DNA Modifications: Function and Applications in Normal and Disease States

 and
and
Abstract
:1. Introduction
2. DNA Methylation
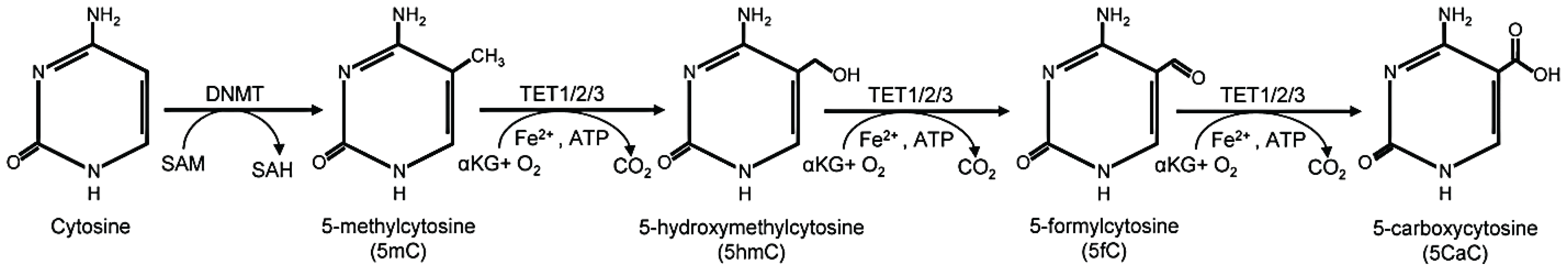
2.1. Five-Methylcytosine: “The Fifth Base”
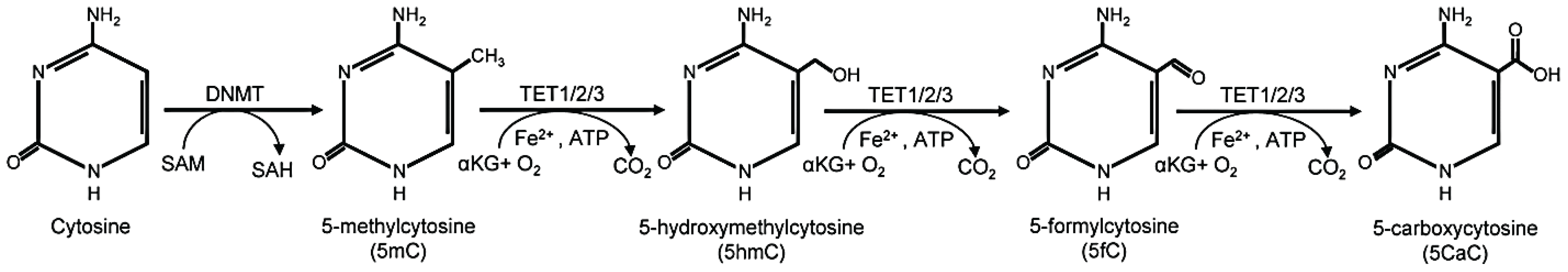
2.2. Five-Hydroxymethylcytosine: “the Sixth Base”
3. Role of DNA Methylation in Biological Processes
3.1. Gene Expression
3.1.1. Direct Hindrance of Transcriptional Activation
3.1.2. Recruitment of Protein Complexes
3.1.3. Cross-Talks with Histone PTMs
3.2. RNA Splicing
3.2.1. Role of MeCP2
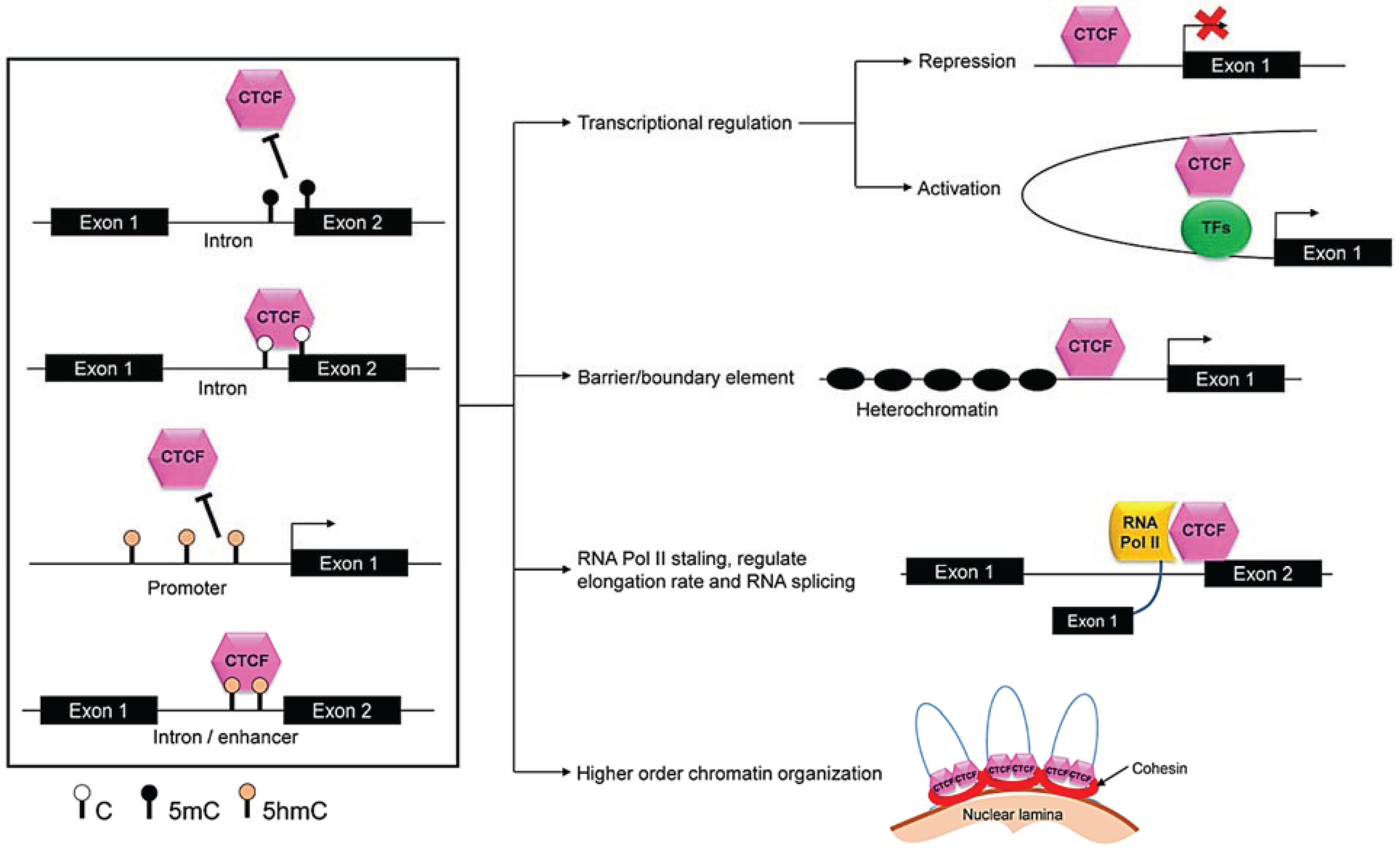
3.2.2. Role of CTCF
3.2.3. Genome Organization
3.2.4. Modulation of Higher Order Chromatin Structure
3.2.5. CTCF and Genome Organization
3.2.6. Nuclear Architecture
3.2.7. Transcription Noise
3.3. Imprinting
3.4. X-chromosome Inactivation
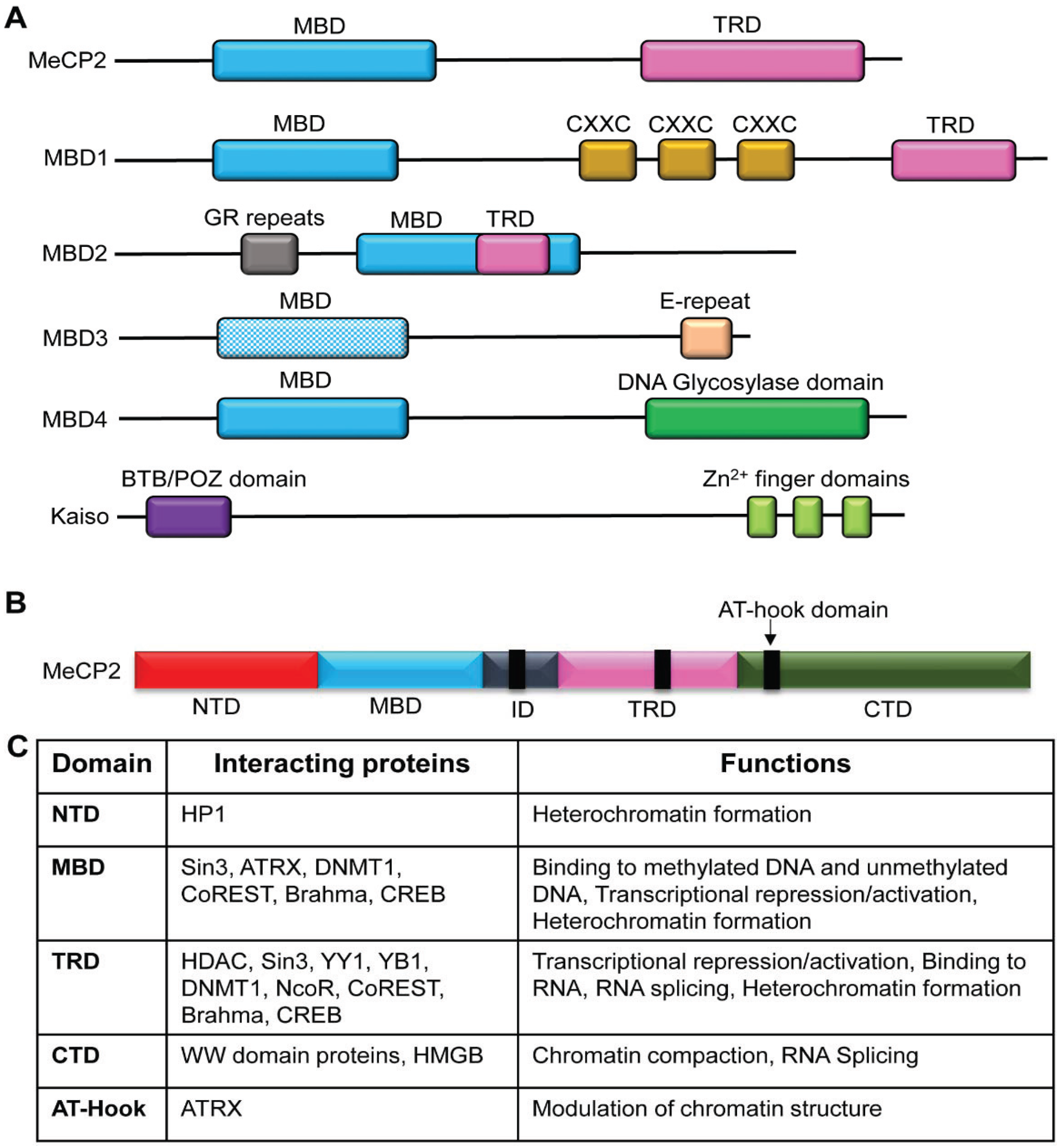
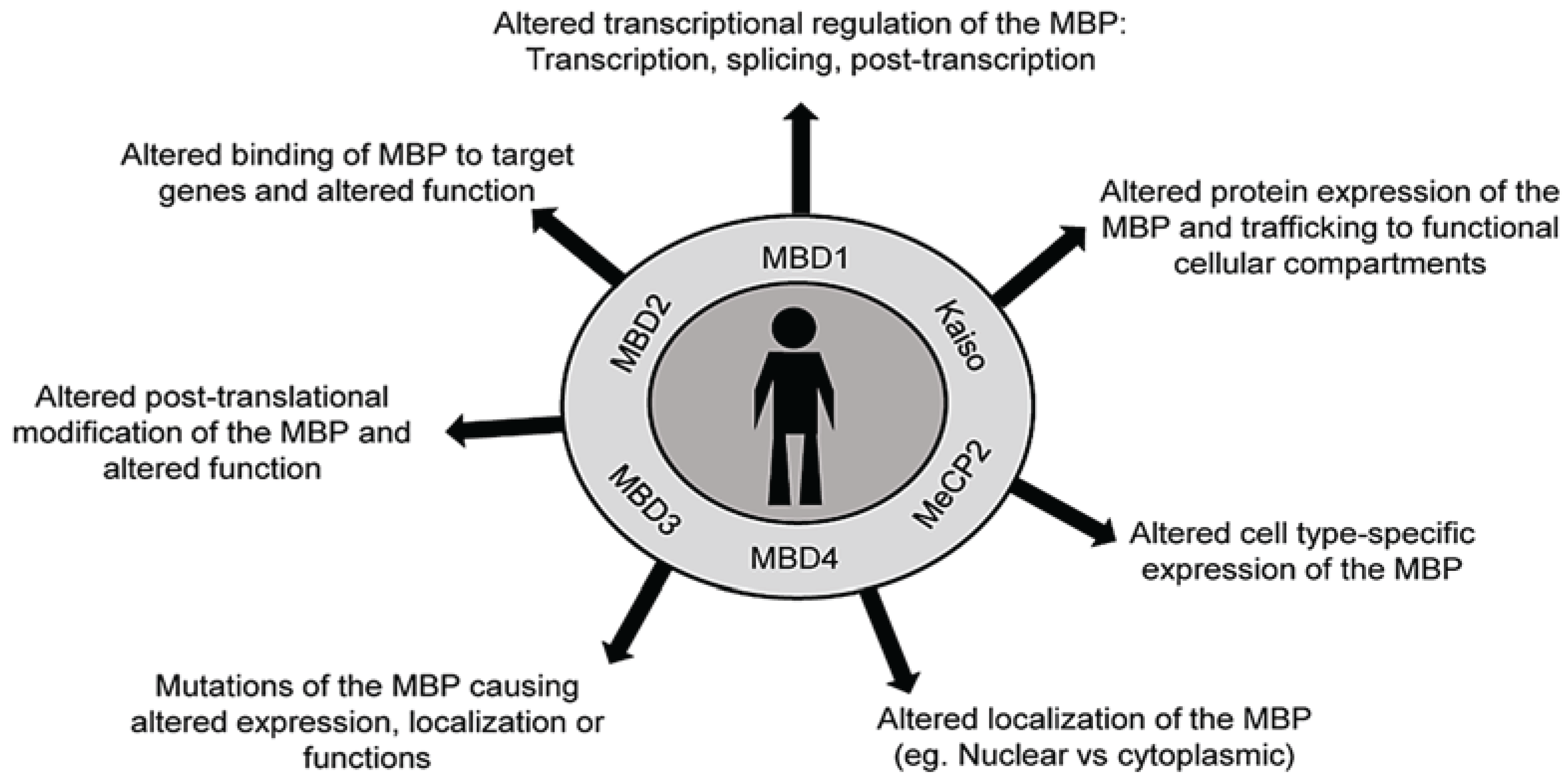
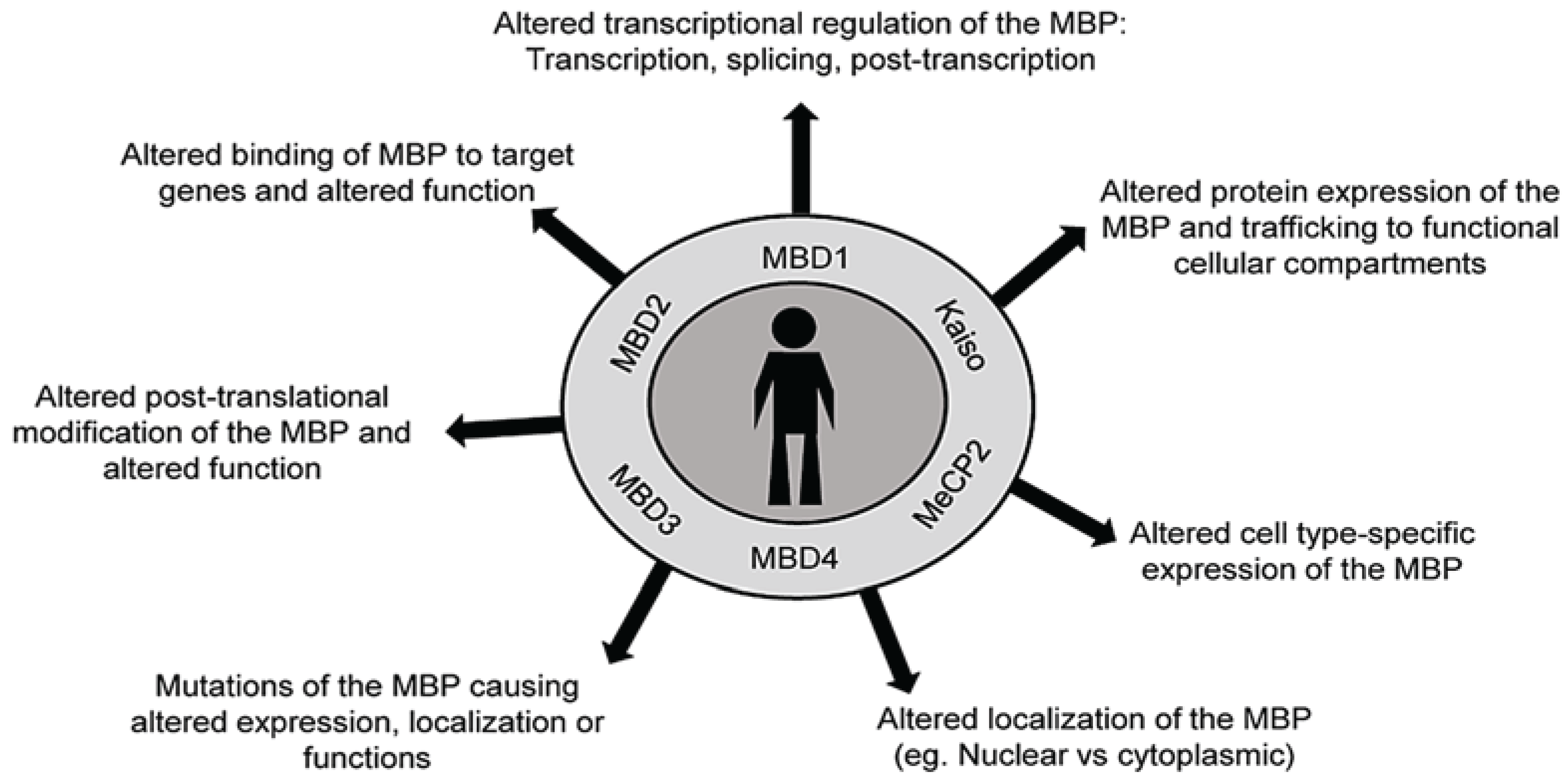
4. Methyl Binding Proteins: “The Methyl Readers”


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methyl Mark | Methyl Binding Protein | Description: Cell/Tissue Type (Genomic Location) |
|---|---|---|
| 5mC | MeCP2 | Mouse ESCs, Mouse Neural precursors, Mouse Brain [114] |
| MBD1 | Mouse ESCs, Mouse Neural precursors, Mouse Brain [114] | |
| MBD2 | Mouse Neural precursors, Mouse Brain [114] | |
| MBD3 | E14 Mouse ESCs (Fgf15) [115] | |
| MBD4 | E14 mouse ESCs (Pax6) [115]; Mouse ESCs, Neural precursors, Brain [114] | |
| 5hmC | MeCP2 | Mouse Brain (Active chromatin); Mouse ESCs [114] |
| MBD3 | E14 mouse ESCs [116] | |
| MBD4 | Mouse Neural precursors [114] | |
| 5fC | MBD3 | E14 mouse ESCs (Fgf15) [115] |
| 5mC | SIN3A | E14 mouse ESCs (Pax6, Fgf15) [115] |
| CREB1 | E14 mouse ESCs (Pax6, Fgf15) [115] | |
| SRSF2 | E14 mouse ESCs (Fgf15) [115] | |
| SRSF3 | E14 mouse ESCs (Pax6) [115] | |
| 5hmC | SRSF2 | E14 mouse ESCs (Fgf15) [115] |
| SRSF3 | E14 mouse ESCs (Pax6) [115] | |
| CREB1 | E14 mouse ESCs (Pax6) [115] | |
| 5fC | SIN3A | E14 mouse ESCs (Fgf15) [115] |
| CREB1 | E14 mouse ESCs (Pax6, Fgf15) [115] | |
| SRSF2 | E14 mouse ESCs (Fgf15) [115] | |
| SRSF3 | E14 mouse ESCs (Pax6) [115] | |
| 5CaC | CTCF | Mouse ESCs [114] |
| EHMT1 | Mouse ESCs [114] | |
| NCOR2 | Mouse ESCs [114] | |
| PRP31 | Mouse ESCs [114] | |
| DNMT1 | Mouse ESCs [114] |
4.1. DNA Methylation and MBPs in Human Diseases
4.1.1. Cancer
4.1.2. Diabetes Mellitus
4.1.3. Imprinted Disorders
 ; Stimulates/Activates
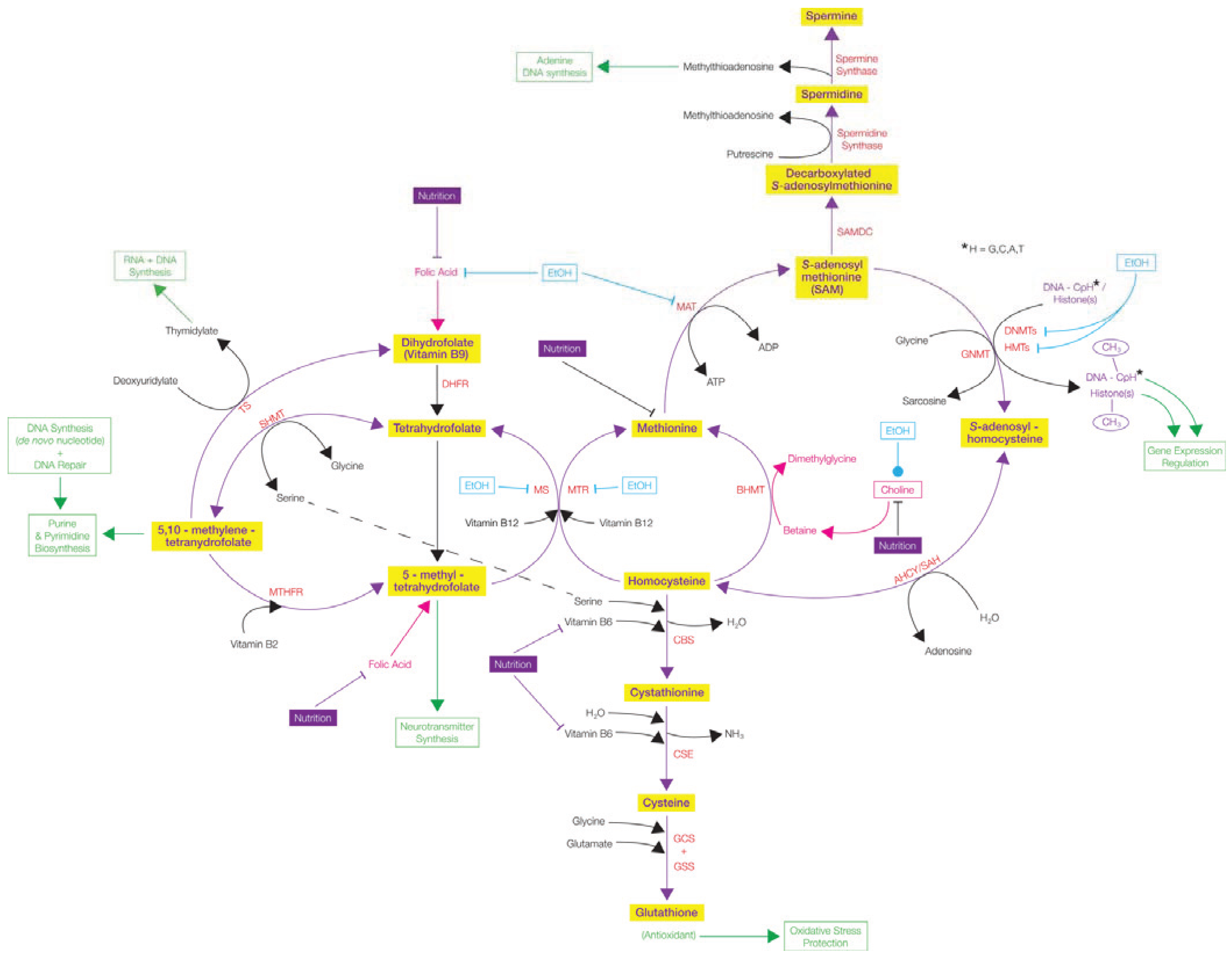
; Stimulates/Activates  ; Enzyme Abbreviations—AHCY/SAH: S-adenosyl-L-homocysteine hydrolase; BHMT: Betaine-homocysteine S-methyltransferase; CBS: Cystathionine-β-synthase; CSE: Ɣ-cystathionase; DHFR: Dihydrofolate reductase; DNMTs: DNA methyltransferases; GCS: Ɣ-Glutamyl cysteine synthetase; GNMT: Glycine N-methyltransferase; GSS: Glutathione synthetase; HMTs: Histone methyltransferases; MAT: Methionine adenosyltransferase; MTHFR: 5,10-methylenetetrahydrofolate reductase; MTR: 5-methyltetrahydrofolate-homocysteine methyltransferase; MS: Methionine synthase; SAMDC: S-adenosylmethionine decarboxylase; SHMT: Serine hydroxymethyltransferase; TS: Thymidylate Synthetase.
; Stimulates/Activates ; Enzyme Abbreviations—AHCY/SAH: S-adenosyl-L-homocysteine hydrolase; BHMT: Betaine-homocysteine S-methyltransferase; CBS: Cystathionine-β-synthase; CSE: Ɣ-cystathionase; DHFR: Dihydrofolate reductase; DNMTs: DNA methyltransferases; GCS: Ɣ-Glutamyl cysteine synthetase; GNMT: Glycine N-methyltransferase; GSS: Glutathione synthetase; HMTs: Histone methyltransferases; MAT: Methionine adenosyltransferase; MTHFR: 5,10-methylenetetrahydrofolate reductase; MTR: 5-methyltetrahydrofolate-homocysteine methyltransferase; MS: Methionine synthase; SAMDC: S-adenosylmethionine decarboxylase; SHMT: Serine hydroxymethyltransferase; TS: Thymidylate Synthetase.
; Enzyme Abbreviations—AHCY/SAH: S-adenosyl-L-homocysteine hydrolase; BHMT: Betaine-homocysteine S-methyltransferase; CBS: Cystathionine-β-synthase; CSE: Ɣ-cystathionase; DHFR: Dihydrofolate reductase; DNMTs: DNA methyltransferases; GCS: Ɣ-Glutamyl cysteine synthetase; GNMT: Glycine N-methyltransferase; GSS: Glutathione synthetase; HMTs: Histone methyltransferases; MAT: Methionine adenosyltransferase; MTHFR: 5,10-methylenetetrahydrofolate reductase; MTR: 5-methyltetrahydrofolate-homocysteine methyltransferase; MS: Methionine synthase; SAMDC: S-adenosylmethionine decarboxylase; SHMT: Serine hydroxymethyltransferase; TS: Thymidylate Synthetase.
; Stimulates/Activates ; Enzyme Abbreviations—AHCY/SAH: S-adenosyl-L-homocysteine hydrolase; BHMT: Betaine-homocysteine S-methyltransferase; CBS: Cystathionine-β-synthase; CSE: Ɣ-cystathionase; DHFR: Dihydrofolate reductase; DNMTs: DNA methyltransferases; GCS: Ɣ-Glutamyl cysteine synthetase; GNMT: Glycine N-methyltransferase; GSS: Glutathione synthetase; HMTs: Histone methyltransferases; MAT: Methionine adenosyltransferase; MTHFR: 5,10-methylenetetrahydrofolate reductase; MTR: 5-methyltetrahydrofolate-homocysteine methyltransferase; MS: Methionine synthase; SAMDC: S-adenosylmethionine decarboxylase; SHMT: Serine hydroxymethyltransferase; TS: Thymidylate Synthetase.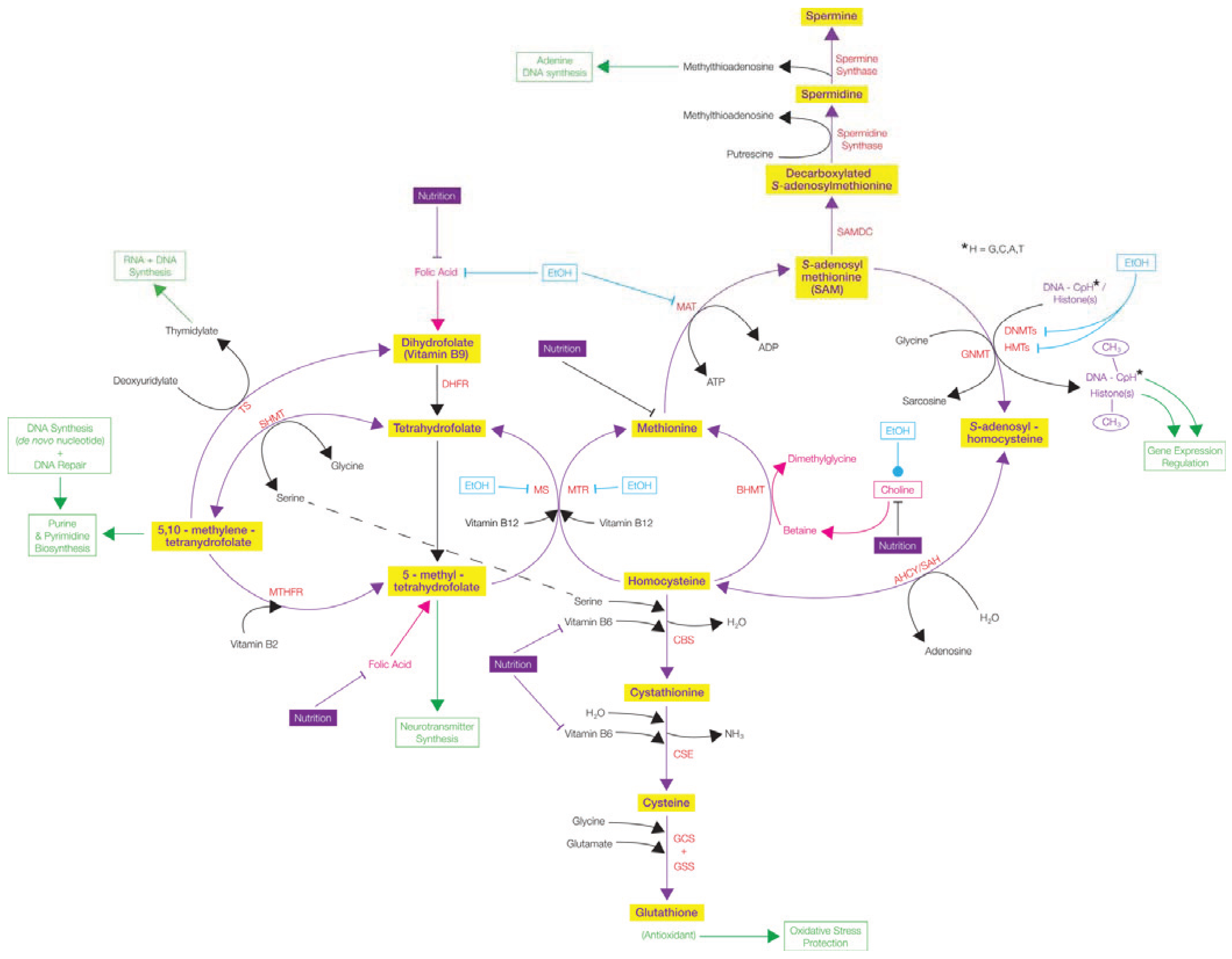
4.1.4. Immune System-Related Disorders
4.1.5. Cardiovascular Diseases and Cerebral Ischemia
4.2. Other DNA Binding Proteins Affected by DNA Methylation and Their Role in Human Diseases
4.2.1. CTCF
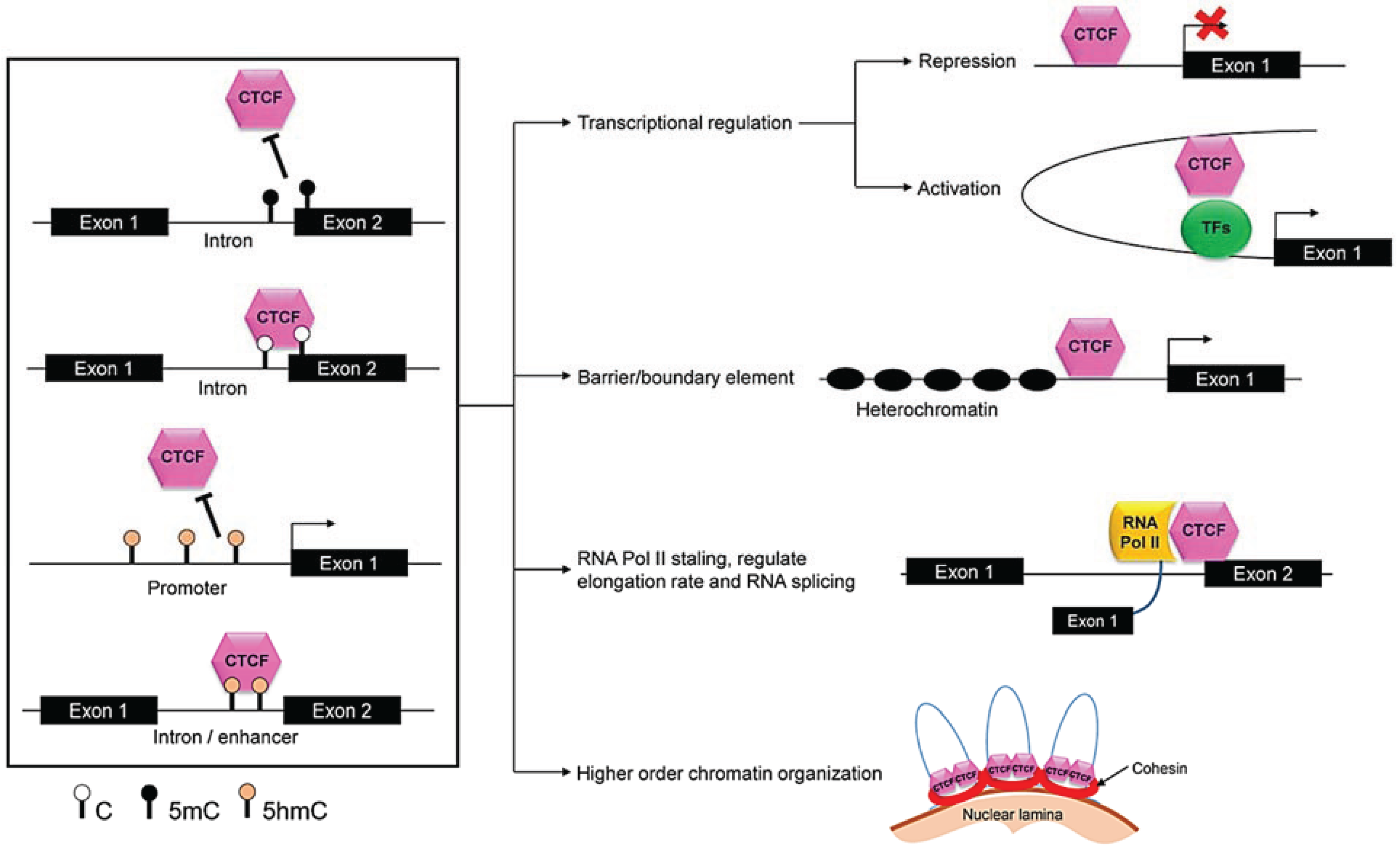
5. DNA Methylation, Teratogens, and Neurodevelopmental Disorders
5.1. Normal Early Brain Development
5.2. DNA Methylation and Brain Development
5.3. Effects of Teratogens on Normal Brain Development
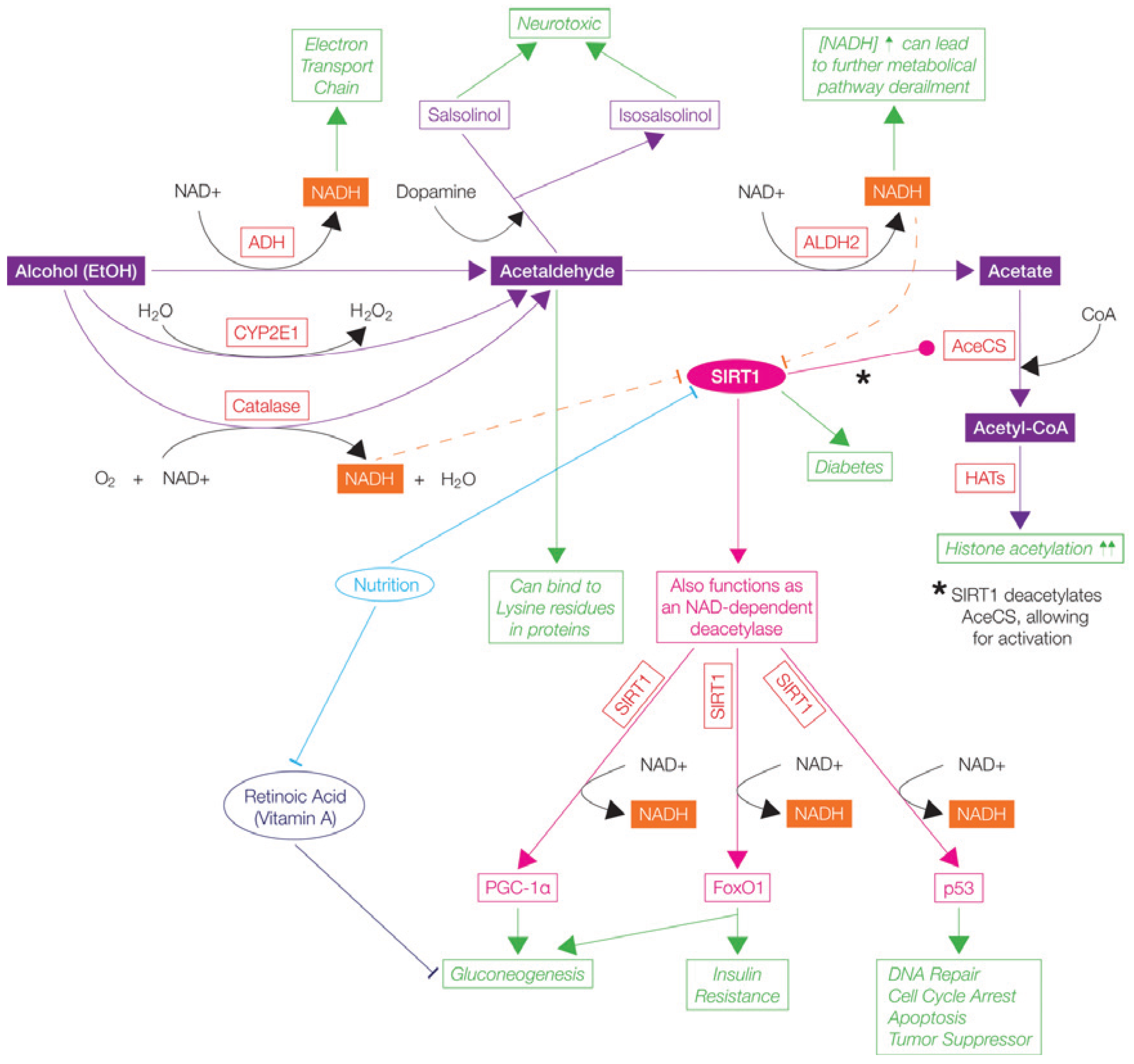
5.3.1. Alcohol (Ethanol)
; Stimulates/Activates ; Enzyme Abbreviations-AceCS: Acetyl-CoA Synthetase; ADH: Alcohol Dehydrogenase; ALDH2: Acetaldehyde dehydrogenase 2; CoA: Coenzyme A; CYP2E1: Cytochrome P40 2E1; Fox01: Forkhead box protein 01; HATs: Histone Acetyltransferases; p53: tumor suppressor protein 53; PGC-1α: Peroxisome proliferation-activated receptor Ɣ coactivator 1α; NAD/NADH: Nicotinamide adenine dinucleotide; NADH: SIRT1: Sirtuin 1/Fast-activated longevity protein sirtuin 1 NAP-dependent deacetylase sirtuitin-1.
; Stimulates/Activates ; Enzyme Abbreviations-AceCS: Acetyl-CoA Synthetase; ADH: Alcohol Dehydrogenase; ALDH2: Acetaldehyde dehydrogenase 2; CoA: Coenzyme A; CYP2E1: Cytochrome P40 2E1; Fox01: Forkhead box protein 01; HATs: Histone Acetyltransferases; p53: tumor suppressor protein 53; PGC-1α: Peroxisome proliferation-activated receptor Ɣ coactivator 1α; NAD/NADH: Nicotinamide adenine dinucleotide; NADH: SIRT1: Sirtuin 1/Fast-activated longevity protein sirtuin 1 NAP-dependent deacetylase sirtuitin-1.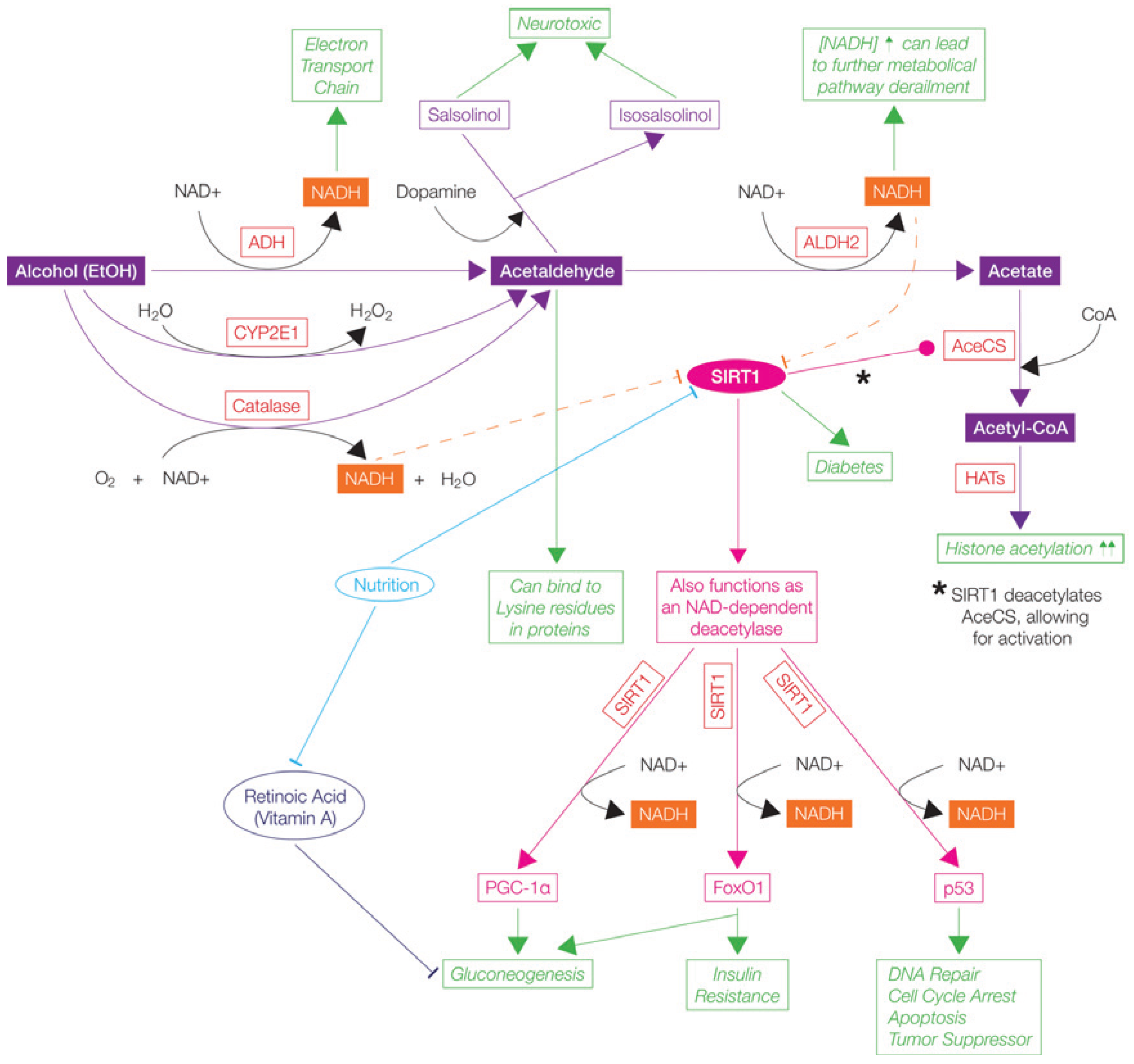
5.3.2. Tobacco (Nicotine)
5.3.3 Cocaine
5.3.4. Other Drugs
5.4. MeCP2 in Neurodevelopmental/Neurological Disorders
5.4.1. Rett Syndrome
5.4.2. Autism Spectrum Disorders
5.4.3. Fetal Alcohol Spectrum Disorders
5.4.4. Other
6. DNA Methylation as a Biomarker for Human Diseases
6.1. Type 2 Diabetes and the DNA Methylome
6.2. DNA Methylation as a Biomarker for Neurological Disorders
Abbreviations
| 5caC | 5-carboxylcytosine |
| 5fC | 5-formylcytosine |
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
| ALDH | acetaldehyde dehydrogenase |
| ADH | alcohol dehydrogenase |
| AS | Angelman syndrome |
| APEX1 | AP endonuclease 1 |
| AP site | apurine/apyramidine site |
| BER | base excision repair |
| BRM | bromodomain |
| CREB1 | cAMP-response element-binding protein-1 |
| CEBPA | CCAAT/enhancer binding protein alpha |
| CTCF | CCCTC-binding factor |
| CNS | central nervous system |
| ChIP | Chromatin immunoprecipitation |
| ChIA-PET | chromatin interaction analysis by paired-end-tag sequencing |
| CTD | C-terminal domain |
| CXXC | Cys-X-X-Cys chromatin-associated binding domain |
| DMRs | Differentially methylated regions |
| DNMTs | DNA methyltransferases |
| DNMT1 | DNA-methyltransferase 1 |
| E | Embryonic day |
| ESCs | Embryonic Stem cells |
| EHMT1 | Euchromatic histone-lysine N-methyltransferase 1 |
| EZH2 | enhancer of zeste homoloque 2 |
| FASD | Fetal Alcohol Spectrum Disorders |
| HCC | hepatocellular carcinoma |
| HP1 | heterochromatin protein 1 |
| HDAC | histone deacetylases |
| SETDB1 | histone-lysine N-methyltransferase |
| ICR | imprint control region |
| ID | inter domain |
| KAT | lysine acetyltransferases |
| MBD | Methyl Binding Domain |
| MeCP2 | Methyl CpG Binding Protein 2 |
| miRNAs | microRNAs |
| ncRNAs | noncoding RNAs |
| NCOR2 | Nuclear receptor co-repressor 2 |
| NuRD | nucleosome remodeling deacetylase |
| SIN3A | Paired amphipathic helix protein Sin3 transcription regulator homolog A |
| PHD | plant homeodomain |
| PRC2 | polycomb repressive complex 2 |
| PWS | Prader-Willi syndrome |
| PGC | primordial germ cells |
| PAR | pseudo-autosomal region |
| RNA | ribonucleic acid |
| SAM | S-adenosyl methionine |
| SRSF | Serine/arginine-rich splicing factor |
| SNPs | single nucleotide polymorphisms |
| TET | ten-eleven translocation |
| TDG | thymine DNA glycosylase |
| TRD | transcription repression domain |
| T2DM | Type 2 diabetes mellitus |
| PRP31 | U4/U6 small nuclear ribonucleoprotein |
| UHRF1 | Ubiquitin-like PHD and RING finger domains 1 |
| UTR | untranslated regions |
| USC | uterine serous carcinoma |
| XIC | X inactivation center |
| XCI | X-chromosome inactivation |
| FGF | Fibroblast growth factor |
| TGF/BMP | Transforming growth factor (Bone morphogenetic portines) |
| SHH | sonic hedgehog |
| Oct4 | octamer-binding transcription factor 4 |
| Map2 | Microtubule-associated protein 2 |
| Crabp1 | Cellular retinoic acid-binding protein 1 |
| Sox2 | SRY/Sex determining region Y-box 2 |
| Nanog | Homeobox protein NANOG |
| GABAA | γ-aminobutyric acid receptor |
| NDMA | N-methyl-D-aspartate |
| NMDAR | N-methyl-D-aspartate Receptor |
| HATs | Histone Acetyltransferases |
| Igf2 | Insulin-like Growth Factor 2 |
| Sox7 | SRY/Sex determining region Y-box 7 |
| Cutl2 | Cut-like homeodomain transcription factor 2 |
| Gria3 | glutamate receptor ionotropic AMPA 3 |
| Adrala | Adrenergic receptor |
| H19 | imprinted maternally expressed non-coding RNA |
| IG | DLK1-GTL2/MEG3 intergenic region |
| Gtl2 | Maternally expressed 3 non-coding RNA |
| Dlk1 | protein delta homolog 1 transmembrane protein |
| Peg1 | Mesoderm-specific transcript homolog protein (MEST) |
| Snrpn | Small nuclear ribonucleoprotein-associated protein N |
| Peg3 | imprinted Paternally-expressed gene 3 protein |
| AluYb8 | short interspersed nucleotide element |
| PTPRO | protein tyrosine phosphatase, receptor type, O |
| LINE1 | long interspersed nucleotide element |
| AXL | AXL receptor tyrosine kinase |
| BDNF | brain derived neurotrophic factor |
| HIE | hypoxic-ischemic encephalopathy |
| AngII | Angiotensin II |
| AT1R, AT2R | AngII type 1 and type 2 receptors |
| PP1c | protein phosphatase-1 catalytic subunit |
| A2AR | Adenosine Receptor 2 |
| fosB | FBJ murine osteosarcoma viral oncogene homolog B |
| DRD2 | dopamine receptor D |
| DMP | DNA methylation program |
| PTMs | Post translational modifications |
| SNPs | small nucleotide polymorphisms |
| METH | methamphetamine |
| NAc | nucleus accumbens |
| THC | Tetrahydrocannabinol |
| RUNX3 | Runt-related transcription factor 3 |
| GFI1 | Growth factor independent 1 |
| AHRR | Aryl-hydrocarbon receptor repressor |
| MYO1G | Myosin 1G |
| CYP1A1 | Cytochrome P450 1A |
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- Dahm, R. Friedrich Miescher and the discovery of DNA. Dev. Biol. 2005, 278, 274–288. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information Pubmed Search “DNA”. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/?term=“DNA” (accessed on 23 March 2014).
- National Human Genome Research Institute (NHGRI) The Human Genome Project. Available online: http://www.genome.gov/25019879 (accessed on 23 March 2014).
- Hallgrimsson, B.; Hall, B.K. Epigenetics: Linking Genotype and Phenotype in Development and Evolution; University of California Press: Oakland, CA, USA, 2011. [Google Scholar]
- Razin, A.; Cedar, H. Distribution of 5-methylcytosine in chromatin. Proc. Natl. Acad. Sci. USA 1977, 74, 2725–2728. [Google Scholar] [CrossRef] [PubMed]
- Pollack, Y.; Stein, R.; Razin, A.; Cedar, H. Methylation of foreign DNA sequences in eukaryotic cells. Proc. Natl. Acad. Sci. USA 1980, 77, 6463–6467. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Riggs, A. DNA methylation and gene function. Science 1980, 2010, 604–610. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information Pubmed Search “DNA Methylation”. Available online: http://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/pubmed/?term=“DNA+methylation” (accessed on 14 May 2014).
- Hashimoto, H.; Vertino, P.M.; Cheng, X. Molecular coupling of DNA methylation and histone methylation. Epigenomics 2010, 2, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Karaulanov, E.; Stapf, U.; Döderlein, G.; Niehrs, C. Ing1 functions in DNA demethylation by directing Gadd45a to H3K4me3. Genes. Dev. 2013, 27, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Bian, C.; Xu, C.; Ruan, J.; Lee, K.K.; Burke, T.L.; Tempel, W.; Barsyte, D.; Li, J.; Wu, M.; Zhou, B.O.; et al. Sgf29 binds histone H3K4me2/3 and is required for SAGA complex recruitment and histone H3 acetylation. EMBO J. 2011, 30, 2829–2842. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.W.; Baas, R.; Jansen, P.W.; Riss, A.; Tora, L.; Vermeulen, M.; Timmers, H.T. A dual role for SAGA-associated factor 29 (SGF29) in ER stress survival by coordination of both histone H3 acetylation and histone H3 lysine-4 trimethylation. PLoS One 2013, 8, e70035. [Google Scholar] [CrossRef] [PubMed]
- Ndlovu, M.N.; Denis, H.; Fuks, F. Exposing the DNA methylome iceberg. Trends Biochem. Sci. 2011, 36, 381–387. [Google Scholar] [PubMed]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; Wei, C.-L. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Auclair, G.; Weber, M. Mechanisms of DNA methylation and demethylation in mammals. Biochimie 2012, 94, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Dahl, C.; Grønbæk, K.; Guldberg, P. Advances in DNA methylation: 5-hydroxymethylcytosine revisited. Clin. Chim. Acta 2011, 412, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Lacey, M. Epigenetic Alterations in Oncogenesis; Karpf, A.R., Ed.; Springer New York: New York, NY, USA, 2013; Volume 754, pp. 31–56. [Google Scholar]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Liang, G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009, 10, 805–811. [Google Scholar] [PubMed]
- Sharma, S.; de Carvalho, D.D.; Jeong, S.; Jones, P.A.; Liang, G. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet. 2011, 7, e1001286. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Liang, G.; Sharma, S.; Lin, J.C.; Choi, S.H.; Han, H.; Yoo, C.B.; Egger, G.; Yang, A.S.; Jones, P.A. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol. 2009, 29, 5366–5376. [Google Scholar] [CrossRef] [PubMed]
- Rose, N.R.; Klose, R.J. Understanding the relationship between DNA and histone lysine methylation. Biochim. Biophys. Acta 2014. [Google Scholar] [CrossRef]
- Nishiyama, A.; Yamaguchi, L.; Sharif, J.; Johmura, Y.; Kawamura, T.; Nakanishi, K.; Shimamura, S.; Arita, K.; Kodama, T.; Ishikawa, F.; et al. Uhrf1-dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication. Nature 2013, 502, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative Sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Ponnaluri, V.K.C.; Maciejewski, J.P.; Mukherji, M. A mechanistic overview of TET-mediated 5-methylcytosine oxidation. Biochem. Biophys. Res. Commun. 2013, 436, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; D’Alessio, A.C.; Ito, S.; Wang, Z.; Cui, K.; Zhao, K.; Sun, Y.E.; Zhang, Y. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes. Dev. 2011, 25, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Feng, S.; Morey Kinney, S.; Pradhan, S.; Jacobsen, S.E. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011, 12, R54. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Shi, Y.G. Tet family proteins and 5-hydroxymethylcytosine in development and disease. Development 2012, 139, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Okashita, N.; Kumaki, Y.; Ebi, K.; Nishi, M.; Okamoto, Y.; Nakayama, M.; Hashimoto, S.; Nakamura, T.; Sugasawa, K.; Kojima, N.; et al. PRDM14 promotes active DNA demethylation through the Ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development 2014, 141, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.P.; Toomire, K.J.; Strauss, P.R. DNA modifications repaired by base excision repair are epigenetic. DNA Repair (Amst.) 2013, 12, 1152–1158. [Google Scholar] [CrossRef]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) gene in hematopoiesis and hematopoietic diseases. Leukemia 2013, 2, 1–12. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.B.; Zachariah, R.M.; Delcuve, G.P.; Davie, J.R.; Rastegar, M. New Developments in Chromatin Research: An Epigenetic Perspective; Simpson, N.M., Stewart, V.J., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2012; pp. 29–58. [Google Scholar]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Baron, B. Breaking the Silence: The Interplay Between Transcription Factors and DNA Methylation. In Methylation—From DNA, RNA and Histones to Diseases and Treatment; Dricu, P.A., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Zhu, W.G.; Srinivasan, K.; Dai, Z.; Duan, W.; Druhan, L.J.; Ding, H.; Yee, L.; Villalona-Calero, M.A.; Plass, C.; Otterson, G.A. Methylation of adjacent CpG sites affects Sp1/Sp3 binding and activity in the p21(Cip1) promoter. Mol. Cell. Biol. 2003, 23, 4056–4065. [Google Scholar] [CrossRef] [PubMed]
- Collings, C.K.; Waddell, P.J.; Anderson, J.N. Effects of DNA methylation on nucleosome stability. Nucl. Acids Res. 2013, 41, 2918–2931. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Lee, T.H. Effects of DNA methylation on the structure of nucleosomes. J. Am. Chem. Soc. 2012, 134, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, R.K.; Feng, S.; Bernatavichute, Y.V.; Chen, P.Y.; Stroud, H.; Yu, Y.; Hetzel, J.A.; Kuo, F.; Kim, J.; Cokus, S.J.; et al. Relationship between nucleosome positioning and DNA methylation. Nature 2010, 466, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Choy, J.S.; Wei, S.; Lee, J.Y.; Tan, S.; Chu, S.; Lee, T.H. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010, 132, 1782–1783. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Noushmehr, H.; Han, H.; Andreu-Vieyra, C.; Liang, G.; Jones, P.A. Gene reactivation by 5-aza-2ꞌ-deoxycytidine-induced demethylation requires SRCAP-mediated H2A.Z insertion to establish nucleosome depleted regions. PLoS Genet. 2012, 8, e1002604. [Google Scholar] [CrossRef] [PubMed]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and Rett syndrome: The emerging novel and challenging concepts in MeCP2 research. Neural Plast. 2012, 2012, 415825. [Google Scholar] [PubMed]
- Hashimoto, H.; Liu, Y.; Upadhyay, A.K.; Chang, Y.; Howerton, S.B.; Vertino, P.M.; Zhang, X.; Cheng, X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucl. Acids Res. 2012, 40, 4841–4849. [Google Scholar] [CrossRef] [PubMed]
- Cartron, P.F.; Nadaradjane, A.; Lepape, F.; Lalier, L.; Gardie, B.; Vallette, F.M. Identification of TET1 partners that control its dna-demethylating function. Genes. Cancer 2013, 4, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Zhubi, A.; Chen, Y.; Dong, E.; Cook, E.H.; Guidotti, A.; Grayson, D.R. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 2014, 4, e349. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, C.; Malousi, A.; Maglaveras, N.; Kouidou, S. Human epigenome data reveal increased CpG methylation in alternatively spliced sites and putative exonic splicing enhancers. DNA Cell Biol. 2011, 30, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.K. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 2010, 11, R70. [Google Scholar] [CrossRef] [PubMed]
- Malousi, A.; Kouidou, S. DNA hypermethylation of alternatively spliced and repeat sequences in humans. Mol. Genet. Genomics 2012, 287, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Long, S.W.; Ooi, J.Y.; Yau, P.M.; Jones, P.L. A brain-derived MeCP2 complex supports a role for MeCP2 in RNA processing. Biosci. Rep. 2011, 31, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Young, J.I.; Hong, E.P.; Castle, J.C.; Crespo-Barreto, J.; Bowman, A.B.; Rose, M.F.; Kang, D.; Richman, R.; Johnson, J.M.; Berget, S.; Zoghbi, H.Y. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. USA 2005, 102, 17551–17558. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R. CTCF: From insulators to alternative splicing regulation. Cell Res. 2012, 22, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Stadhouders, R.; Thongjuea, S.; Andrieu-Soler, C.; Palstra, R.J.; Bryne, J.C.; van den Heuvel, A.; Stevens, M.; de Boer, E.; Kockx, C.; van der Sloot, A.; et al. Dynamic long-range chromatin interactions control Myb proto-oncogene transcription during erythroid development. EMBO J. 2012, 31, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Paredes, S.H.; Melgar, M.F.; Sethupathy, P. Promoter-proximal CCCTC-factor binding is associated with an increase in the transcriptional pausing index. Bioinformatics 2013, 29, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Kubiura, M.; Okano, M.; Kimura, H.; Kawamura, F.; Tada, M. Chromosome-wide regulation of euchromatin-specific 5mC to 5hmC conversion in mouse ES cells and female human somatic cells. Chromosom. Res. 2012, 20, 837–848. [Google Scholar] [CrossRef]
- Alioui, A.; Wheldon, L.M.; Abakir, A.; Ferjentsik, Z.; Johnson, A.D.; Ruzov, A. 5-Carboxylcytosine is localized to euchromatic regions in the nuclei of follicular cells in axolotl ovary. Nucleus 2012, 3, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Wu, H.; Diep, D.; Yamaguchi, S.; D’Alessio, A.C.; Fung, H.L.; Zhang, K.; Zhang, Y. Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 2013, 153, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.P.; Horowitz-Scherer, R.A.; Nikitina, T.; Shlyakhtenko, L.S.; Woodcock, C.L. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol. Cell. Biol. 2010, 30, 4656–4670. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, T.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Grigoryev, S.A.; Woodcock, C.L. MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 2007, 282, 28237–28245. [Google Scholar] [CrossRef] [PubMed]
- Yasui, D.H.; Peddada, S.; Bieda, M.C.; Vallero, R.O.; Hogart, A.; Nagarajan, R.P.; Thatcher, K.N.; Farnham, P.J.; Lasalle, J.M. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. USA 2007, 104, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, T.; Shi, X.; Ghosh, R.P.; Horowitz-Scherer, R.A.; Hansen, J.C.; Woodcock, C.L. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 2007, 27, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.E.; Corces, V.G. CTCF: Master weaver of the genome. Cell 2009, 137, 1194–1211. [Google Scholar] [CrossRef] [PubMed]
- Majumder, P.; Gomez, J.A.; Chadwick, B.P.; Boss, J.M. The insulator factor CTCF controls MHC class II gene expression and is required for the formation of long-distance chromatin interactions. J. Exp. Med. 2008, 205, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, S.J.; de Laat, W. CTCF: The protein, the binding partners, the binding sites and their chromatin loops. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013, 368, 20120369. [Google Scholar] [CrossRef] [PubMed]
- Handoko, L.; Xu, H.; Li, G.; Ngan, C.Y.; Chew, E.; Schnapp, M.; Lee, C.W.; Ye, C.; Ping, J.L.; Mulawadi, F.; et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011, 43, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Botta, M.; Haider, S.; Leung, I.X.; Lio, P.; Mozziconacci, J. Intra- and inter-chromosomal interactions correlate with CTCF binding genome wide. Mol. Syst. Biol. 2010, 6, 426. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, N.; Thomson, I.; Boyle, S.; Allan, J.; Ramsahoye, B.; Bickmore, W.A. DNA methylation affects nuclear organization, histone modifications, and linker histone binding but not chromatin compaction. J. Cell Biol. 2007, 177, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Singleton, M.K.; Gonzales, M.L.; Leung, K.N.; Yasui, D.H.; Schroeder, D.I.; Dunaway, K.; LaSalle, J.M. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol. Dis. 2011, 43, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Matarazzo, M.R.; Boyle, S.; D’Esposito, M.; Bickmore, W.A. Chromosome territory reorganization in a human disease with altered DNA methylation. Proc. Natl. Acad. Sci. USA 2007, 104, 16546–16551. [Google Scholar] [CrossRef] [PubMed]
- Huh, I.; Zeng, J.; Park, T.; Yi, S.V. DNA methylation and transcriptional noise. Epigenetics Chromatin 2013, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Della Ragione, F.; Filosa, S.; Scalabri, F.; D’Esposito, M. MeCP2 as a genome-wide modulator: The renewal of an old story. Front. Genet. 2012, 3, 181. [Google Scholar] [PubMed]
- Miura, A.; Yonebayashi, S.; Watanabe, K.; Toyama, T.; Shimada, H.; Kakutani, T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 2001, 411, 212–214. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, R.K.; Martienssen, R. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007, 8, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.; Du, T.; Wagner, U.; Xie, W.; Lee, A.Y.; Goyal, P.; Li, Y.; Szulwach, K.E.; Jin, P.; Lorincz, M.C.; Ren, B. Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. Proc. Natl. Acad. Sci. 2014, 111, 6690–6695. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Inoue, K.; Ono, R.; Ogonuki, N.; Kohda, T.; Kaneko-Ishino, T.; Ogura, A.; Ishino, F. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development 2002, 129, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, S.; Hong, K.; Liu, R.; Inoue, A.; Shen, L.; Zhang, K.; Zhang, Y. Dynamics of 5-methylcytosine and 5-hydroxymethylcytosine during germ cell reprogramming. Cell Res. 2013, 23, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.A.; Sengupta, R.; Zylicz, J.J.; Murakami, K.; Lee, C.; Down, T.A.; Surani, M.A. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science 2013, 339, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Huang, Z.; Hoyo, C. Differentially methylated regions of imprinted genes in prenatal, perinatal and postnatal human tissues. PLoS One 2012, 7, e40924. [Google Scholar] [CrossRef] [PubMed]
- Coolen, M.W.; Statham, A.L.; Qu, W.; Campbell, M.J.; Henders, A.K.; Montgomery, G.W.; Martin, N.G.; Clark, S.J. Impact of the genome on the epigenome is manifested in DNA methylation patterns of imprinted regions in monozygotic and dizygotic twins. PLoS One 2011, 6, e25590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kernohan, K.D.; Bérubé, N.G. Genetic and epigenetic dysregulation of imprinted genes in the brain. Epigenomics 2010, 2, 743–763. [Google Scholar] [CrossRef] [PubMed]
- Court, F.; Martin-Trujillo, A.; Romanelli, V.; Garin, I.; Iglesias-Platas, I.; Salafsky, I.; Guitart, M.; Perez de Nanclares, G.; Lapunzina, P.; Monk, D. Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes. Hum. Mutat. 2013, 34, 595–602. [Google Scholar] [PubMed]
- Soubry, A.; Murphy, S.K.; Wang, F.; Huang, Z.; Vidal, A.C.; Fuemmeler, B.F.; Kurtzberg, J.; Murtha, A.; Jirtle, R.L.; Schildkraut, J.M.; et al. Newborns of obese parents have altered DNA methylation patterns at imprinted genes. Int. J. Obes. 2013. [Google Scholar] [CrossRef]
- LaSalle, J.M. The Odyssey of MeCP2 and parental imprinting. Epigenetics 2007, 2, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Flashner, B.M.; Russo, M.E.; Boileau, J.E.; Leong, D.W.; Gallicano, G.I. Epigenetic factors and autism spectrum disorders. NeuroMol. Med. 2013, 15, 339–350. [Google Scholar] [CrossRef]
- Lewis, A.; Murrell, A. Genomic imprinting: CTCF protects the boundaries. Curr. Biol. 2004, 14, R284–R286. [Google Scholar] [PubMed]
- Prickett, A.R.; Barkas, N.; McCole, R.B.; Hughes, S.; Amante, S.M.; Schulz, R.; Oakey, R.J. Genome-wide and parental allele-specific analysis of CTCF and cohesin DNA binding in mouse brain reveals a tissue-specific binding pattern and an association with imprinted differentially methylated regions. Genome Res. 2013, 23, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Sheng, J.; Lau, H.-T.; McDonald, C.M.; Andrade, M.; Cullen, D.E.; Bell, F.T.; Iacovino, M.; Kyba, M.; Xu, G.; et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J. Biol. Chem. 2012, 287, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Quenneville, S.; Verde, G.; Corsinotti, A.; Kapopoulou, A.; Jakobsson, J.; Offner, S.; Baglivo, I.; Pedone, P.V.; Grimaldi, G.; Riccio, A.; et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell 2011, 44, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Pontier, D.B.; Gribnau, J. Xist regulation and function explored. Hum. Genet. 2011, 130, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Marks, H.; Chow, J.C.; Denissov, S.; Françoijs, K.-J.; Brockdorff, N.; Heard, E.; Stunnenberg, H.G. High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res. 2009, 19, 1361–1373. [Google Scholar] [PubMed]
- Ahn, J.Y.; Lee, J.T. X chromosome: X inactivation. Nat. Educ. 2008, 1, 24. [Google Scholar]
- Escamilla-Del-Arenal, M.; da Rocha, S.T.; Heard, E. Evolutionary diversity and developmental regulation of X-chromosome inactivation. Hum. Genet. 2011, 130, 307–327. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Stathaki, E.; Migliavacca, E.; Brahmachary, M.; Montgomery, S.B.; Dupre, Y.; Antonarakis, S.E. DNA methylation profiles of human active and inactive X chromosomes. Genome Res. 2011, 21, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.; Chess, A. Gene body-specific methylation on the active X chromosome. Science 2007, 315, 1141–1143. [Google Scholar] [CrossRef] [PubMed]
- Migeon, B.R.; Chowdhury, A.K.; Dunston, J.A.; McIntosh, I. Identification of TSIX, encoding an RNA antisense to human XIST, reveals differences from its murine counterpart: Implications for X inactivation. Am. J. Hum. Genet. 2001, 69, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Krepelova, A.; Incarnato, D.; Maldotti, M.; Parlato, C.; Galvagni, F.; Matarese, F.; Stunnenberg, H.G.; Oliviero, S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell 2013, 155, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Gu, H.; Bartels, S.J.; Zhang, Y.; Matarese, F.; Simmer, F.; Marks, H.; Bock, C.; Gnirke, A.; Meissner, A.; et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012, 22, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, T.B.; Popova, B.C.; Cobb, B.S.; Norton, S.; Senner, C.E.; Tang, Y.A.; Spruce, T.; Rodriguez, T.A.; Sado, T.; Merkenschlager, M.; et al. Dicer regulates Xist promoter methylation in ES cells indirectly through transcriptional control of Dnmt3a. Epigenetics Chromatin 2008, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Pichard, S.; Ciaudo, C.; Avner, P.; Rougeulle, C. Tsix transcription across the Xist gene alters chromatin conformation without affecting Xist transcription: Implications for X-chromosome inactivation. Genes Dev. 2005, 19, 1474–1484. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.; Huynh, K.D.; Spencer, R.J.; Davidow, L.S.; Lee, J.T. CTCF, a candidate trans-acting factor for X-inactivation choice. Science 2002, 295, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Teng, F. Epigenetic re-programming during mammalian preimplantation embryogenesis and pgc development. J. IVF Reprod Med. Genet. 2013, 1, 114. [Google Scholar]
- Lan, J.; Hua, S.; He, X.; Zhang, Y. DNA methyltransferases and methyl-binding proteins of mammals. Acta Biochim. Biophys. Sin. 2010, 42, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Ishikawa, F. The mCpG-binding domain of human MBD3 does not bind to mCpG but interacts with NuRD/Mi2 components HDAC1 and MTA2. J. Biol. Chem. 2002, 277, 35434–35439. [Google Scholar] [CrossRef] [PubMed]
- Prokhortchouk, A.; Hendrich, B.; Jorgensen, H.; Ruzov, A.; Wilm, M.; Georgiev, G.; Bird, A.; Prokhortchouk, E. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001, 15, 1613–1618. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Rastegar, M. Rett Syndrome and MeCP2. Neuromol. Med. 2014, 16, 231–264. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Montoya, G.; Taysavang, P.; Wade, P.A.; Esteller, M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucl. Acids Res. 2003, 31, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Sarraf, S.A.; Schmiedeberg, L.; McDermott, S.M.; Stancheva, I.; Bird, A.P. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell 2005, 19, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; de Las Heras, J.I.; Merusi, C.; Stancheva, I. Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucl. Acids Res. 2010, 38, 4620–4634. [Google Scholar] [CrossRef] [PubMed]
- Scarsdale, J.N.; Webb, H.D.; Ginder, G.D.; Williams, D.C., Jr. Solution structure and dynamic analysis of chicken MBD2 methyl binding domain bound to a target-methylated DNA sequence. Nucl. Acids Res. 2011, 39, 6741–6752. [Google Scholar] [CrossRef] [PubMed]
- Spruijt, C.G.; Gnerlich, F.; Smits, A.H.; Pfaffeneder, T.; Jansen, P.W.; Bauer, C.; Munzel, M.; Wagner, M.; Muller, M.; Khan, F.; et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013, 152, 1146–1159. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, M.; Ficz, G.; Oxley, D.; Raiber, E.A.; Bachman, M.; Booth, M.J.; Andrews, S.; Balasubramanian, S.; Reik, W. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol 2013, 14, R119. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, O.; Li, R.; Hung, J.H.; Chen, P.B.; Dong, X.; Ee, L.S.; Weng, Z.; Rando, O.J.; Fazzio, T.G. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 2011, 147, 1498–1510. [Google Scholar] [CrossRef] [PubMed]
- Baubec, T.; Ivanek, R.; Lienert, F.; Schubeler, D. Methylation-dependent and -independent genomic targeting principles of the MBD protein family. Cell 2013, 153, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.C.; Ghosh, R.P.; Woodcock, C.L. Binding of the Rett syndrome protein, MeCP2, to methylated and unmethylated DNA and chromatin. IUBMB Life 2010, 62, 732–738. [Google Scholar] [CrossRef]
- Ghosh, R.P.; Nikitina, T.; Horowitz-Scherer, R.A.; Gierasch, L.M.; Uversky, V.N.; Hite, K.; Hansen, J.C.; Woodcock, C.L. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 2010, 49, 4395–4410. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, H.F.; Ben-Porath, I.; Bird, A.P. Mbd1 is recruited to both methylated and nonmethylated CpGs via distinct DNA binding domains. Mol. Cell. Biol. 2004, 24, 3387–3395. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.M.; Scarsdale, J.N.; Walavalkar, N.M.; Buchwald, W.A.; Ginder, G.D.; Williams, D.C., Jr. Probing the dynamic distribution of bound states for methylcytosine-binding domains on DNA. J. Biol. Chem. 2014, 289, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Parry, L.; Clarke, A.R. The Roles of the Methyl-CpG Binding Proteins in Cancer. Genes Cancer 2011, 2, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, T.A.; Olsson, A.H.; Volkov, P.; Almgren, P.; Ronn, T.; Ling, C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia 2013, 56, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Taqi, M.M.; Bazov, I.; Watanabe, H.; Sheedy, D.; Harper, C.; Alkass, K.; Druid, H.; Wentzel, P.; Nyberg, F.; Yakovleva, T.; Bakalkin, G. Prodynorphin CpG-SNPs associated with alcohol dependence: Elevated methylation in the brain of human alcoholics. Addict. Biol. 2011, 16, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Chik, F.; Szyf, M.; Rabbani, S.A. Role of epigenetics in cancer initiation and progression. Adv. Exp. Med. Biol. 2011, 720, 91–104. [Google Scholar] [PubMed]
- Yaqinuddin, A.; Abbas, F.; Naqvi, S.Z.; Bashir, M.U.; Qazi, R.; Qureshi, S.A. Silencing of MBD1 and MeCP2 in prostate-cancer-derived PC3 cells produces differential gene expression profiles and cellular phenotypes. Biosci. Rep. 2008, 28, 319–326. [Google Scholar] [CrossRef]
- Fournier, A.; Sasai, N.; Nakao, M.; Defossez, P.A. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Br. Funct. Genomics. 2012, 11, 251–264. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Zhang, Y.; Srivenugopal, K.S.; Lim, S.H. SPAN-XB core promoter sequence is regulated in myeloma cells by specific CpG dinucleotides associated with the MeCP2 protein. Int. J. Cancer 2006, 119, 2878–2884. [Google Scholar] [CrossRef] [PubMed]
- Pontes, T.B.; Chen, E.S.; Gigek, C.O.; Calcagno, D.Q.; Wisnieski, F.; Leal, M.F.; Demachki, S.; Assumpcao, P.P.; Artigiani, R.; Lourenco, L.G.; et al. Reduced mRNA expression levels of MBD2 and MBD3 in gastric carcinogenesis. Tumour Biol. 2013, 35, 3447–3453. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, Y.; Robson, P.; Lai, R.; Cass, C.E.; Mackey, J.R.; Damaraju, S. A two-stage association study identifies methyl-CpG-binding domain protein 2 gene polymorphisms as candidates for breast cancer susceptibility. Eur. J. Hum. Genet. 2012, 20, 682–689. [Google Scholar] [PubMed]
- Stefanska, B.; Suderman, M.; Machnes, Z.; Bhattacharyya, B.; Hallett, M.; Szyf, M. Transcription onset of genes critical in liver carcinogenesis is epigenetically regulated by methylated DNA-binding protein MBD2. Carcinogenesis 2013, 34, 2738–2749. [Google Scholar] [CrossRef]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef] [PubMed]
- Hendrich, B.; Guy, J.; Ramsahoye, B.; Wilson, V.A.; Bird, A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001, 15, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Caballero, I.M.; MacLeod, R.; Nichols, J.; Wilson, V.A.; Hendrich, B. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat. Cell Biol. 2006, 8, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.D.; Luo, X.P.; Liu, X.; Jing, X.; Zeng, L.Q.; Lei, M.; Hong, X.S.; Chen, Y. The MBD4 Glu346Lys polymorphism is associated with the risk of cervical cancer in a Chinese population. Int. J. Gynecol. Cancer 2012, 22, 1552–1556. [Google Scholar] [PubMed]
- Jones, J.; Wang, H.; Karanam, B.; Theodore, S.; Dean-Colomb, W.; Welch, D.R.; Grizzle, W.; Yates, C. Nuclear localization of Kaiso promotes the poorly differentiated phenotype and EMT in infiltrating ductal carcinomas. Clin. Exp. Metastasis 2014, 31, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, J.F.; van de Ven, R.A.; Ercan, C.; van der Groep, P.; van der Wall, E.; Bult, P.; Christgen, M.; Lehmann, U.; Daniel, J.; van Diest, P.J.; et al. Nuclear Kaiso expression is associated with high grade and triple-negative invasive breast cancer. PLoS One 2012, 7, e37864. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.D.; Wang, Y.; Jiang, G.Y.; Zhang, P.X.; Dong, X.J.; Wei, Q.; Xu, H.T.; Li, Q.C.; Zhao, C.; Wang, E.H. Kaiso is expressed in lung cancer: Its expression and localization is affected by p120ctn. Lung Cancer 2010, 67, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef]
- Shaikh, M.K.; Devrajani, B.R.; Shaikh, A.; Shah, S.Z.A.; Shaikh, S.; Singh, D. Plasma homocysteine level in patients with diabetes mellitus. World Appl. Sci. J. 2012, 16, 1269–1273. [Google Scholar]
- Zheng, M.; Zhang, M.; Yang, J.; Zhao, S.; Qin, S.; Chen, H.; Gao, Y.; Huang, G. Relationship between blood levels of methyl donor and folate and mild cognitive impairment in Chinese patients with type 2 diabetes: A case-control study. J. Clin. Biochem. Nutr. 2014, 54, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Al-Maskari, M.Y.; Waly, M.I.; Ali, A.; Al-Shuaibi, Y.S.; Ouhtit, A. Folate and vitamin B12 deficiency and hyperhomocysteinemia promote oxidative stress in adult type 2 diabetes. Nutrition 2012, 28, e23–e26. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, R.O.; Marschoff, E.R.; Guareschi, E.M.; Famulari, A.L.; Pagano, M.A.; Serra, J.A. Homocysteine, vitamin B12 and folate in Alzheimer’s and vascular dementias: The paradoxical effect of the superimposed type II diabetes mellitus condition. Clin. Chim. Acta 2005, 359, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Belot, M.-P.; Fradin, D.; Mai, N.; Le Fur, S.; Zélénika, D.; Kerr-Conte, J.; Pattou, F.; Lucas, B.; Bougnères, P. CpG methylation changes within the IL2RA promoter in type 1 diabetes of childhood onset. PLoS One 2013, 8, e68093. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; del Guerra, S.; Lupi, R.; Ronn, T.; Granhall, C.; Luthman, H.; Masiello, P.; Marchetti, P.; Groop, L.; del Prato, S. Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 2008, 51, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Hall, E.; Dayeh, T.; Kirkpatrick, C.L.; Wollheim, C.B.; Dekker Nitert, M.; Ling, C. DNA methylation of the glucagon-like peptide 1 receptor (GLP1R) in human pancreatic islets. BMC Med. Genet. 2013, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.T.; Dayeh, T.A.; Volkov, P.A.; Kirkpatrick, C.L.; Malmgren, S.; Jing, X.; Renstrom, E.; Wollheim, C.B.; Nitert, M.D.; Ling, C. Increased DNA methylation and decreased expression of PDX-1 in pancreatic islets from patients with type 2 diabetes. Mol. Endocrinol. 2012, 26, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Cai, X.; Yi, B.; Huang, J.; Wang, J.; Sun, J. Correlation of CTGF gene promoter methylation with CTGF expression in type 2 diabetes mellitus with or without nephropathy. Mol. Med. Rep. 2014, 9, 2138–2144. [Google Scholar] [PubMed]
- Kuroda, A.; Rauch, T.A.; Todorov, I.; Ku, H.T.; Al-Abdullah, I.H.; Kandeel, F.; Mullen, Y.; Pfeifer, G.P.; Ferreri, K. Insulin gene expression is regulated by DNA methylation. PLoS One 2009, 4, e6953. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, M.R.; Ward, C.S.; Arvide, E.M.; Chapleau, C.A.; Pozzo-Miller, L.; Hoeflich, A.; Sivaramakrishnan, M.; Saenger, S.; Metzger, F.; Neul, J.L. Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum. Mol. Genet. 2013, 22, 2626–2633. [Google Scholar] [PubMed]
- Akin, L.; Adal, E.; Akin, M.A.; Kurtoglu, S. A case of diabetes mellitus associated with Rett syndrome. J. Pediatr. Endocrinol. Metab. 2012, 25, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Rekik, N.M.; Kamoun, M.; Mnif, F.; Charfi, N.; Mnif, M.F.; Abid, M. Type 1 diabetes mellitus and Rett syndrome: Is there a link? J. Endocrinol. Invest. 2010, 33, 851. [Google Scholar] [CrossRef]
- Kurtoglu, S.; Atabek, M.E.; Kumandas, S.; Keskin, M. Diabetes mellitus type 1: Association with Rett syndrome. Pediatr. Int. 2005, 47, 90–91. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Genomic imprinting disorders in humans: A mini-review. J. Assist. Reprod. Genet. 2009, 26, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Migliavacca, E.; Dupre, Y.; Stathaki, E.; Sailani, M.R.; Baumer, A.; Schinzel, A.; Mackay, D.J.; Robinson, D.O.; Cobellis, G.; et al. Methylation profiling in individuals with uniparental disomy identifies novel differentially methylated regions on chromosome 15. Genome Res. 2010, 20, 1271–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, R.P.; Bogdarina, I.; Cazier, J.B.; Buchanan, C.; Donaldson, M.C.; Johnston, L.B.; Hokken-Koelega, A.C.; Clark, A.J. Multiple segmental uniparental disomy associated with abnormal DNA methylation of imprinted Loci in silver-russell syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E2188–E2193. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Gennarino, V.A.; Lee, Y.; Pang, K.; Hashimoto-Torii, K.; Choufani, S.; Raju, C.S.; Oldham, M.C.; Weksberg, R.; Rakic, P.; Liu, Z.; Zoghbi, H.Y. Human-specific regulation of MeCP2 levels in fetal brains by microRNA miR-483-5p. Genes Dev. 2013, 27, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Drewell, R.A.; Goddard, C.J.; Thomas, J.O.; Surani, M.A. Methylation-dependent silencing at the H19 imprinting control region by MeCP2. Nucl. Acids Res. 2002, 30, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Makedonski, K.; Abuhatzira, L.; Kaufman, Y.; Razin, A.; Shemer, R. MeCP2 deficiency in Rett syndrome causes epigenetic aberrations at the PWS/AS imprinting center that affects UBE3A expression. Hum. Mol. Genet. 2005, 14, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Samaco, R.C.; Hogart, A.; LaSalle, J.M. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum. Mol. Genet. 2005, 14, 483–492. [Google Scholar] [CrossRef]
- Horike, S.; Cai, S.; Miyano, M.; Cheng, J.F.; Kohwi-Shigematsu, T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 2005, 37, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.D.; Velazquez, J.; French, S.W.; Lu, S.C.; Ticku, M.K.; Zakhari, S. Emerging role of epigenetics in the actions of alcohol. Alcohol. Clin. Exp. Res. 2008, 32, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nandakumar, V.; Jiang, X.X.; Jones, L.; Yang, A.G.; Huang, X.F.; Chen, S.Y. The control of hematopoietic stem cell maintenance, self-renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Beerman, I.; Bock, C.; Garrison, B.S.; Smith, Z.D.; Gu, H.; Meissner, A.; Rossi, D.J. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 2013, 12, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Hogart, A.; Lichtenberg, J.; Ajay, S.S.; Anderson, S.; Margulies, E.H.; Bodine, D.M. Genome-wide DNA methylation profiles in hematopoietic stem and progenitor cells reveal overrepresentation of ETS transcription factor binding sites. Genome Res. 2012, 22, 1407–1418. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Alvarez, B.; Rodriguez, R.M.; Fraga, M.F.; Lopez-Larrea, C. DNA methylation: A promising landscape for immune system-related diseases. Trends Genet. 2012, 28, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Friez, M.J.; Jones, J.R.; Clarkson, K.; Lubs, H.; Abuelo, D.; Bier, J.A.; Pai, S.; Simensen, R.; Williams, C.; Giampietro, P.F.; Schwartz, C.E.; Stevenson, R.E. Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28. Pediatrics 2006, 118, e1687–e1695. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Ramocki, M.B.; Neul, J.L.; Lu, W.; Roberts, L.; Knight, J.; Ward, C.S.; Zoghbi, H.Y.; Kheradmand, F.; Corry, D.B. Overexpression of methyl-CpG binding protein 2 impairs T(H)1 responses. Sci. Transl. Med. 2012, 4, 1–10. [Google Scholar] [CrossRef]
- Sawalha, A.H.; Webb, R.; Han, S.; Kelly, J.A.; Kaufman, K.M.; Kimberly, R.P.; Alarcon-Riquelme, M.E.; James, J.A.; Vyse, T.J.; Gilkeson, G.S.; et al. Common variants within MECP2 confer risk of systemic lupus erythematosus. PLoS One 2008, 3, e1727. [Google Scholar] [CrossRef] [PubMed]
- Sawalha, A.H. Overexpression of methyl-CpG-binding protein 2 and autoimmunity: Evidence from MECP2 duplication syndrome, lupus, MECP2 transgenic and Mecp2 deficient mice. Lupus 2013, 22, 870–872. [Google Scholar] [CrossRef] [PubMed]
- Cobb, B.L.; Fei, Y.; Jonsson, R.; Bolstad, A.I.; Brun, J.G.; Rischmueller, M.; Lester, S.E.; Witte, T.; Illei, G.; Brennan, M.; et al. Genetic association between methyl-CpG binding protein 2 (MECP2) and primary Sjogren’s syndrome. Ann. Rheum. Dis. 2010, 69, 1731–1732. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M. Epigenetic therapeutics in autoimmune disease. Clin. Rev. Allergy Immunol. 2010, 39, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Balada, E.; Ordi-Ros, J.; Vilardell-Tarres, M. DNA methylation and systemic lupus erythematosus. Ann. N. Y. Acad. Sci. 2007, 1108, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Balada, E.; Ordi-Ros, J.; Serrano-Acedo, S.; Martinez-Lostao, L.; Vilardell-Tarres, M. Transcript overexpression of the MBD2 and MBD4 genes in CD4+ T cells from systemic lupus erythematosus patients. J. Leukoc. Biol. 2007, 81, 1609–1616. [Google Scholar] [CrossRef] [PubMed]
- Udali, S.; Guarini, P.; Moruzzi, S.; Choi, S.W.; Friso, S. Cardiovascular epigenetics: From DNA methylation to microRNAs. Mol. Aspects Med. 2013, 34, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Baccarelli, A.; Wright, R.; Bollati, V.; Litonjua, A.; Zanobetti, A.; Tarantini, L.; Sparrow, D.; Vokonas, P.; Schwartz, J. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 2010, 21, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Meisel, A.; Biniszkiewicz, D.; Namura, S.; Prass, K.; Ruscher, K.; Lipski, A.; Jaenisch, R.; Moskowitz, M.A.; Dirnagl, U. DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 2000, 20, 3175–3181. [Google Scholar] [PubMed]
- Endres, M.; Fan, G.; Meisel, A.; Dirnagl, U.; Jaenisch, R. Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport 2001, 12, 3763–3766. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-T.; Hsi, E.; Lin, H.-F.; Liao, Y.-C.; Wang, Y.-S.; Juo, S.-H. LINE-1 methylation is associated with an increased risk of ischemic stroke in men. Curr. Neurovasc. Res. 2014, 11, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, S.; Meisel, A.; Märschenz, S. Epigenetic mechanisms in cerebral ischemia. J. Cereb. Blood Flow Metab. 2013, 33, 1335–1346. [Google Scholar] [CrossRef]
- Lusardi, T.A.; Farr, C.D.; Faulkner, C.L.; Pignataro, G.; Yang, T.; Lan, J.; Simon, R.P.; Saugstad, J.A. Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. J. Cereb. Blood Flow Metab. 2010, 30, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.; Wen, L.; Liao, S.; Lin, X.; Ma, T.; Liu, J.; Song, C.X.; Wang, M.; He, C.; Han, C.; et al. Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat. Commun. 2013, 4, 1995. [Google Scholar] [PubMed]
- Klenova, E.M.; Nicolas, R.H.; Paterson, H.F.; Carne, A.F.; Heath, C.M.; Goodwin, G.H.; Neiman, P.E.; Lobanenkov, V.V. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell. Biol. 1993, 13, 7612–7624. [Google Scholar] [PubMed]
- Szabo, P.E.; Tang, S.H.; Silva, F.J.; Tsark, W.M.; Mann, J.R. Role of CTCF binding sites in the Igf2/H19 imprinting control region. Mol. Cell. Biol. 2004, 24, 4791–4800. [Google Scholar] [CrossRef] [PubMed]
- Zlatanova, J.; Caiafa, P. CTCF and its protein partners: Divide and rule? J. Cell Sci. 2009, 122, 1275–1284. [Google Scholar] [CrossRef]
- Gause, M.; Schaaf, C.A.; Dorsett, D. Cohesin and CTCF: Cooperating to control chromosome conformation? Bioessays 2008, 30, 715–718. [Google Scholar] [CrossRef]
- Chernukhin, I.; Shamsuddin, S.; Kang, S.Y.; Bergstrom, R.; Kwon, Y.W.; Yu, W.; Whitehead, J.; Mukhopadhyay, R.; Docquier, F.; Farrar, D.; et al. CTCF interacts with and recruits the largest subunit of RNA polymerase II to CTCF target sites genome-wide. Mol. Cell. Biol. 2007, 27, 1631–1648. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, M.E.; Zhang, L.F.; Xu, N.; Shi, Y.; Lee, J.T. Identification of a Ctcf cofactor, YY1, for the X chromosome binary switch. Mol. Cell 2007, 25, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Aulmann, S.; Blaker, H.; Penzel, R.; Rieker, R.J.; Otto, H.F.; Sinn, H.P. CTCF gene mutations in invasive ductal breast cancer. Breast Cancer Res. Treat. 2003, 80, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Docquier, F.; Farrar, D.; D’Arcy, V.; Chernukhin, I.; Robinson, A.F.; Loukinov, D.; Vatolin, S.; Pack, S.; Mackay, A.; Harris, R.A.; et al. Heightened expression of CTCF in breast cancer cells is associated with resistance to apoptosis. Cancer Res. 2005, 65, 5112–5122. [Google Scholar] [CrossRef] [PubMed]
- Paradowska, A.; Fenic, I.; Konrad, L.; Sturm, K.; Wagenlehner, F.; Weidner, W.; Steger, K. Aberrant epigenetic modifications in the CTCF binding domain of the IGF2/H19 gene in prostate cancer compared with benign prostate hyperplasia. Int. J. Oncol. 2009, 35, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.P.; Macaluso, M.; Miranda, F.; Montanari, M.; Russo, A.; Bagella, L.; Giordano, A. CTCF and BORIS regulate Rb2/p130 gene transcription: A novel mechanism and a new paradigm for understanding the biology of lung cancer. Mol. Cancer Res. 2011, 9, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Chadwick, R.B.; Peltomaki, P.; Plass, C.; Nakamura, Y.; de La Chapelle, A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Weksberg, R.; Smith, A.C.; Squire, J.; Sadowski, P. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum. Mol. Genet. 2003, 12, R61–R68. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, R.P.; Patzel, K.A.; Martin, M.; Yasui, D.H.; Swanberg, S.E.; Hertz-Picciotto, I.; Hansen, R.L.; van de Water, J.; Pessah, I.N.; Jiang, R.; Robinson, W.P.; LaSalle, J.M. MECP2 promoter methylation and X chromosome inactivation in autism. Autism Res. 2008, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Stiles, J.; Jernigan, T.L. The basics of brain development. Neuropsychol. Rev. 2010, 20, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Sadler, T.W. Embryology of neural tube development. Am. J. Med. Genet. C. Semin. Med. Genet. 2005, 135C, 2–8. [Google Scholar] [CrossRef]
- O’Rahilly, R.R.; Muller, F. The Embryonic Human Brain: An Atlas of Developmental Stages, 2rd ed.; Wiley-Liss: Toronto, Canada, 1999. [Google Scholar]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed]
- Seisenberger, S.; Peat, J.R.; Hore, T.A.; Santos, F.; Dean, W.; Reik, W. Reprogramming DNA methylation in the mammalian life cycle: Building and breaking epigenetic barriers. Philasophical Trans. R. Soc. Biol. Sci. 2013, 368, 1–11. [Google Scholar]
- Zhou, F.C. DNA methylation program during development. Front. Biol. (Beijing) 2012, 7, 485–494. [Google Scholar] [CrossRef]
- Chen, Y.; Damayanti, N.P.; Irudayaraj, J.; Dunn, K.; Zhou, F.C. Diversity of two forms of DNA methylation in the brain. Front. Genet. 2014, 5, 46. [Google Scholar] [PubMed]
- Chen, Y.; Ozturk, N.C.; Zhou, F.C. DNA methylation program in developing hippocampus and its alteration by alcohol. PLoS One 2013, 8, e60503. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics 2011, 6, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Israel, Y.; Rivera-Meza, M.; Karahanian, E.; Quintanilla, M.E.; Tampier, L.; Morales, P.; Herrera-Marschitz, M. Gene specific modifications unravel ethanol and acetaldehyde actions. Front. Behav. Neurosci. 2013, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Behnke, M.; Smith, V.C. Prenatal substance abuse: Short- and long-term effects on the exposed fetus. Pediatrics 2013, 131, e1009–e1024. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhao, M.; Li, M.; Song, T.; Zhang, M.; Quan, L.; Li, S.; Sun, Z.S. Reversal of cocaine-conditioned place preference through methyl supplementation in mice: Altering global DNA methylation in the prefrontal cortex. PLoS One 2012, 7, e33435. [Google Scholar] [CrossRef] [PubMed]
- Satta, R.; Maloku, E.; Zhubi, A.; Pibiri, F.; Hajos, M.; Costa, E.; Guidotti, A. Nicotine decreases DNA methyltransferase 1 expression and glutamic acid decarboxylase 67 promoter methylation in GABAergic interneurons. PNAS 2008, 105, 16356–16361. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Balaraman, Y.; Wang, G.; Nephew, K.P.; Zhou, F.C. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009, 4, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Balaraman, Y.; Teng, M.; Liu, Y.; Singh, R.P.; Nephew, K.P. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol. Clin. Exp. Res. 2011, 35, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Varela-Rey, M.; Woodhoo, A.; Martinez-Chantar, M.-L.; Mato, J.M.; Lu, S.C. Alcohol, DNA methylation, and cancer. Alcohol Res. Curr. Rev. 2011, 25–35. [Google Scholar]
- Kruman, I.; Fowler, A.-K. One-carbon metabolism and risk factors for its disturbance. J. Neurochem. 2014, 129, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Mantha, K.; Kleiber, M.L.; Diehl, E.J.; Addison, S.M.F.; Singh, S.M. Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis. Model. Mech. 2013, 6, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, D.M.; Zaher, F.M.; Svinarich, D.M.; Abel, E.L. Paternal alcohol exposure affects sperm cytosine methyltransferase messenger RNA levels. Alcohol. Clin. Exp. Res. 2002, 26, 347–351. [Google Scholar] [CrossRef]
- Ouko, L.A.; Shantikumar, K.; Knezovich, J.; Haycock, P.; Schnugh, D.J.; Ramsay, M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes: Implications for fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2009, 33, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Stouder, C.; Somm, E.; Paoloni-Giacobino, A. Prenatal exposure to ethanol: A specific effect on the H19 gene in sperm. Reprod. Toxicol. 2011, 31, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Knezovich, J.G.; Ramsay, M. The effect of preconception paternal alcohol exposure on epigenetic remodeling of the h19 and rasgrf1 imprinting control regions in mouse offspring. Front. Genet. 2012, 3, 10. [Google Scholar] [CrossRef]
- Maccani, J.Z.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Kelsey, K.T. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics 2013, 5, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.R.; Håberg, S.E.; Nilsen, R.M.; Wang, X.; Vollset, S.E.; Murphy, S.K.; Nystad, W.; Bell, D.A.; Peddada, S.D.; London, S.J. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012, 120, 1425–1432. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, D.; Dasgupta, C.; Xiong, F.; Tong, W.; Yang, S.; Zhang, L. Perinatal nicotine exposure increases vulnerability of hypoxic-ischemic brain injury in neonatal rats: Role of angiotensin II receptors. Stroke 2012, 43, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Breton, C.V.; Byun, H.-M.; Wenten, M.; Pan, F.; Yang, A.; Gilliland, F.D. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009, 180, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Rodriguez, M.; Lotfipour, S.; Leonard, G.; Perron, M.; Richer, L.; Veillette, S.; Pausova, Z.; Paus, T. Maternal smoking during pregnancy is associated with epigenetic modifications of the brain-derived neurotrophic factor-6 exon in adolescent offspring. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2010, 153B, 1350–1354. [Google Scholar] [CrossRef]
- Chung, W.C.; Auger, A.P. Gender differences in neurodevelopment and epigenetics. Pflugers Arch. 2013, 465, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.E.; Rodgers, A.B.; Morgan, C.P.; Bale, T.L. Epigenetic mechanisms in pubertal brain maturation. Neuroscience 2014, 264, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Pagani, L.S. Environmental tobacco smoke exposure and brain development: The case of attention deficit/hyperactivity disorder. Neurosci. Biobehav. Rev. 2014. [Google Scholar] [CrossRef]
- Novikova, S.I.; He, F.; Bai, J.; Cutrufello, N.J.; Lidow, M.S.; Undieh, A.S. Maternal cocaine administration in mice alters DNA methylation and gene expression in hippocampal neurons of neonatal and prepubertal offspring. PLoS One 2008, 3, e1919. [Google Scholar] [CrossRef] [PubMed]
- Anier, K.; Malinovskaja, K.; Aonurm-Helm, A.; Zharkovsky, A.; Kalda, A. DNA methylation regulates cocaine-induced behavioral sensitization in mice. Neuropsychopharmacology 2010, 35, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; James, S.R.; Kazim, L.; Karpf, A.R. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Chem. 2005, 77, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Itzhak, Y.; Ergui, I.; Young, J.I. Long-term parental methamphetamine exposure of mice influences behavior and hippocampal DNA methylation of the offspring. Mol. Psychiatry 2014. [Google Scholar] [CrossRef]
- Fragou, D.; Zanos, P.; Kouidou, S.; Njau, S.; Kitchen, I.; Bailey, A.; Kovatsi, L. Effect of chronic heroin and cocaine administration on global DNA methylation in brain and liver. Toxicol. Lett. 2013, 218, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.; Palta, P.; Joyce, C.J.; Scott, C.; Grundberg, E.; Deloukas, P.; Palotie, A.; Coffey, A.J. A comparison of the whole genome approach of MeDIP-seq to the targeted approach of the Infinium HumanMethylation450 BeadChip® for methylome profiling. PLoS One 2012, 7, e50233. [Google Scholar] [CrossRef] [PubMed]
- Olynik, B.M.; Rastegar, M. The genetic and epigenetic journey of embryonic stem cells into mature neural cells. Front. Genet. 2012, 3, 81. [Google Scholar] [CrossRef] [PubMed]
- Ezeonwuka, C.; Rastegar, M. MeCP2-related diseases and animal models. Diseases 2014, 2, 45–70. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Antalffy, B.; Armstrong, D.L.; Zoghbi, H.Y. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 2002, 11, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.O.; Zachariah, R.M.; Ezeonwuka, C.D.; Liyanage, V.R.; Rastegar, M. Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS One 2014, 9, e90645. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Zachariah, R.M.; Rastegar, M. Decitabine alters the expression of Mecp2 isoforms via dynamic DNA methylation at the Mecp2 regulatory elements in neural stem cells. Mol. Autism 2013, 4, 46. [Google Scholar] [CrossRef] [PubMed]
- Rastegar, M.; Hotta, A.; Pasceri, P.; Makarem, M.; Cheung, A.Y.; Elliott, S.; Park, K.J.; Adachi, M.; Jones, F.S.; Clarke, I.D.; et al. MECP2 isoform-specific vectors with regulated expression for Rett syndrome gene therapy. PLoS One 2009, 4, e6810. [Google Scholar] [CrossRef] [PubMed]
- Renieri, A.; Meloni, I.; Longo, I.; Ariani, F.; Mari, F.; Pescucci, C.; Cambi, F. Rett syndrome: The complex nature of a monogenic disease. J. Mol. Med. 2003, 81, 346–354. [Google Scholar] [PubMed]
- Amir, R.E.; van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.H.; Gabel, H.W.; Robinson, N.D.; Kastan, N.R.; Hu, L.S.; Cohen, S.; Navarro, A.J.; Lyst, M.J.; Ekiert, R.; Bird, A.P.; et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 2013, 499, 341–345. [Google Scholar] [PubMed]
- Nagarajan, R.P.; Hogart, A.R.; Gwye, Y.; Martin, M.R.; LaSalle, J.M. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006, 1, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, Y.; Kamio, Y.; Kawai, T.; Katsuura, S.; Inada, N.; Takaki, A.; Rokutan, K. Autism-associated gene expression in peripheral leucocytes commonly observed between subjects with autism and healthy women having autistic children. PLoS One 2011, 6, e24723. [Google Scholar] [CrossRef] [PubMed]
- Carney, R.M.; Wolpert, C.M.; Ravan, S.A.; Shahbazian, M.; Ashley-Koch, A.; Cuccaro, M.L.; Vance, J.M.; Pericak-Vance, M.A. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr. Neurol. 2003, 28, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.S.; Blasi, F.; Bacchelli, E.; Klauck, S.M.; Maestrini, E.; Poustka, A. Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum. Genet. 2002, 111, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.W.; Yeung, W.L.; Ko, C.H.; Poon, P.M.; Tong, S.F.; Chan, K.Y.; Lo, I.F.; Chan, L.Y.; Hui, J.; Wong, V.; et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J. Med. Genet. 2000, 37, E41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loat, C.S.; Curran, S.; Lewis, C.M.; Duvall, J.; Geschwind, D.; Bolton, P.; Craig, I.W. Methyl-CpG-binding protein 2 polymorphisms and vulnerability to autism. Genes Brain Behav. 2008, 7, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, A.; Cook, E.H., Jr.; Feng, J.; Glanzmann, C.; Yan, J.; Craddock, N.; Jones, I.R.; Goldman, D.; Heston, L.L.; Sommer, S.S. MECP2 structural and 3ꞌ-UTR variants in schizophrenia, autism and other psychiatric diseases: A possible association with autism. Am. J. Med. Genet B Neuropsychiatr. Genet. 2004, 128B, 50–53. [Google Scholar] [CrossRef]
- Coutinho, A.M.; Oliveira, G.; Katz, C.; Feng, J.; Yan, J.; Yang, C.; Marques, C.; Ataide, A.; Miguel, T.S.; Borges, L.; et al. MECP2 coding sequence and 3ꞌUTR variation in 172 unrelated autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144B, 475–483. [Google Scholar] [CrossRef]
- Peters, S.U.; Hundley, R.J.; Wilson, A.K.; Warren, Z.; Vehorn, A.; Carvalho, C.M.; Lupski, J.R.; Ramocki, M.B. The behavioral phenotype in MECP2 duplication syndrome: A comparison with idiopathic autism. Autism Res. 2013, 6, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xu, Q.; Zhang, Y.; Zhang, X.; Cheng, T.; Wu, B.; Ding, Y.; Lu, P.; Zheng, J.; Zhang, M.; Qiu, Z.; Yu, X. A case report of Chinese brothers with inherited MECP2-containing duplication: Autism and intellectual disability, but not seizures or respiratory infections. BMC Med. Genet. 2012, 13, 75. [Google Scholar] [CrossRef] [PubMed]
- Zoll, B.; Huppke, P.; Wessel, A.; Bartels, I.; Laccone, F. Fetal alcohol syndrome in association with Rett syndrome. Genet. Couns. 2004, 15, 207–212. [Google Scholar] [PubMed]
- Baker, S.A.; Chen, L.; Wilkins, A.D.; Yu, P.; Lichtarge, O.; Zoghbi, H.Y. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 2013, 152, 984–996. [Google Scholar] [CrossRef]
- Kim, P.; Park, J.H.; Choi, C.S.; Choi, I.; Joo, S.H.; Kim, M.K.; Kim, S.Y.; Kim, K.C.; Park, S.H.; Kwon, K.J.; et al. Effects of ethanol exposure during early pregnancy in hyperactive, inattentive and impulsive behaviors and MeCP2 expression in rodent offspring. Neurochem. Res. 2013, 38, 620–631. [Google Scholar] [CrossRef] [PubMed]
- Romano-Lopez, A.; Mendez-Diaz, M.; Ruiz-Contreras, A.E.; Carrisoza, R.; Prospero-Garcia, O. Maternal separation and proclivity for ethanol intake: A potential role of the endocannabinoid system in rats. Neuroscience 2012, 223, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Repunte-Canonigo, V.; Chen, J.; Lefebvre, C.; Kawamura, T.; Kreifeldt, M.; Basson, O.; Roberts, A.J.; Sanna, P.P. MeCP2 regulates ethanol sensitivity and intake. Addict. Biol. 2013, 19, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Tunc-Ozcan, E.; Ullmann, T.M.; Shukla, P.K.; Redei, E.E. Low-dose thyroxine attenuates autism-associated adverse effects of fetal alcohol in male offspring’s social behavior and hippocampal gene expression. Alcohol. Clin. Exp. Res. 2013, 37, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Choi, C.S.; Park, J.H.; Joo, S.H.; Kim, S.Y.; Ko, H.M.; Kim, K.C.; Jeon, S.J.; Park, S.H.; Han, S.H.; et al. Chronic exposure to ethanol of male mice before mating produces attention deficit hyperactivity disorder-like phenotype along with epigenetic dysregulation of dopamine transporter expression in mouse offspring. J. Neurosci. Res. 2014, 92, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol. Clin. Exp. Res. 2013, 37, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Subbanna, S.; Nagre, N.N.; Shivakumar, M.; Umapathy, N.S.; Psychoyos, D.; Basavarajappa, B.S. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 2014, 258, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Joyner, A.H.; Roddey, J.C.; Bloss, C.S.; Bakken, T.E.; Rimol, L.M.; Melle, I.; Agartz, I.; Djurovic, S.; Topol, E.J.; Schork, N.J.; et al. A common MECP2 haplotype associates with reduced cortical surface area in humans in two independent populations. Proc. Natl. Acad. Sci. USA 2009, 106, 15483–15488. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, F.; Racagni, G.; Riva, M.A. The expanding role of BDNF: A therapeutic target for Alzheimer’s disease? Pharmacogenomics J. 2006, 6, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Couvert, P.; Bienvenu, T.; Aquaviva, C.; Poirier, K.; Moraine, C.; Gendrot, C.; Verloes, A.; Andres, C.; le Fevre, A.C.; Souville, I.; et al. MECP2 is highly mutated in X-linked mental retardation. Hum. Mol. Genet. 2001, 10, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.; Black, G.; Ramsden, S.; Barrow, M.; Super, M.; Kerr, B.; Clayton-Smith, J. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J. Med. Genet. 2001, 38, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Olkhov-Mitsel, E.; Bapat, B. Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers. Cancer Med. 2012, 1, 237–260. [Google Scholar] [CrossRef] [PubMed]
- Montano, C.M.; Irizarry, R.A.; Kaufmann, W.E.; Talbot, K.; Gur, R.E.; Feinberg, A.P.; Taub, M.A. Measuring cell-type specific differential methylation in human brain tissue. Genome Biol. 2013, 14, R94. [Google Scholar] [CrossRef] [PubMed]
- Guintivano, J.; Aryee, M.J.; Kaminsky, Z.A. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 2013, 8, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Mainous, A., 3rd; Tanner, R.; Baker, R.; Zayas, C.; Harle, C. Prevalence of prediabetes in England from 2003 to 2011: Population-based, cross-sectional study. BMJ Open 2014, 4, e005002. [Google Scholar] [CrossRef] [PubMed]
- Volkmar, M.; Dedeurwaerder, S.; Cunha, D.A.; Ndlovu, M.N.; Defrance, M.; Deplus, R.; Calonne, E.; Volkmar, U.; Igoillo-Esteve, M.; Naamane, N.; et al. DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J. 2012, 31, 1405–1426. [Google Scholar] [CrossRef] [PubMed]
- Dayeh, T.; Volkov, P.; Salö, S.; Hall, E.; Nilsson, E.; Olsson, A.H.; Kirkpatrick, C.L.; Wollheim, C.B.; Eliasson, L.; Rönn, T.; et al. Genome-wide DNA methylation analysis of human pancreatic islets from type 2 diabetic and non-diabetic donors identifies candidate genes that influence insulin secretion. PLoS Genet. 2014, 10, e1004160. [Google Scholar] [CrossRef] [PubMed]
- Toperoff, G.; Aran, D.; Kark, J.D.; Rosenberg, M.; Dubnikov, T.; Nissan, B.; Wainstein, J.; Friedlander, Y.; Levy-Lahad, E.; Glaser, B.; et al. Genome-wide survey reveals predisposing diabetes type 2-related DNA methylation variations in human peripheral blood. Hum. Mol. Genet. 2012, 21, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Finer, S.; Lindgren, C.M.; Wilson, G.A.; Rakyan, V.K.; Teschendorff, A.E.; Akan, P.; Stupka, E.; Down, T.A.; Prokopenko, I.; et al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS One 2010, 5, e14040. [Google Scholar] [CrossRef] [PubMed]
- Levenson, V.V.; Melnikov, A.A. DNA methylation as clinically useful biomarkers-light at the end of the tunnel. Pharmaceuticals 2012, 5, 94–113. [Google Scholar] [CrossRef] [PubMed]
- Levenson, V.V. DNA methylation as a universal biomarker. Expert Rev. Mol. Diagn. 2010, 10, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, E.A.; Karagrigoriou, A.; Tsaliki, E.; Velissariou, V.; Carter, N.P.; Patsalis, P.C. Fetal-specific DNA methylation ratio permits noninvasive prenatal diagnosis of trisomy 21. Nat. Med. 2011, 17, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Marsit, C.J.; Lambertini, L.; MacCani, M.A.; Koestler, D.C.; Houseman, E.A.; Padbury, J.F.; Lester, B.M.; Chen, J. Placenta-imprinted gene expression association of infant neurobehavior. J. Pediatr. 2012, 160, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Marsit, C.J.; Maccani, M.A.; Padbury, J.F.; Lester, B.M. Placental 11-beta hydroxysteroid dehydrogenase methylation is associated with newborn growth and a measure of neurobehavioral outcome. PLoS One 2012, 7, e33794. [Google Scholar] [CrossRef] [PubMed]
- Maccani, M.A.; Padbury, J.F.; Lester, B.M.; Knopik, V.S.; Marsit, C.J. Placental miRNA expression profiles are associated with measures of infant neurobehavioral outcomes. Pediatr. Res. 2013, 74, 272–278. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liyanage, V.R.B.; Jarmasz, J.S.; Murugeshan, N.; Del Bigio, M.R.; Rastegar, M.; Davie, J.R. DNA Modifications: Function and Applications in Normal and Disease States. Biology 2014, 3, 670-723. https://0-doi-org.brum.beds.ac.uk/10.3390/biology3040670
Liyanage VRB, Jarmasz JS, Murugeshan N, Del Bigio MR, Rastegar M, Davie JR. DNA Modifications: Function and Applications in Normal and Disease States. Biology. 2014; 3(4):670-723. https://0-doi-org.brum.beds.ac.uk/10.3390/biology3040670
Chicago/Turabian StyleLiyanage, Vichithra R. B., Jessica S. Jarmasz, Nanditha Murugeshan, Marc R. Del Bigio, Mojgan Rastegar, and James R. Davie. 2014. "DNA Modifications: Function and Applications in Normal and Disease States" Biology 3, no. 4: 670-723. https://0-doi-org.brum.beds.ac.uk/10.3390/biology3040670