Trypanosomatid Aquaporins: Roles in Physiology and Drug Response

Abstract
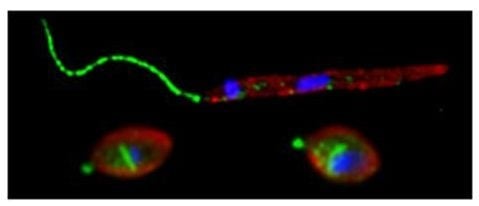
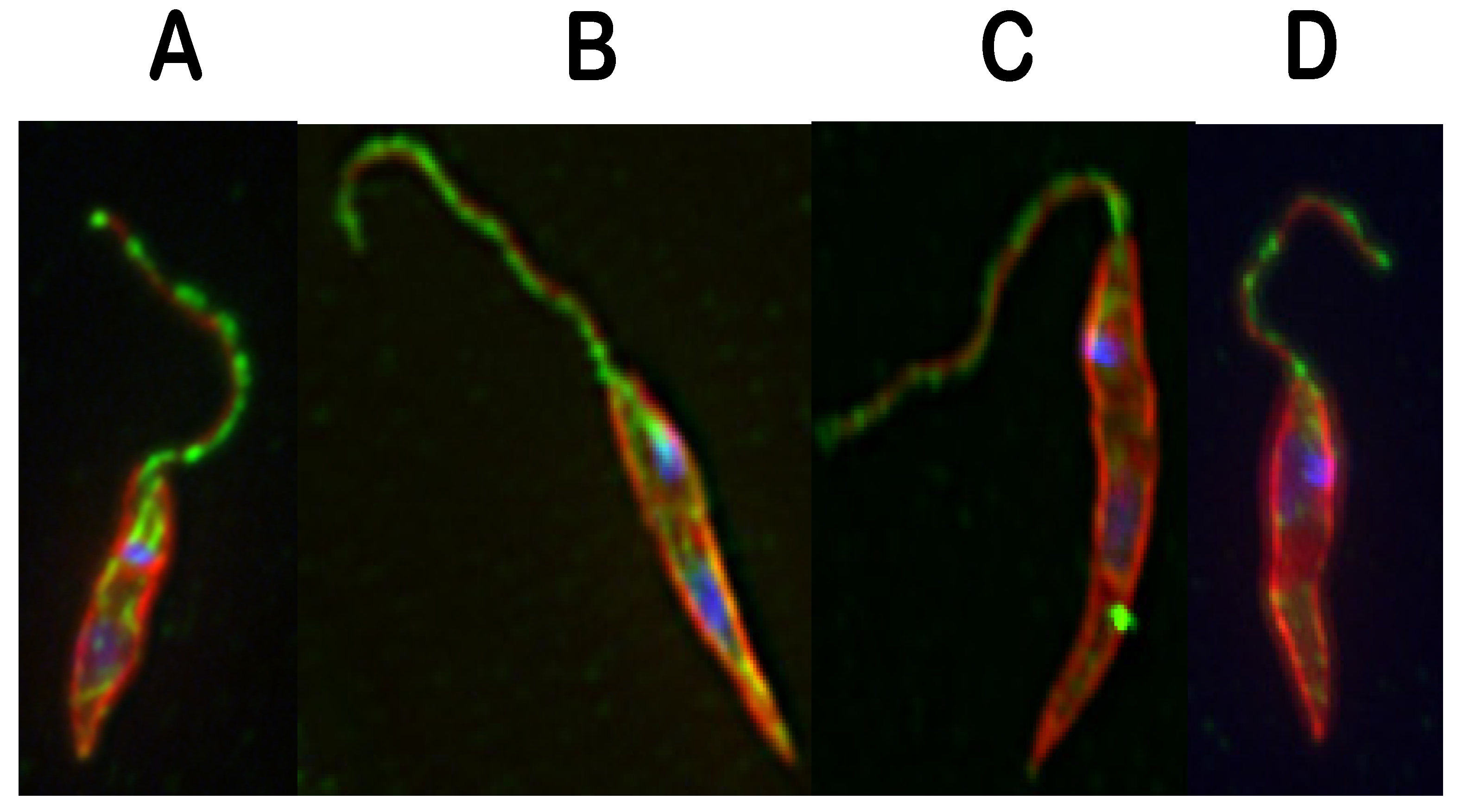
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Leishmania spp.: Leishmaniasis
2.1. Life Cycle
2.2. Disease
2.3. Treatment and Drug Resistance

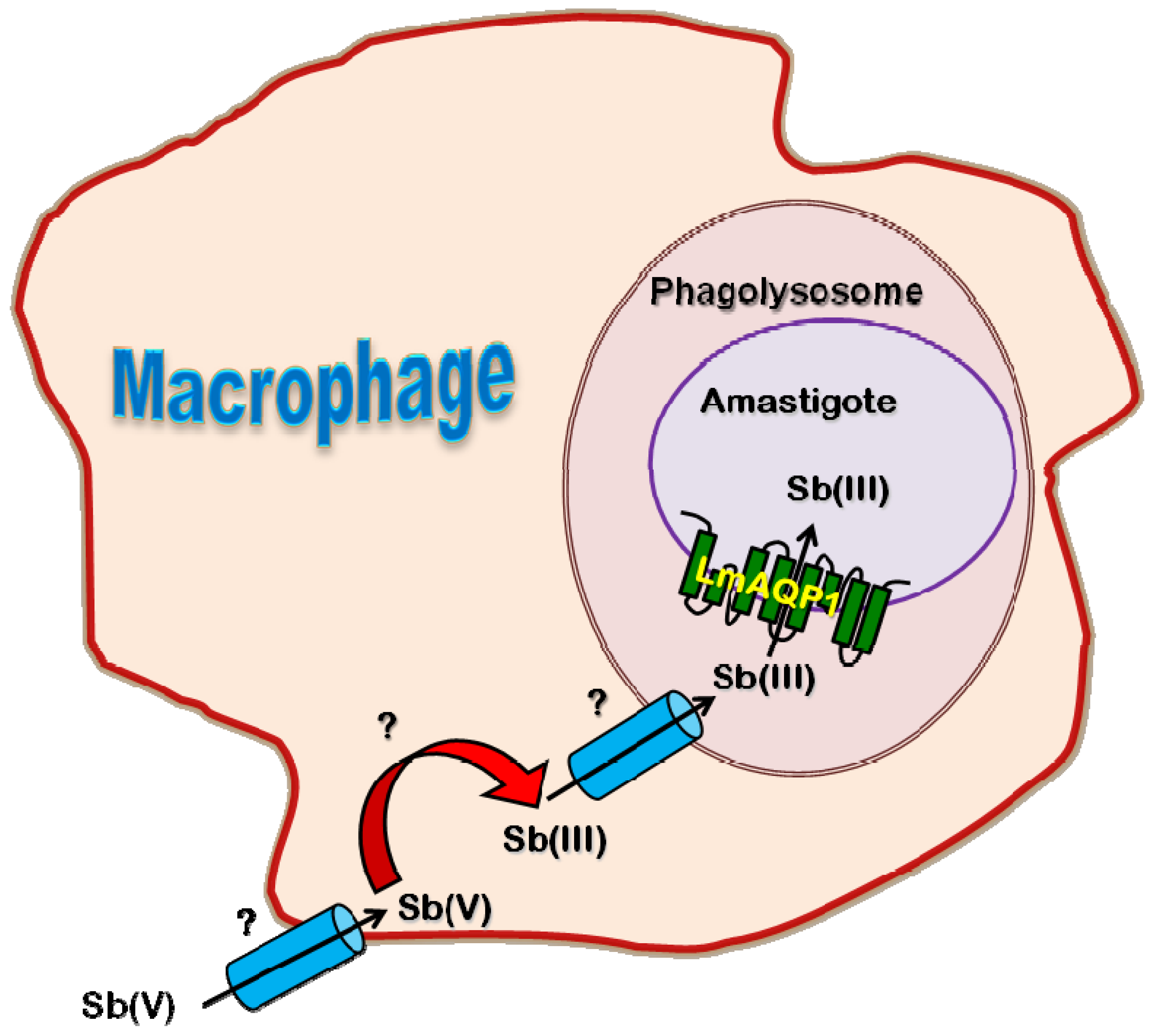
2.4. Role of AQP in Parasite Physiology and Drug Response
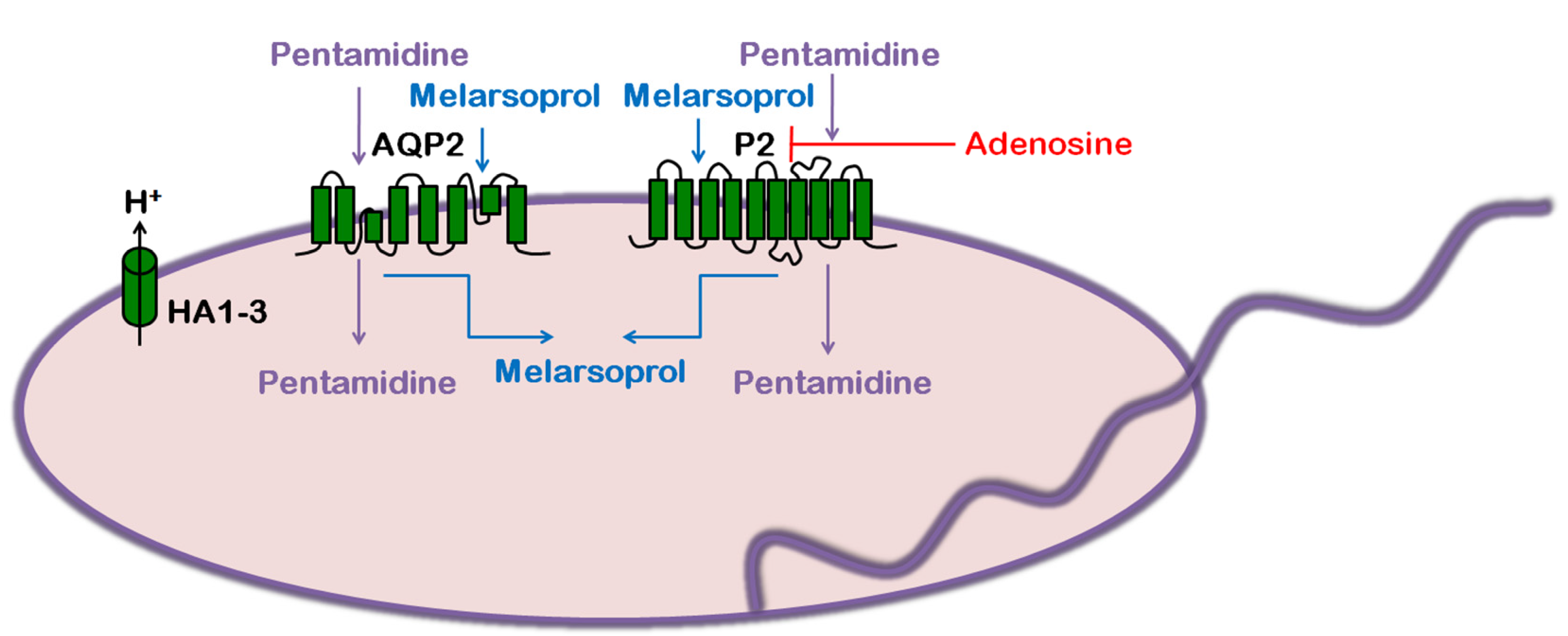
3. Trypanosoma brucei: Human African Trypanosomiasis (HAT)
3.1. Life Cycle
3.2. Disease
3.3. Treatment and Drug Resistance
3.4. Role of AQP in Parasite Physiology and Drug Response

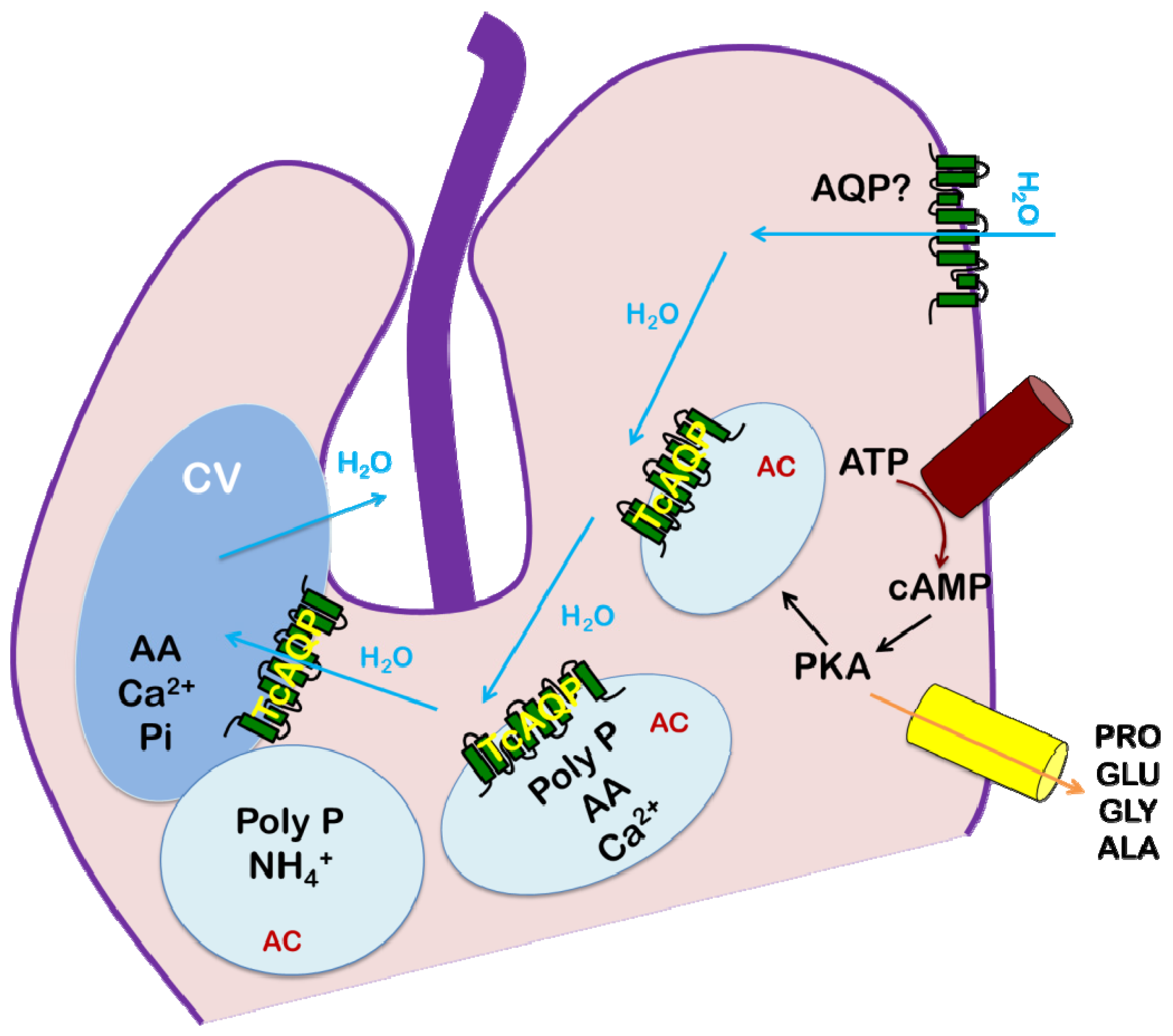
4. Trypanosoma cruzi: Chagas Disease
4.1. Life Cycle
4.2. Disease
4.3. Treatment and Drug Resistance
4.4. Role of AQP in Parasite Physiology and Drug Response

5. Conclusions
Acknowledgments
Conflicts of Interest
References
- den Boer, M.; Argaw, D.; Jannin, J.; Alvar, J. Leishmaniasis impact and treatment access. Clin. Microbiol. Infect. 2011, 17, 1471–1477. [Google Scholar] [CrossRef]
- Aït-Oudhia, K.; Gazanion, E.; Vergnes, B.; Oury, B.; Sereno, D. Leishmania antimony resistance: What we know what we can learn from the field. Parasitol. Res. 2011, 109, 1225–1232. [Google Scholar] [CrossRef]
- Human African trypanosomiasis. Available online: http://www.who.int/trypanosomiasis_african/en/ (accessed on 27 November 2013).
- Kennedy, P.G. Clinical features, Diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013, 12, 186–194. [Google Scholar]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Gull, K. The biology of kinetoplastid parasites: Insights and challenges from genomics and post-genomics. Int. J. Parasitol. 2001, 31, 443–452. [Google Scholar] [CrossRef]
- Rohloff, P.; Docampo, R.A. Contractile vacuole complex is involved in osmoregulation in Trypanosoma cruzi. Exp. Parasitol. 2008, 118, 17–24. [Google Scholar] [CrossRef]
- Ishibashi, K.; Kondo, S.; Hara, S.; Morishita, Y. The evolutionary aspects of aquaporin family. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R566–R576. [Google Scholar] [CrossRef]
- Verkman, A.S. Aquaporins at a glance. J. Cell. Sci. 2011, 124, 2107–2112. [Google Scholar] [CrossRef]
- Preston, G.M.; Carroll, T.P.; Guggino, W.B.; Agre, P. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 1992, 256, 385–387. [Google Scholar]
- Chrispeels, M.J.; Agre, P. Aquaporins: Water channel proteins of plant and animal cells. Trends Biochem. Sci. 1994, 19, 421–425. [Google Scholar] [CrossRef]
- Loitto, V.M.; Karlsson, T.; Magnusson, K.E. Water flux in cell motility: Expanding the mechanisms of membrane protrusion. Cell Motil. Cytoskeleton 2009, 66, 237–247. [Google Scholar] [CrossRef]
- Borgnia, M.; Nielsen, S.; Engel, A.; Agre, P. Cellular and molecular biology of the aquaporin water channels. Annu. Rev. Biochem. 1999, 68, 425–458. [Google Scholar] [CrossRef]
- Gourbal, B.; Sonuc, N.; Bhattacharjee, H.; Legare, D.; Sundar, S.; Ouellette, M.; Rosen, B.P.; Mukhopadhyay, R. Drug uptake and modulation of drug resistance in Leishmania by an aquaglyceroporin. J. Biol. Chem. 2004, 279, 31010–31017. [Google Scholar] [CrossRef]
- Musa-Aziz, R.; Chen, L.M.; Pelletier, M.F.; Boron, W.F. Relative CO2/NH3 selectivities of AQP1, AQP4, AQP5, AmtB, and RhAG. Proc. Natl. Acad. Sci. USA 2009, 106, 5406–5411. [Google Scholar] [CrossRef]
- Yool, A.J. Functional domains of aquaporin-1: Keys to physiology, and targets for drug discovery. Curr. Pharm. Des. 2007, 13, 3212–3221. [Google Scholar] [CrossRef]
- Engel, A.; Fujiyoshi, Y.; Agre, P. The importance of aquaporin water channel protein structures. EMBO J. 2000, 19, 800–806. [Google Scholar] [CrossRef]
- Song, J.; Mak, E.; Wu, B.; Beitz, E. Parasite aquaporins: Current developments in drug facilitation and resistance. Biochim. Biophys. Acta 2013, in press. [Google Scholar]
- Uzcategui, N.L.; Szallies, A.; Pavlovic-Djuranovic, S.; Palmada, M.; Figarella, K.; Boehmer, C.; Lang, F.; Beitz, E.; Duszenko, M. Cloning, Heterologous expression, and characterization of three aquaglyceroporins from Trypanosoma brucei. J. Biol. Chem. 2004, 279, 42669–42676. [Google Scholar] [CrossRef]
- Beitz, E. Aquaporins from pathogenic protozoan parasites: Structure, Function and potential for chemotherapy. Biol. Cell 2005, 97, 373–383. [Google Scholar] [CrossRef]
- Montalvetti, A.; Rohloff, P.; Docampo, R. A functional aquaporin co-localizes with the vacuolar proton pyrophosphatase to acidocalcisomes and the contractile vacuole complex of Trypanosoma cruzi. J. Biol. Chem. 2004, 279, 38673–38682. [Google Scholar] [CrossRef]
- Kirk, K. Channels and transporters as drug targets in the Plasmodium-infected erythrocyte. Acta Trop. 2004, 89, 285–298. [Google Scholar] [CrossRef]
- Pavlovic-Djuranovic, S.; Kun, J.F.; Schultz, J.E.; Beitz, E. Dihydroxyacetone and methylglyoxal as permeants of the Plasmodium aquaglyceroporin inhibit parasite proliferation. Biochim. Biophys. Acta 2006, 1758, 1012–1017. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Beitz, E. Metalloid transport by aquaglyceroporins: Consequences in the treatment of human diseases. Adv. Exp. Med. Biol. 2010, 679, 57–69. [Google Scholar] [CrossRef]
- Bhattacharjee, H.; Mukhopadhyay, R.; Thiyagarajan, S.; Rosen, B.P. Aquaglyceroporins: Ancient channels for metalloids. J. Biol. 2008, 7, 33. [Google Scholar] [CrossRef]
- Dostálová, A.; Volf, P. Leishmania development in sand flies: Parasite-vector interactions overview. Parasit. Vectors 2012, 5, 276. [Google Scholar] [CrossRef]
- Rogers, M.E.; Chance, M.L.; Bates, P.A. The role of promastigote secretory gel in the origin and transmission of the infective stage of Leishmania mexicana by the sandfly Lutzomyia longipalpis. Parasitology 2002, 124, 495–507. [Google Scholar]
- Walters, L.I. Leishmania differentiation in natural and unnatural sand fly hosts. J. Eukaryot. Microbiol. 1993, 40, 196–206. [Google Scholar] [CrossRef]
- Kamhawi, S. Phlebotomine sand flies and Leishmania parasites: Friends or foes? Trends Parasitol. 2006, 22, 439–445. [Google Scholar] [CrossRef]
- Esch, K.J.; Petersen, C.A. Transmission and epidemiology of zoonotic protozoal diseases of companion animals. Clin. Microbiol. Rev. 2013, 26, 58–85. [Google Scholar] [CrossRef]
- Convit, J.; Castellanos, P.L.; Ulrich, M.; Castés, M.; Rondón, A.; Pinardi, M.E.; Rodríquez, N.; Bloom, B.R.; Formica, S.; Valecillos, L.; et al. Immunotherapy of localized, Intermediate, and diffuse forms of American cutaneous leishmaniasis. J. Infect. Dis. 1989, 160, 104–115. [Google Scholar] [CrossRef]
- Convit, J.; Ulrich, M.; Fernández, C.T.; Tapia, F.J.; Cáceres-Dittmar, G.; Castés, M.; Rondón, A.J. The clinical and immunological spectrum of American cutaneous leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 1993, 87, 444–448. [Google Scholar] [CrossRef]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M. WHO leishmaniasis control team, Leishmaniasis Worldwide and global estimates of its incidence. PLoS One 2012, 7, e35671. [Google Scholar]
- Alvar, J.; Aparicio, P.; Aseffa, A.; Den Boer, M.; Cañavate, C.; Dedet, J.P.; Gradoni, L.; Ter Horst, R.; López-Vélez, R.; Moreno, J. The relationship between leishmaniasis and AIDS: the second 10 years. Clin. Microbiol. Rev. 2008, 21, 334–359. [Google Scholar] [CrossRef]
- Schwartz, E.; Hatz, C.; Blum, J. New world cutaneous leishmaniasis in travellers. Lancet Infect. Dis. 2006, 6, 342–349. [Google Scholar] [CrossRef]
- Garcia, L.S. Trypanosomiasis. In Diagnostic Medical Parasitology, 5th ed.; American Society of Microbiology: Washington, DC, USA, 2007; pp. 218–243. [Google Scholar]
- Ives, A.; Ronet, C.; Prevel, F.; Ruzzante, G.; Fuertes-Marraco, S.; Schutz, F.; Zangger, H.; Revaz-Breton, M.; Lye, L.F.; Hickerson, S.M.; Beverley, S.M.; et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 2011, 331, 775–778. [Google Scholar] [CrossRef]
- van Griensven, J.; Diro, E. Visceral leishmaniasis. Infect. Dis. Clin. North Am. 2012, 26, 309–322. [Google Scholar] [CrossRef]
- Ashutosh.; Sundar, S.; Goyal, N. Molecular mechanisms of antimony resistance in Leishmania. J. Med. Microbiol. 2007, 56, 143–153. [Google Scholar] [CrossRef]
- Sundar, S.; Chakravarty, J. Recent advances in the diagnosis and treatment of kala-azar. Natl. Med. J. India 2012, 25, 85–89. [Google Scholar]
- Ephros, M.; Bitnun, A.; Shaked, P.; Waldman, E.; Zilberstein, D. Stage-specific activity of pentavalent antimony against Leishmania donovani axenic amastigotes. Antimicrob. Agents Chemother. 1999, 43, 278–282. [Google Scholar]
- Shaked-Mishan, P.; Ulrich, N.; Ephros, M.; Zilberstein, D. Novel intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. J. Biol. Chem. 2001, 276, 3971–3976. [Google Scholar]
- Singh, N.; Kumar, M.; Singh, R.K. Leishmaniasis: Current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 2012, 5, 485–497. [Google Scholar] [CrossRef]
- Marquis, N.; Gourbal, B.; Rosen, B.P.; Mukhopadhyay, R.; Ouellette, M. Modulation in aquaglyceroporin AQP1 gene transcript levels in drug-resistant Leishmania. Mol. Microbiol. 2005, 57, 1690–1699. [Google Scholar] [CrossRef]
- Mandal, S.; Maharjan, M.; Singh, S.; Chatterjee, M.; Madhubala, R. Assessing aquaglyceroporin gene status and expression profile in antimony-susceptible and -resistant clinical isolates of Leishmania donovani from India. J. Antimicrob. Chemother. 2010, 65, 496–507. [Google Scholar] [CrossRef]
- Rai, S.; Bhaskar; Goel, S.K.; Nath Dwivedi, U.; Sundar, S.; Goyal, N. Role of efflux pumps and intracellular thiols in natural antimony resistant isolates of Leishmania donovani. PLoS One. 2013, 8, e74862. [Google Scholar]
- Figarella, K.; Uzcategui, N.L.; Zhou, Y.; LeFurgey, A.; Ouellette, M.; Bhattacharjee, H.; Mukhopadhyay, R. Biochemical characterization of Leishmania major aquaglyceroporin LmAQP1: Possible role in volume regulation and osmotaxis. Mol Microbiol. 2007, 65, 1006–1017. [Google Scholar] [CrossRef]
- Bhattacharjee, H.; Rosen, B.P.; Mukhopadhyay, R. Aquaglyceroporins and metalloid transport: implications in human diseases. Handb. Exp. Pharmacol. 2009, 190, 309–325. [Google Scholar] [CrossRef]
- Uzcategui, N.L.; Zhou, Y.; Figarella, K.; Ye, J.; Mukhopadhyay, R.; Bhattacharjee, H. Alteration in glycerol and metalloid permeability by a single mutation in the extracellular C-loop of Leishmania major aquaglyceroporin LmAQP1. Mol. Microbiol. 2008, 70, 1477–1486. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Mandal, G.; Atluri, V.S.; Figarella, K.; Uzcategui, N.L.; Zhou, Y.; Beitz, E.; Ajees, A.A.; Bhattacharjee, H. The role of alanine 163 in solute permeability of Leishmania major aquaglyceroporin LmAQP1. Mol. Biochem. Parasitol. 2011, 175, 83–90. [Google Scholar] [CrossRef]
- Mandal, G.; Sharma, M.; Kruse, M.; Sander-Juelch, C.; Munro, L.A.; Wang, Y.; Vilg, J.V.; Tamás, M.J.; Bhattacharjee, H.; Wiese, M.; et al. Modulation of Leishmania major aquaglyceroporin activity by a mitogen-activated protein kinase. Mol. Microbiol. 2012, 85, 1204–1218. [Google Scholar] [CrossRef]
- Malvy, D.; Chappuis, F. Sleeping sickness. Clin. Microbiol. Infect. 2011, 17, 986–995. [Google Scholar] [CrossRef] [Green Version]
- Neglected tropical diseases. Available online: http://www.who.int/neglected_diseases/diseases/en/ (accessed on 27 November 2013).
- Mandal, G.; Mukhopadhyay, R. The role of aquaporins in pathogenic protozoan parasites: putative target for chemotherapy. J. Med. Res. Sci. 2011, 1, 29–47. [Google Scholar]
- Namangala, B. Contribution of innate immune responses towards resistance to African trypanosome infections. Scand J. Immunol. 2012, 75, 5–15. [Google Scholar] [CrossRef]
- Nok, A.J. Arsenicals (melarsoprol), pentamidine and suramin in the treatment of human African trypanosomiasis. Parasitol. Res. 2003, 90, 71–79. [Google Scholar] [CrossRef]
- Baker, N.; de Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: the melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef]
- Burri, C. Chemotherapy against human African trypanosomiasis: Is there a road to success? Parasitology 2010, 137, 1987–1994. [Google Scholar] [CrossRef]
- Mäser, P.; Lüscher, A.; Kaminsky, R. Drug transport and drug resistance in African trypanosomes. Drug Resist. Updat. 2003, 6, 281–290. [Google Scholar] [CrossRef]
- Bassarak, B.; Uzcátegui, N.L.; Schönfeld, C.; Duszenko, M. Functional characterization of three aquaglyceroporins from Trypanosoma brucei in osmoregulation and glycerol transport. Cell. Physiol. Biochem. 2011, 27, 411–420. [Google Scholar] [CrossRef]
- Baker, N.; Glover, L.; Munday, J.C.; Aguinaga Andrés, D.; Barrett, M.P.; de Koning, H.P.; Horn, D. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl. Acad. Sci. USA 2012, 109, 10996–11001. [Google Scholar]
- Uzcátegui, N.L.; Figarella, K.; Bassarak, B.; Meza, N.W.; Mukhopadhyay, R.; Ramirez, J.L.; Duszenko, M. Trypanosoma brucei aquaglyceroporins facilitate the uptake of arsenite and antimonite in a pH dependent way. Cell. Physiol. Biochem. 2013, 32, 880–888. [Google Scholar] [CrossRef]
- Deisenhammer, F.; Bartos, A.; Egg, R.; Gilhus, N.E.; Giovannoni, G.; Rauer, S.; Sellebjerg, F. EFNS Task Force. Guidelines on routine cerebrospinal fluid analysis. Report from an EFNS task force. Eur. J. Neurol. 2006, 13, 913–922. [Google Scholar] [CrossRef]
- Schmidt, H.; Stuertz, K.; Chen, V.; Stringaris, A.K.; Brück, W.; Nau, R. Glycerol does not reduce neuronal damage in experimental Streptococcus pneumoniae meningitis in rabbits. Inflammopharmacology 1998, 6, 19–26. [Google Scholar] [CrossRef]
- Johanson, C.E.; Stopa, E.G.; McMillan, P.N. The blood-cerebrospinal fluid barrier: Structure and functional significance. Methods Mol. Biol. 2011, 686, 101–131. [Google Scholar] [CrossRef]
- Edwards, A.S.; Scott, J.D. A-kinase anchoring proteins: protein kinase A and beyond. Curr. Opin. Cell Biol. 2000, 12, 217–221. [Google Scholar] [CrossRef]
- Luu, D.T.; Maurel, C. Aquaporin trafficking in plant cells: an emerging membrane-protein model. Traffic 2013, 14, 629–635. [Google Scholar] [CrossRef]
- Moeller, H.B.; Olesen, E.T.; Fenton, R.A. Regulation of the water channel aquaporin-2 by posttranslational modification. Am. J. Physiol. Renal Physiol. 2011, 300, F1062–F1073. [Google Scholar] [CrossRef]
- Rump, K.; Brendt, P.; Frey, U.H.; Schäfer, S.T.; Siffert, W.; Peters, J.; Adamzik, M. Aquaporin 1 and 5 expression evoked by the β2 adrenoreceptor agonist terbutaline and lipopolysaccharide in mice and in the human monocytic cell line THP-1 is differentially regulated. Shock 2013, 40, 430–436. [Google Scholar] [CrossRef]
- Morand, S.; Renggli, C.K.; Roditi, I.; Vassella, E. MAP kinase kinase 1 (MKK1) is essential for transmission of Trypanosoma brucei by Glossina morsitans. Mol. Biochem. Parasitol. 2012, 186, 73–76. [Google Scholar] [CrossRef]
- Domenicali Pfister, D.; Burkard, G.; Morand, S.; Renggli, C.K.; Roditi, I.; Vassella, E. A Mitogen-activated protein kinase controls differentiation of bloodstream forms of Trypanosoma brucei. Eukaryot. Cell 2006, 5, 1126–1135. [Google Scholar] [CrossRef]
- Fenn, K.; Matthews, K.R. The cell biology of Trypanosoma brucei differentiation. Curr. Opin. Microbiol. 2007, 10, 539–546. [Google Scholar] [CrossRef]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar]
- Graf, F.E.; Ludin, P.; Wenzler, T.; Kaiser, M.; Brun, R.; Pyana, P.P.; Büscher, P.; de Koning, H.P.; Horn, D.; Mäser, P. Aquaporin 2 mutations in Trypanosoma brucei gambiense field isolates correlate with decreased susceptibility to pentamidine and melarsoprol. PLoS Negl. Trop. Dis. 2013, 7, e2475. [Google Scholar] [CrossRef]
- Uzcátegui, N.L.; Carmona-Gutiérrez, D.; Denninger, V.; Schoenfeld, C.; Lang, F.; Figarella, K.; Duszenko, M. Antiproliferative effect of dihydroxyacetone on Trypanosoma brucei bloodstream forms: Cell cycle progression, Subcellular alterations, and cell death. Antimicrob. Agents Chemother. 2007, 51, 3960–3968. [Google Scholar] [CrossRef]
- Uzcátegui, N.L.; Denninger, V.; Merkel, P.; Schoenfeld, C.; Figarella, K.; Duszenko, M. Dihydroxyacetone induced autophagy in African Trypanosomes. Autophagy 2007, 3, 626–629. [Google Scholar]
- Rassi, A., Jr.; Rassi, A.; de Rezende, J.M. American trypanosomiasis (chagas disease). Infect. Dis. Clin. North Am. 2012, 26, 275–291. [Google Scholar] [CrossRef]
- Kollien, A.H.; Schaub, G.A. The development of Trypanosoma cruzi in triatominae. Parasitol. Today 2000, 16, 381–387. [Google Scholar] [CrossRef]
- Control and prevention of Chagas disease in Europe. Available online: http://www.fac.org.ar/1/comites/chagas/Chagas_WHO_Technical%20Report_16_06_10.pdf (accessed on 27 November 2013).
- Araújo, A.; Jansen, A.M.; Reinhard, K.; Ferreira, L.F. Paleoparasitology of chagas disease-a review. Mem. Inst. Oswaldo. Cruz. 2009, 104 Suppl 1, S9–S16. [Google Scholar] [CrossRef]
- Chagas disease (American trypanosomiasis). Available online: http://www.who.int/mediacentre/factsheets/fs340/en/ (accessed on 27 November 2013).
- Lescure, F.X.; Le Loup, G.; Freilij, H.; Develoux, M.; Paris, L.; Brutus, L.; Pialoux, G. Chagas disease: Changes in knowledge and management. Lancet Infect Dis. 2010, 10, 556–570. [Google Scholar] [CrossRef]
- Congenital Transmission of Chagas Disease — Virginia, 2010. Available online: http://www.cdc.gov/chagas/epi.html (accessed on 27 November 2013).
- Wilkinson, S.R.; Taylor, M.C.; Horn, D.; Kelly, J.M.; Cheeseman, I. A mechanism for cross-resistance to nifurtimox and benznidazole in trypanosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 5022–5027. [Google Scholar]
- Portal, P.; Fernández Villamil, S.; Alonso, G.D.; De Vas, M.G.; Flawiá, M.M.; Torres, H.N.; Paveto, C. Multiple NADPH-cytochrome P450 reductases from Trypanosoma cruzi suggested role on drug resistance. Mol. Biochem. Parasitol. 2008, 160, 42–51. [Google Scholar] [CrossRef]
- Campos, M.C.; Castro-Pinto, D.B.; Ribeiro, G.A.; Berredo-Pinho, M.M.; Gomes, L.H.; da Silva Bellieny, M.S.; Goulart, C.M.; Echevarria, A.; Leon, L.L. P-glycoprotein efflux pump plays an important role in Trypanosoma cruzi drug resistance. Parasitol. Res. 2013, 112, 2341–2351. [Google Scholar] [CrossRef]
- Kollien, A.H.; Grospietsch, T.; Kleffmann, T.; Zerbst-Boroffka, I.; Schaub, G.A. Ionic composition of the rectal contents and excreta of the reduviid bug Triatoma infestans. J. Insect. Physiol. 2001, 47, 739–747. [Google Scholar] [CrossRef]
- Docampo, R.; Moreno, S.N. Acidocalcisomes. Cell Calcium 2011, 50, 113–119. [Google Scholar] [CrossRef]
- Miranda, K.; de Souza, W.; Plattner, H.; Hentschel, J.; Kawazoe, U.; Fang, J.; Moreno, S.N. Acidocalcisomes in Apicomplexan parasites. Exp. Parasitol. 2008, 118, 2–9. [Google Scholar] [CrossRef]
- Ruiz, F.A.; Marchesini, N.; Seufferheld, M.; Govindjee; Docampo, R. The polyphosphate bodies of Chlamydomonas reinhardtii possess a proton-pumping pyrophosphatase and are similar to acidocalcisomes. J. Biol. Chem. 2001, 276, 46196–46203. [Google Scholar]
- Marchesini, N.; Ruiz, F.A.; Vieira, M.; Docampo, R. Acidocalcisomes are functionally linked to the contractile vacuole of Dictyostelium discoideum. J. Biol. Chem. 2002, 277, 8146–8153. [Google Scholar] [CrossRef]
- Seufferheld, M.; Vieira, M.C.; Ruiz, F.A.; Rodrigues, C.O.; Moreno, S.N.; Docampo, R. Identification of organelles in bacteria similar to acidocalcisomes of unicellular eukaryotes. J. Biol. Chem. 2003, 278, 29971–29978. [Google Scholar]
- Docampo, R.; Ulrich, P.; Moreno, S.N. Evolution of acidocalcisomes and their role in polyphosphate storage and osmoregulation in eukaryotic microbes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 775–784. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mandal, G.; Orta, J.F.; Sharma, M.; Mukhopadhyay, R. Trypanosomatid Aquaporins: Roles in Physiology and Drug Response. Diseases 2014, 2, 3-23. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases2010003
Mandal G, Orta JF, Sharma M, Mukhopadhyay R. Trypanosomatid Aquaporins: Roles in Physiology and Drug Response. Diseases. 2014; 2(1):3-23. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases2010003
Chicago/Turabian StyleMandal, Goutam, Jose F. Orta, Mansi Sharma, and Rita Mukhopadhyay. 2014. "Trypanosomatid Aquaporins: Roles in Physiology and Drug Response" Diseases 2, no. 1: 3-23. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases2010003