honeybee colonies. Ecol. Genet. 2017, 15, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Momeni, J.; Parejo, M.; Nielsen, R.O.; Langa, J.; Montes, I.; Papoutsis, L.; Farajzadeh, L.; Bendixen, C.; Căuia, E.; Charrière, J.D.; et al. Authoritative subspecies diagnosis tool for European honey bees based on ancestry informative SNPs. BMC Genom. 2021, 22, 101. [Google Scholar] [CrossRef]
- Orgel, L.E.; Crick, F.H.C. Selfish DNA: The ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Oliver, K.R.; Greene, W.K. Transposable elements and viruses as factors in adaptation and evolution: An expansion and strengthening of the TE-Thrust hypothesis. Ecol. Evol. 2012, 2, 2912–2933. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, M.I.; Hollister, J.D.; Gaut, B.S. A triptych of the evolution of plant transposable elements. Trends Plant. Sci. 2010, 15, 471–478. [Google Scholar] [CrossRef]
- Gao, B.; Shen, D.; Xue, S.; Chen, C.; Cui, H.; Song, C. The contribution of transposable elements to size variations between four teleost genomes. Mob. DNA 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, J.L.; Peyton, J.T.; Fiston-Lavier, A.S.; Teets, N.M.; Yee, M.C.; Johnston, J.S.; Bustamante, C.D.; Lee, R.E.; Denlinger, D.L. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment. Nat. Commun. 2014, 5, 4611. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Fang, X.; Yang, P.; Jiang, X.; Jiang, F.; Zhao, D.; Li, B.; Cui, F.; Wei, J.; Ma, C.; et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 2014, 5, 2957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chénais, B.; Caruso, A.; Hiard, S.; Casse, N. The impact of transposable elements on eukaryotic genomes: From genome size increase to genetic adaptation to stressful environments. Gene 2012, 509, 7–15. [Google Scholar] [CrossRef]
- Rebollo, R.; Zhang, Y.; Mager, D.L. Transposable elements: Not as quiet as a mouse. Genome Biol. 2012, 13, 159. [Google Scholar] [CrossRef]
- Feschotte, C.; Pritham, E.J. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007, 41, 331–368. [Google Scholar] [CrossRef] [Green Version]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Jacobson, J.W.; Medhora, M.M.; Hartl, D.L. Molecular structure of a somatically unstable transposable element in Drosophila. Proc. Natl. Acad. Sci. USA 1986, 83, 8684–8688. [Google Scholar] [CrossRef] [Green Version]
- Emmons, S.W.; Yesner, L.; Ruan, K.s.; Katzenberg, D. Evidence for a transposon in caenorhabditis elegans. Cell 1983, 32, 55–65. [Google Scholar] [CrossRef]
- Palomeque, T.; Sanllorente, O.; Maside, X.; Vela, J.; Mora, P.; Torres, M.I.; Periquet, G.; Lorite, P. Evolutionary history of the Azteca-like mariner transposons and their host ants. Sci. Nat. 2015, 102, 44. [Google Scholar] [CrossRef] [PubMed]
- Coy, M.R.; Tu, Z. Gambol and Tc1 are two distinct families of DD34E transposons: Analysis of the Anopheles gambiae genome expands the diversity of the IS630-Tc1-mariner superfamily. Insect Mol. Biol. 2005, 14, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Hernandez, E.M.; Fernández-Medina, R.D.; Navarro-Escalante, L.; Nuñez, J.; Benavides-Machado, P.; Carareto, C.M.A. Genome-wide analysis of transposable elements in the coffee berry borer Hypothenemus hampei (Coleoptera: Curculionidae): Description of novel families. Mol. Genet. Genom. 2017, 292, 565–583. [Google Scholar] [CrossRef] [PubMed]
- De Melo, E.S.; Wallau, G.L. Mosquito genomes are frequently invaded by transposable elements through horizontal transfer. PLoS Genet. 2020, 16, e1008946. [Google Scholar] [CrossRef]
- Subramanian, R.A.; Akala, O.O.; Adejinmi, J.O.; O’Brochta, D.A. Topi, an IS630/Tc1/mariner-type transposable element in the African malaria mosquito, Anopheles gambiae. Gene 2008, 423, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Zong, W.; Miskey, C.; Ullah, N.; Diaby, M.; Chen, C.; Wang, X.; Ivics, Z.; Song, C. Intruder (DD38E), a recently evolved sibling family of DD34E/Tc1 transposons in animals. Mob. DNA 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Sang, Y.; Gao, B.; Diaby, M.; Zong, W.; Chen, C.; Shen, D.; Wang, S.; Wang, Y.; Ivics, Z.; Song, C. Incomer, a DD36E family of Tc1/mariner transposons newly discovered in animals. Mob. DNA 2019, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Gao, B.; Miskey, C.; Chen, C.; Sang, Y.; Zong, W.; Wang, S.; Wang, Y.; Wang, X.; Ivics, Z.; et al. Multiple invasions of visitor, a DD41D family of tc1/mariner transposons, throughout the evolution of vertebrates. Genome Biol. Evol. 2020, 12, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Diaby, M.; Puzakov, M.; Ullah, N.; Wang, Y.; Danley, P.; Chen, C.; Wang, X.; Gao, B.; Song, C. Divergent evolution profiles of DD37D and DD39D families of Tc1/mariner transposons in eukaryotes. Mol. Phylogenet. Evol. 2021, 161, 107143. [Google Scholar] [CrossRef]
- Shao, H.; Tu, Z. Expanding the diversity of the IS630-Tc1-mariner superfamily: Discovery of a unique DD37E transposon and reclassification of the DD37D and DD39D transposons. Genetics 2001, 159, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 29, e05378. [Google Scholar] [CrossRef] [PubMed]
- Vos, J.C.; van Luenen, H.G.; Plasterk, R.H. Characterization of the Caenorhabditis elegans Tc1 transposase in vivo and in vitro. Genes Dev. 1993, 7, 1244–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radice, A.D.; Bugaj, B.; Fitch, D.H.A.; Emmons, S.W. Widespread occurrence of the Tc1 transposon family: Tc1-like transposons from teleost fish. Mol. Gen. Genet. 1994, 244, 606–612. [Google Scholar] [CrossRef]
- Lam, W.L.; Seo, P.; Robison, K.; Virk, S.; Gilbert, W. Discovery of amphibian Tc1-like transposon families. J. Mol. Biol. 1996, 257, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Sinzelle, L.; Pollet, N.; Bigot, Y.; Mazabraud, A. Characterization of multiple lineages of Tc1-like elements within the genome of the amphibian Xenopus tropicalis. Gene 2005, 349, 187–196. [Google Scholar] [CrossRef]
- Robertson, H.M. The mariner transposable element is widespread in insects. Nature 1993, 362, 241–245. [Google Scholar] [CrossRef]
- Plasterk, R.H.A.; Izsvák, Z.; Ivics, Z. Resident aliens the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999, 15, 326–332. [Google Scholar] [CrossRef]
- Arkhipova, I.R.; Meselson, M. Diverse DNA transposons in rotifers of the class Bdelloidea. Proc. Natl. Acad. Sci. USA 2005, 102, 11781–11786. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.H.; Hermann, D.; Caruso, A.; Tastard, E.; Marchand, J.; Rouault, J.D.; Denis, F.; Thiriet-Rupert, S.; Casse, N.; Morant-Manceau, A. First Evidence of Mariner-like Transposons in the Genome of the Marine Microalga Amphora acutiuscula (Bacillariophyta). Protist 2014, 165, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.; Gao, B.; Diaby, M.; Shen, D.; Wang, S.; Wang, Y.; Sang, Y.; Chen, C.; Wang, X.; Song, C. Traveler, a new DD35E family of Tc1/mariner transposons, invaded vertebrates very recently. Genome Biol. Evol. 2020, 12, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, H.M.; Asplund, M.L. Bmmar1: A basal lineage of the Mariner family of transposable elements in the silkworm moth, Bombyx mori. Insect Biochem. Mol. Biol. 1996, 26, 945–954. [Google Scholar] [CrossRef]
- Gilchrist, A.S.; Shearman, D.C.A.; Frommer, M.; Raphael, K.A.; Deshpande, N.P.; Wilkins, M.R.; Sherwin, W.B.; Sved, J.A. The draft genome of the pest tephritid fruit fly Bactrocera tryoni: Resources for the genomic analysis of hybridising species. BMC Genom. 2014, 15, 1153. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Li, G.Y.; Xiong, X.M.; Han, M.J.; Dai, F.Y. TRT, a Vertebrate and Protozoan Tc1-Like Transposon: Current Activity and Horizontal Transfer. Genome Biol. Evol. 2016, 8, 2994–3005. [Google Scholar] [CrossRef] [Green Version]
- Tudor, M.; Lobocka, M.; Goodell, M.; Pettitt, J.; O’Hare, K. The pogo transposable element family of Drosophila melanogaster. Mol. Gen. Genet. MGG 1992, 232, 126–134. [Google Scholar] [CrossRef]
- Smit, A.F.A.; Riggs, A.D. Tiggers and other DNA transposon fossils in the human genome. Proc. Natl. Acad. Sci. USA 1996, 93, 1443–1448. [Google Scholar] [CrossRef] [Green Version]
- Dufresne, M.; Lespinet, O.; Daboussi, M.J.; Hua-Van, A. Genome-wide comparative analysis of pogo-like transposable elements in different Fusarium species. J. Mol. Evol. 2011, 73, 230–243. [Google Scholar] [CrossRef]
- Hey, P.; Robson, G.; Birch, M.; Bromley, M. Characterisation of Aft1 a Fot1/Pogo type transposon of Aspergillus fumigatus. Fungal Genet. Biol. 2008, 45, 117–126. [Google Scholar] [CrossRef]
- Nyyssönen, E.; Amutan, M.; Enfield, L.; Stubbs, J.; Dunn-Coleman, N.S. The transposable element Tan1 of Aspergillus niger var. awamori, a new member of the Fot1 family. Mol. Gen. Genet. 1996, 253, 50–56. [Google Scholar] [CrossRef]
- Levis, C.; Fortini, D.; Brygoo, Y. Flipper, a mobile Fot1-like transposable element in Botrytis cinerea. MGG Mol. Gen. Genet. 1997, 254, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, P.; Leong, S.A.; Chattoo, B.B. Pot2, an inverted repeat transposon from the rice blast fungus Magnaporthe grisea. MGG Mol. Gen. Genet. 1994, 245, 339–348. [Google Scholar] [CrossRef]
- Daboussi, M.J.; Langin, T.; Brygoo, Y. Fot1, a new family of fungal transposable elements. MGG Mol. Gen. Genet. 1992, 232, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Feschotte, C.; Mouchès, C. Evidence that a family of miniature inverted-repeat transposable elements (MITEs) from the Arabidopsis thaliana genome has arisen from a pogo- like DNA transposon. Mol. Biol. Evol. 2000, 17, 730–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, B.; Chen, W.; Shen, D.; Wang, S.; Chen, C.; Zhang, L.; Wang, W.; Wang, X.; Song, C. Characterization of autonomous families of Tc1/mariner transposons in neoteleost genomes. Mar. Genom. 2017, 34, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Wang, Y.; Diaby, M.; Zong, W.; Shen, D.; Wang, S.; Chen, C.; Wang, X. Evolution of pogo, a separate superfamily of IS630-Tc1-mariner transposons, revealing recurrent domestication events in vertebrates. Mob. DNA 2020, 11, 25. [Google Scholar] [CrossRef]
- Sumitani, M.; Lee, J.M.; Hatakeyama, M.; Oishi, K. Cloning and characterization of Acmar1, a mariner-like element in the Asiatic honey bee, Apis cerana japonica (Hymenoptera, Apocrita). Arch. Insect Biochem. Physiol. 2002, 50, 183–190. [Google Scholar] [CrossRef]
- Rouleux-Bonnin, F.; Petit, A.; Demattei, M.V.; Bigot, Y. Evolution of full-length and deleted forms of the Mariner-like element, Botmar1, in the genome of the bumble bee, Bombus terrestris (hymenoptera: Apidae). J. Mol. Evol. 2005, 60, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Li, G.Y.; Xiong, X.M.; Han, M.J.; Dai, F.Y. Horizontal transfer of a novel Helentron in insects. Mol. Genet. Genom. 2017, 292, 243–250. [Google Scholar] [CrossRef]
- Zhang, H.H.; Xu, H.E.; Shen, Y.H.; Han, M.J.; Zhang, Z. The origin and evolution of six miniature inverted-repeat transposable elements in Bombyx mori and Rhodnius prolixus. Genome Biol. Evol. 2013, 5, 2020–2031. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.K.; Kelley, S.T.; Spiegelman, G.B.; Pace, N.R. The genetic core of the universal ancestor. Genome Res. 2003, 13, 407–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Dunemann, S.M.; Wasmuth, J.D. Horizontal transfer of a retrotransposon between parasitic nematodes and the common shrew. Mob. DNA 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, W.; Jurka, M.G.; Kapitonov, V.V.; Jurka, J. New superfamilies of eukaryotic DNA tyransposons and their internal divisions. Mol. Biol. Evol. 2009, 26, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.; Zhang, H.; Huang, K.; Zhang, X.; Han, M.; Zhang, Z. Repeated horizontal transfers of four DNA transposons in invertebrates and bats. Mob. DNA 2015, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Oppenheim, S.; Cao, X.; Rueppel, O.; Krongdang, S.; Phokasem, P.; Desalle, R.; Goodwin, S.; Xing, J.; Chantawannakul, P.; Rosenfeld, J.A. Whole Genome Sequencing and Assembly of the Asian Honey Bee Apis dorsata. Genome Biol. Evol. 2020, 12, 3677–3683. [Google Scholar] [CrossRef] [Green Version]
- Haddad, N.J.; Adjlane, N.; Saini, D.; Menon, A.; Krishnamurthy, V.; Jonklaas, D.; Tomkins, J.P.; Loucif-Ayad, W.; Horth, L. Whole-genome sequencing of north African honey bee Apis mellifera intermissa to assess its beneficial traits. Entomol. Res. 2018, 48, 174–186. [Google Scholar] [CrossRef]
- Wallberg, A.; Bunikis, I.; Pettersson, O.V.; Mosbech, M.B.; Childers, A.K.; Evans, J.D.; Mikheyev, A.S.; Robertson, H.M.; Robinson, G.E.; Webster, M.T. A hybrid de novo genome assembly of the honeybee, Apis mellifera, with chromosome-length scaffolds. BMC Genom. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Elsik, C.G.; Worley, K.C.; Bennett, A.K.; Beye, M.; Camara, F.; Childers, C.P.; de Graaf, D.C.; Debyser, G.; Deng, J.; Devreese, B.; et al. Finding the missing honey bee genes: Lessons learned from a genome upgrade. BMC Genom. 2014, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.L.; Zhu, Y.Q.; Yan, Q.; Yan, W.Y.; Zheng, H.J.; Zeng, Z.J. A Chromosome-Scale Assembly of the Asian Honeybee Apis cerana Genome. Front. Genet. 2020, 11, 279. [Google Scholar] [CrossRef]
- Park, D.; Jung, W.W.; Choi, B.S.; Jayakodi, M.; Lee, J.; Lim, J.; Yu, Y.; Choi, Y.S.; Lee, M.L.; Park, Y.; et al. Uncovering the novel characteristics of Asian honey bee, Apis cerana, by whole genome sequencing. BMC Genom. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Sadd, B.; Barribeau, S.; Bloch, G.; de Graaf, D.; Dearden, P.; Elsik, C.; Gadau, J.; Grimmelikhuijzen, C.; Hasselmann, M.; Lozier, J.; et al. The genomes of two key bumblebee species with primitive eusocial organization. Genome Biol. 2015, 16, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Kaminker, J.S.; Bergman, C.M.; Kronmiller, B.; Carlson, J.; Svirskas, R.; Patel, S.; Frise, E.; Wheeler, D.A.; Lewis, S.E.; Rubin, G.M.; et al. The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biol. 2002, 3, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Suen, G.; Teiling, C.; Li, L.; Holt, C.; Abouheif, E.; Bornberg-Bauer, E.; Bouffard, P.; Caldera, E.J.; Cash, E.; Cavanaugh, A.; et al. The genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 2011, 7, e1002007. [Google Scholar] [CrossRef] [Green Version]
- Werren, J.H.; Richards, S.; Desjardins, C.A.; Niehuis, O.; Gadau, J.; Colbourne, J.K.; Beukeboom, L.W.; Desplan, C.; Elsik, C.G.; Grimmelikhuijzen, C.J.P.; et al. Functional and evolutionary insights from the genomes of three parasitoid nasonia species. Science 2010, 327, 343–348. [Google Scholar] [CrossRef]
- Richards, S.; Gibbs, R.A.; Gerardo, N.M.; Moran, N.; Nakabachi, A.; Stern, D.; Tagu, D.; Wilson, A.C.C.; Muzny, D.; Kovar, C.; et al. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010, 8, e1000313. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.C.; Bastida, F.; Bidwell, S.L.; Johnson, P.J.; Carlton, J.M. A potentially functional mariner transposable element in the protist Trichomonas vaginalis. Mol. Biol. Evol. 2005, 22, 126–134. [Google Scholar] [CrossRef]
- Langin, T.; Capy, P.; Daboussi, M.J. The transposable element impala, a fungal member of the Tc1-mariner superfamily. MGG Mol. Gen. Genet. 1995, 246, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Halaimia-Toumi, N.; Casse, N.; Demattei, M.V.; Renault, S.; Pradier, E.; Bigot, Y.; Laulier, M. The GC-rich transposon Bytmar1 from the deep-sea hydrothermal crab, Bythograea thermydron, may encode three transposase isoforms from a single ORF. J. Mol. Evol. 2004, 59, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Auge-Gouillou, C.; Bigot, Y.; Pollet, N.; Hamelin, M.H.; Meunier-Rotival, M.; Periquet, G. Human and other mammalian genomes contain transposons of the mariner family. FEBS Lett. 1995, 368, 541–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvik, T.; Lark, K.G. Characterization of Soymar1, a mariner element in soybean. Genetics 1998, 149, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Pujolar, J.M.; Astolfi, L.; Boscari, E.; Vidotto, M.; Barbisan, F.; Bruson, A.; Congiu, L. Tana1, a new putatively active Tc1-like transposable element in the genome of sturgeons. Mol. Phylogenet. Evol. 2013, 66, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Plant, J.D.; Paulus, H.F. Evolution and phylogeny of bees—Review and cladistic analysis in light of morphological evidence (Hymenoptera, Apoidea). Zoologica 2016, 161, 7–21. [Google Scholar]
- Weinstock, G.M.; Robinson, G.E.; Gibbs, R.A.; Worley, K.C.; Evans, J.D.; Maleszka, R.; Robertson, H.M.; Weaver, D.B.; Beye, M.; Bork, P.; et al. Insights into social insects from the genome of the honeybee Apis mellifera. Nature 2006, 443, 931–949. [Google Scholar] [CrossRef]
- Waterhouse, P.M.; Wang, M.B.; Lough, T. Gene silencing as an adaptive defence against viruses. Nature 2001, 411, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.L.; Lohe, A.R.; Lozovskaya, E.R. Modern thoughts on an ancyent marinere: Function, evolution, regulation. Annu. Rev. Genet. 1997, 31, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-López, M.; Garcia-Pérez, J.L. DNA transposons: Nature and applications in genomics. Curr. Genom. 2010, 11, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Casse, N.; Bui, Q.T.; Nicolas, V.; Renault, S.; Bigot, Y.; Laulier, M. Species sympatry and horizontal transfers of Mariner transposons in marine crustacean genomes. Mol. Phylogenet. Evol. 2006, 40, 609–619. [Google Scholar] [CrossRef]
- Robertson, H.M.; Lampe, D.J. Recent horizontal transfer of a mariner transposable element among and between Diptera and Neuroptera. Mol. Biol. Evol. 1995, 12, 850–862. [Google Scholar] [CrossRef] [Green Version]
- Lampe, D.J. Recent Horizontal Transfer of Mellifera Subfamily Mariner Transposons into Insect Lineages Representing Four Different Orders Shows that Selection Acts Only During Horizontal Transfer. Mol. Biol. Evol. 2003, 20, 554–562. [Google Scholar] [CrossRef]
- Laha, T.; Loukas, A.; Wattanasatitarpa, S.; Somprakhon, J.; Kewgrai, N.; Sithithaworn, P.; Kaewkes, S.; Mitreva, M.; Brindley, P.J. The bandit, a new DNA transposon from a hookworm possible horizontal genetic transfer between host and parasite. PLoS Negl. Trop. Dis. 2007, 1, e35. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DD29–36D Tigger | DD35–36D Fot | DD34D mariner | DD37D maT | DD39D GT | DD41D VS | DD34E Tc1 | DD36E IC | DD38E IT | |
|---|---|---|---|---|---|---|---|---|---|
| Copy number | 1–67 | 1–5 | 1–320 | 1–6 | 3 | 1–401 | 1–61 | 3–24 | 3–50 |
| Intact copy number | 1–4 | 1–2 | 1–28 | 1–2 | 0 | 1–3 | 1–5 | 0 | 1–2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Zong, W.; Diaby, M.; Lin, Z.; Wang, S.; Gao, B.; Ji, T.; Song, C. Diversity and Evolution of pogo and Tc1/mariner Transposons in the Apoidea Genomes. Biology 2021, 10, 940. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090940
Liu Y, Zong W, Diaby M, Lin Z, Wang S, Gao B, Ji T, Song C. Diversity and Evolution of pogo and Tc1/mariner Transposons in the Apoidea Genomes. Biology. 2021; 10(9):940. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090940
Chicago/Turabian StyleLiu, Yibing, Wencheng Zong, Mohamed Diaby, Zheguang Lin, Saisai Wang, Bo Gao, Ting Ji, and Chengyi Song. 2021. "Diversity and Evolution of pogo and Tc1/mariner Transposons in the Apoidea Genomes" Biology 10, no. 9: 940. https://0-doi-org.brum.beds.ac.uk/10.3390/biology10090940