Role of MTDH, FOXM1 and microRNAs in Drug Resistance in Hepatocellular Carcinoma

{kind=link}
{kind=link}
Abstract
:1. PI3K/Akt Pathway Is Involved in the Sorafenib Resistance
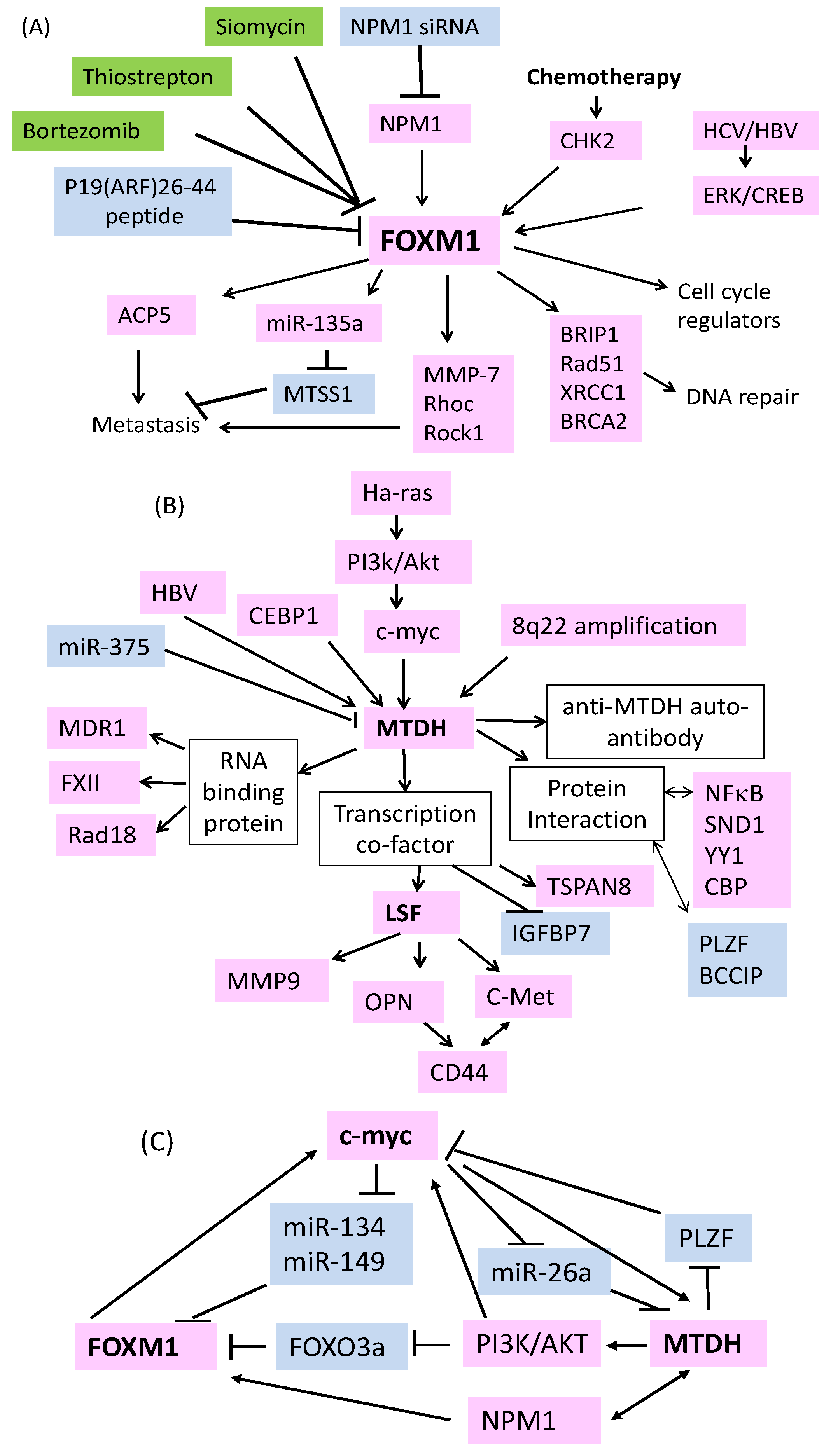
2. FOXM1 is Essential for the Development of HCC
3. FOXM1 Acts as a Therapeutic Target of HCC

4. Role of MTDH as an RNA Binding Protein in Drug Resistance
5. Mechanisms of MTDH Overexpression in HCC
6. Signal Transduction Pathways Activated by MTDH
7. MTDH Regulation of Downstream Genes in HCC
8. MicroRNAs in HCC: Drug Resistance and Beyond

9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meerzaman, D.M.; Yan, C.; Chen, Q.R.; Edmonson, M.N.; Schaefer, C.F.; Clifford, R.J.; Dunn, B.K.; Dong, L.; Finney, R.P.; Cultraro, C.M.; et al. Genome-wide transcriptional sequencing identifies novel mutations in metabolic genes in human hepatocellular carcinoma. Cancer Genomics Proteomics 2014, 11, 1–12. [Google Scholar]
- Qin, Y.; Lu, Y.; Wang, R.; Li, W.; Qu, X. SL1122-37, a novel derivative of sorafenib, has greater effects than sorafenib on the inhibition of human hepatocellular carcinoma (HCC) growth and prevention of angiogenesis. Biosci. Trends 2013, 7, 237–244. [Google Scholar]
- Scartozzi, M.; Faloppi, L.; Svegliati Baroni, G.; Loretelli, C.; Piscaglia, F.; Iavarone, M.; Toniutto, P.; Fava, G.; De Minicis, S.; Mandolesi, A.; et al. VEGF and VEGFR genotyping in the prediction of clinical outcome for HCC patients receiving sorafenib: The ALICE-1 study. Int. J. Cancer 2014, 125, 1247–1256. [Google Scholar]
- Greten, T.F.; Manns, M.P.; Malek, N. Sorafenib for the treatment of HCC--the beginning of a new era in the treatment of HCC. Z. Gastroenterol. 2009, 47, 55–60. [Google Scholar] [CrossRef]
- Wong, H.; Tang, Y.F.; Yao, T.J.; Chiu, J.; Leung, R.; Chan, P.; Cheung, T.T.; Chan, A.C.; Pang, R.W.; Poon, R.; et al. The outcomes and safety of single-agent sorafenib in the treatment of elderly patients with advanced hepatocellular carcinoma (HCC). Oncologist 2011, 16, 1721–1728. [Google Scholar] [CrossRef]
- Chow, A.K.; Ng, L.; Lam, C.S.; Wong, S.K.; Wan, T.M.; Cheng, N.S.; Yau, T.C.; Poon, R.T.; Pang, R.W. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS One 2013, 8, e78675. [Google Scholar]
- Gedaly, R.; Angulo, P.; Hundley, J.; Daily, M.F.; Chen, C.; Evers, B.M. PKI-587 and sorafenib targeting PI3K/AKT/mTOR and Ras/Raf/MAPK pathways synergistically inhibit HCC cell proliferation. J. Surg. Res. 2012, 176, 542–548. [Google Scholar] [CrossRef]
- Chen, K.F.; Chen, H.L.; Tai, W.T.; Feng, W.C.; Hsu, C.H.; Chen, P.J.; Cheng, A.L. Activation of phosphatidylinositol 3-kinase/Akt signaling pathway mediates acquired resistance to sorafenib in hepatocellular carcinoma cells. J. Pharmacol. Exp. Ther. 2011, 337, 155–161. [Google Scholar] [CrossRef]
- Wu, J.M.; Sheng, H.; Saxena, R.; Skill, N.J.; Bhat-Nakshatri, P.; Yu, M.; Nakshatri, H.; Maluccio, M.A. NF-kappaB inhibition in human hepatocellular carcinoma and its potential as adjunct to sorafenib based therapy. Cancer Lett. 2009, 278, 145–155. [Google Scholar] [CrossRef]
- Urbanik, T.; Kohler, B.C.; Boger, R.J.; Worns, M.A.; Heeger, S.; Otto, G.; Hovelmeyer, N.; Galle, P.R.; Schuchmann, M.; Waisman, A.; et al. Down- regulation of CYLD as a trigger for NF-kappaB activation and a mechanism of apoptotic resistance in hepatocellular carcinoma cells. Int. J. Oncol. 2011, 38, 121–131. [Google Scholar]
- Wang, W.H.; Chiang, I.T.; Liu, Y.C.; Hsu, F.T.; Chen, H.W.; Chen, C.L.; Lee, Y.J.; Lin, W.J.; Hwang, J.J. Simultaneous imaging of temporal changes of NF-kappaB activity and viable tumor cells in Huh7/NF-kappaB-tk-luc2/rfp tumor-bearing mice. In Vivo 2013, 27, 339–350. [Google Scholar]
- Zhai, B.; Sun, X.Y. Mechanisms of resistance to sorafenib and the corresponding strategies in hepatocellular carcinoma. World J. Hepatol. 2013, 5, 345–352. [Google Scholar]
- Wang, B.; Hikosaka, K.; Sultana, N.; Sharkar, M.T.; Noritake, H.; Kimura, W.; Wu, Y.X.; Kobayashi, Y.; Uezato, T.; Miura, N. Liver tumor formation by a mutant retinoblastoma protein in the transgenic mice is caused by an upregulation of c-Myc target genes. Biochem. Biophys. Res. Commun. 2012, 417, 601–606. [Google Scholar] [CrossRef]
- Xia, L.; Mo, P.; Huang, W.; Zhang, L.; Wang, Y.; Zhu, H.; Tian, D.; Liu, J.; Chen, Z.; Zhang, Y.; et al. The TNF-alpha/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis 2012, 33, 2250–2259. [Google Scholar] [CrossRef]
- Grant, G.D.; Brooks, L., 3rd; Zhang, X.; Mahoney, J.M.; Martyanov, V.; Wood, T.A.; Sherlock, G.; Cheng, C.; Whitfield, M.L. Identification of cell cycle-regulated genes periodically expressed in U2OS cells and their regulation by FOXM1 and E2F transcription factors. Mol. Biol. Cell 2013, 24, 3634–3650. [Google Scholar] [CrossRef]
- Barsotti, A.M.; Prives, C. Pro-proliferative FoxM1 is a target of p53-mediated repression. Oncogene 2009, 28, 4295–4305. [Google Scholar] [CrossRef]
- Pandit, B.; Halasi, M.; Gartel, A.L. p53 negatively regulates expression of FoxM1. Cell Cycle 2009, 8, 3425–3427. [Google Scholar] [CrossRef]
- Millour, J.; de Olano, N.; Horimoto, Y.; Monteiro, L.J.; Langer, J.K.; Aligue, R.; Hajji, N.; Lam, E.W. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011, 10, 1046–1058. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Luo, J.; Fu, Z.; Ying, J.; Yu, Y.; Yu, W. miR-134 inhibits epithelial to mesenchymal transition by targeting FOXM1 in non-small cell lung cancer cells. FEBS Lett. 2012, 586, 3761–3765. [Google Scholar] [CrossRef]
- Ke, Y.; Zhao, W.; Xiong, J.; Cao, R. miR-149 Inhibits Non-Small-Cell Lung Cancer Cells EMT by Targeting FOXM1. Biochem. Res. Int. 2013, 2013, 506731. [Google Scholar]
- Ying, H.; Lv, J.; Ying, T.; Jin, S.; Shao, J.; Wang, L.; Xu, H.; Yuan, B.; Yang, Q. Gene-gene interaction network analysis of ovarian cancer using TCGA data. J. Ovarian Res. 2013, 6, 88. [Google Scholar] [CrossRef]
- Parikh, N.; Hilsenbeck, S.; Creighton, C.J.; Dayaram, T.; Shuck, R.; Shinbrot, E.; Xi, L.; Gibbs, R.A.; Wheeler, D.A.; Donehower, L.A. Effects of TP53 mutational status on gene expression patterns across 10 human cancer types. J. Pathol. 2014, 232, 522–533. [Google Scholar] [CrossRef]
- Kalinichenko, V.V.; Major, M.L.; Wang, X.; Petrovic, V.; Kuechle, J.; Yoder, H.M.; Dennewitz, M.B.; Shin, B.; Datta, A.; Raychaudhuri, P.; et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004, 18, 830–850. [Google Scholar] [CrossRef]
- Park, H.J.; Gusarova, G.; Wang, Z.; Carr, J.R.; Li, J.; Kim, K.H.; Qiu, J.; Park, Y.D.; Williamson, P.R.; Hay, N.; et al. Deregulation of FoxM1b leads to tumour metastasis. EMBO Mol. Med. 2011, 3, 21–34. [Google Scholar] [CrossRef]
- Xia, L.; Huang, W.; Tian, D.; Zhu, H.; Zhang, Y.; Hu, H.; Fan, D.; Nie, Y.; Wu, K. Upregulated FoxM1 expression induced by hepatitis B virus X protein promotes tumor metastasis and indicates poor prognosis in hepatitis B virus-related hepatocellular carcinoma. J. Hepatol. 2012, 57, 600–612. [Google Scholar] [CrossRef]
- Xia, L.; Huang, W.; Tian, D.; Chen, Z.; Zhang, L.; Li, Y.; Hu, H.; Liu, J.; Chen, Z.; Tang, G.; et al. ACP5, a direct transcriptional target of FoxM1, promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Oncogene 2014, 33, 1395–1406. [Google Scholar] [CrossRef]
- Liu, S.; Guo, W.; Shi, J.; Li, N.; Yu, X.; Xue, J.; Fu, X.; Chu, K.; Lu, C.; Zhao, J.; et al. MicroRNA-135a contributes to the development of portal vein tumor thrombus by promoting metastasis in hepatocellular carcinoma. J. Hepatol. 2012, 56, 389–396. [Google Scholar]
- Bhat, U.G.; Zipfel, P.A.; Tyler, D.S.; Gartel, A.L. Novel anticancer compounds induce apoptosis in melanoma cells. Cell Cycle 2008, 7, 1851–1855. [Google Scholar] [CrossRef]
- Costa, R.H.; Kalinichenko, V.V.; Major, M.L.; Raychaudhuri, P. New and unexpected: Forkhead meets ARF. Curr. Opin. Genet. Dev. 2005, 15, 42–48. [Google Scholar] [CrossRef]
- Gusarova, G.A.; Wang, I.C.; Major, M.L.; Kalinichenko, V.V.; Ackerson, T.; Petrovic, V.; Costa, R.H. A cell-penetrating ARF peptide inhibitor of FoxM1 in mouse hepatocellular carcinoma treatment. J. Clin. Invest. 2007, 117, 99–111. [Google Scholar] [CrossRef]
- Bhat, U.G.; Jagadeeswaran, R.; Halasi, M.; Gartel, A.L. Nucleophosmin interacts with FOXM1 and modulates the level and localization of FOXM1 in human cancer cells. J. Biol. Chem. 2011, 286, 41425–41433. [Google Scholar]
- Park, Y.Y.; Jung, S.Y.; Jennings, N.B.; Rodriguez-Aguayo, C.; Peng, G.; Lee, S.R.; Kim, S.B.; Kim, K.; Leem, S.H.; Lin, S.Y.; et al. FOXM1 mediates Dox resistance in breast cancer by enhancing DNA repair. Carcinogenesis 2012, 33, 1843–1853. [Google Scholar] [CrossRef]
- Zhang, N.; Wu, X.; Yang, L.; Xiao, F.; Zhang, H.; Zhou, A.; Huang, Z.; Huang, S. FoxM1 inhibition sensitizes resistant glioblastoma cells to temozolomide by downregulating the expression of DNA-repair gene Rad51. Clin. Cancer Res. 2012, 18, 5961–5971. [Google Scholar] [CrossRef]
- Khongkow, P.; Karunarathna, U.; Khongkow, M.; Gong, C.; Gomes, A.R.; Yague, E.; Monteiro, L.J.; Kongsema, M.; Zona, S.; Man, E.P.; et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 2013. [Google Scholar] [CrossRef]
- Monteiro, L.J.; Khongkow, P.; Kongsema, M.; Morris, J.R.; Man, C.; Weekes, D.; Koo, C.Y.; Gomes, A.R.; Pinto, P.H.; Varghese, V.; et al. The Forkhead Box M1 protein regulates BRIP1 expression and DNA damage repair in epirubicin treatment. Oncogene 2013, 32, 4634–4645. [Google Scholar] [CrossRef]
- Srivastava, J.; Siddiq, A.; Emdad, L.; Santhekadur, P.K.; Chen, D.; Gredler, R.; Shen, X.N.; Robertson, C.L.; Dumur, C.I.; Hylemon, P.B.; et al. Astrocyte elevated gene-1 promotes hepatocarcinogenesis: novel insights from a mouse model. Hepatology 2012, 56, 1782–1791. [Google Scholar] [CrossRef]
- Sarkar, D. AEG-1/MTDH/LYRIC in liver cancer. Adv. Cancer Res. 2013, 120, 193–221. [Google Scholar] [CrossRef]
- Meng, X.; Zhu, D.; Yang, S.; Wang, X.; Xiong, Z.; Zhang, Y.; Brachova, P.; Leslie, K.K. Cytoplasmic Metadherin (MTDH) provides survival advantage under conditions of stress by acting as RNA-binding protein. J. Biol. Chem. 2012, 287, 4485–4491. [Google Scholar]
- Yoo, B.K.; Chen, D.; Su, Z.Z.; Gredler, R.; Yoo, J.; Shah, K.; Fisher, P.B.; Sarkar, D. Molecular mechanism of chemoresistance by astrocyte elevated gene-1. Cancer Res. 2010, 70, 3249–3258. [Google Scholar] [CrossRef]
- Meng, X.; Thiel, K.W.; Leslie, K.K. Drug resistance mediated by AEG- 1/MTDH/LYRIC. Adv. Cancer Res. 2013, 120, 135–157. [Google Scholar] [CrossRef]
- Yoo, B.K.; Emdad, L.; Su, Z.Z.; Villanueva, A.; Chiang, D.Y.; Mukhopadhyay, N.D.; Mills, A.S.; Waxman, S.; Fisher, R.A.; Llovet, J.M.; et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J. Clin. Invest. 2009, 119, 465–477. [Google Scholar] [CrossRef]
- Wang, K.; Lim, H.Y.; Shi, S.; Lee, J.; Deng, S.; Xie, T.; Zhu, Z.; Wang, Y.; Pocalyko, D.; Yang, W.J.; et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology 2013, 58, 706–717. [Google Scholar]
- Ahn, S.; Hyeon, J.; Park, C.K. Metadherin is a prognostic predictor of hepatocellular carcinoma after curative hepatectomy. Gut Liver 2013, 7, 206–212. [Google Scholar] [CrossRef]
- Lee, S.G.; Su, Z.Z.; Emdad, L.; Sarkar, D.; Fisher, P.B. Astrocyte elevated gene-1 (AEG-1) is a target gene of oncogenic Ha-ras requiring phosphatidylinositol 3-kinase and c-Myc. Proc. Natl. Acad. Sci. USA 2006, 103, 17390–17395. [Google Scholar]
- Thirkettle, H.J.; Mills, I.G.; Whitaker, H.C.; Neal, D.E. Nuclear LYRIC/AEG-1 interacts with PLZF and relieves PLZF-mediated repression. Oncogene 2009, 28, 3663–3670. [Google Scholar] [CrossRef]
- He, X.X.; Chang, Y.; Meng, F.Y.; Wang, M.Y.; Xie, Q.H.; Tang, F.; Li, P.Y.; Song, Y.H.; Lin, J.S. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene 2012, 31, 3357–3369. [Google Scholar] [CrossRef]
- Zheng, J.; Li, C.; Wu, X.; Yang, Y.; Hao, M.; Sheng, S.; Sun, Y.; Zhang, H.; Long, J.; Hu, C. Astrocyte elevated gene-1 is a novel biomarker of epithelial-mesenchymal transition and progression of hepatocellular carcinoma in two China regions. Tumour Biol. 2014, 35, 2265–2269. [Google Scholar] [CrossRef]
- Zhu, K.; Dai, Z.; Pan, Q.; Wang, Z.; Yang, G.H.; Yu, L.; Ding, Z.B.; Shi, G.M.; Ke, A.W.; Yang, X.R.; et al. Metadherin promotes hepatocellular carcinoma metastasis through induction of epithelial- mesenchymal transition. Clin. Cancer Res. 2011, 17, 7294–7302. [Google Scholar] [CrossRef]
- Sarkar, D.; Fisher, P.B. Advances in Cancer Research. AEG-1/MTDH/LYRIC Implicated in Multiple Human Cancers; Academic Press (ELSEVIER): San Diego, USA, 2013. [Google Scholar]
- Sarkar, D.; Fisher, P.B. AEG-1/MTDH/LYRIC: Clinical significance. Adv. Cancer Res. 2013, 120, 39–74. [Google Scholar] [CrossRef]
- Gong, Z.; Liu, W.; You, N.; Wang, T.; Wang, X.; Lu, P.; Zhao, G.; Yang, P.; Wang, D.; Dou, K. Prognostic significance of metadherin overexpression in hepatitis B virus- related hepatocellular carcinoma. Oncol. Rep. 2012, 27, 2073–2079. [Google Scholar]
- Robertson, C.L.; Srivastava, J.; Siddiq, A.; Gredler, A.; Rajasekaran, D.; Akiel, M.; Shen, X.N.; Arkun, K.; Ghosh, S.; Mark, M.A.; et al. Analyzing the role of Astrocyte Elevated Gene-1 (AEG-1) in hepatocarcinogenesis using a knockout mouse model. In Presented at the 2014 Annual of the American Association of cancer Research (AACR), San Diego, CA, USA, 5–9 April 2014.
- Chen, X.; Dong, K.; Long, M.; Lin, F.; Wang, X.; Wei, J.; Ren, J.; Zhang, H. Serum anti-AEG-1 auto-antibody is a potential novel biomarker for malignant tumors. Oncol. Lett. 2012, 4, 319–323. [Google Scholar]
- Medine, E.I.; Odaci, D.; Gacal, B.N.; Gacal, B.; Sakarya, S.; Unak, P.; Timur, S.; Yagci, Y. A new approach for in vitro imaging of breast cancer cells by anti-metadherin targeted PVA-pyrene. Macromol. Biosci. 2010, 10, 657–663. [Google Scholar] [CrossRef]
- Emdad, L.; Das, S.K.; Dasgupta, S.; Hu, B.; Sarkar, D.; Fisher, P.B. AEG-1/MTDH/LYRIC: Signaling pathways, downstream genes, interacting proteins, and regulation of tumor angiogenesis. Adv. Cancer Res. 2013, 120, 75–111. [Google Scholar] [CrossRef]
- Sarkar, D.; Park, E.S.; Emdad, L.; Lee, S.G.; Su, Z.Z.; Fisher, P.B. Molecular basis of nuclear factor-kappaB activation by astrocyte elevated gene-1. Cancer Res. 2008, 68, 1478–1484. [Google Scholar] [CrossRef]
- Yoo, B.K.; Emdad, L.; Gredler, R.; Fuller, C.; Dumur, C.I.; Jones, K.H.; Jackson-Cook, C.; Su, Z.Z.; Chen, D.; Saxena, U.H.; et al. Transcription factor Late SV40 Factor (LSF) functions as an oncogene in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2010, 107, 8357–8362. [Google Scholar] [CrossRef]
- Yoo, B.K.; Gredler, R.; Chen, D.; Santhekadur, P.K.; Fisher, P.B.; Sarkar, D. c-Met activation through a novel pathway involving osteopontin mediates oncogenesis by the transcription factor LSF. J. Hepatol. 2011, 55, 1317–1324. [Google Scholar]
- Santhekadur, P.K.; Rajasekaran, D.; Siddiq, A.; Gredler, R.; Chen, D.; Schaus, S.E.; Hansen, U.; Fisher, P.B.; Sarkar, D. The transcription factor LSF: A novel oncogene for hepatocellular carcinoma. Am. J. Cancer Res. 2012, 2, 269–285. [Google Scholar]
- Chen, D.; Yoo, B.K.; Santhekadur, P.K.; Gredler, R.; Bhutia, S.K.; Das, S.K.; Fuller, C.; Su, Z.Z.; Fisher, P.B.; Sarkar, D. Insulin-like growth factor-binding protein-7 functions as a potential tumor suppressor in hepatocellular carcinoma. Clin. Cancer Res. 2011, 17, 6693–6701. [Google Scholar] [CrossRef]
- Hauptman, N.; Glavac, D. MicroRNAs and long non-coding RNAs: Prospects in diagnostics and therapy of cancer. Radiol. Oncol. 2013, 47, 311–318. [Google Scholar]
- Xu, Y.; Xia, F.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, J.; Cui, Y.; Bian, X.; Bie, P.; et al. MicroRNA-122 sensitizes HCC cancer cells to adriamycin and vincristine through modulating expression of MDR and inducing cell cycle arrest. Cancer Lett. 2013, 310, 160–169. [Google Scholar]
- Zhou, L.; Liu, J.; Wang, B.; Gao, M.; Huang, A. Differential miRNA expression profiles in hepatocellular cells and drug-resistant sublines. Oncol. Rep. 2012, 29, 555–562. [Google Scholar]
- Buendia, M.A.; Bourre, L.; Cairo, S. Myc target miRs and liver cancer: Small molecules to get Myc sick. Gastroenterology 2012, 142, 214–218. [Google Scholar] [CrossRef]
- Hou, J.; Lin, L.; Zhou, W.; Wang, Z.; Ding, G.; Dong, Q.; Qin, L.; Wu, X.; Zheng, Y.; Yang, Y.; et al. Identification of miRNomes in human liver and hepatocellular carcinoma reveals miR-199a/b-3p as therapeutic target for hepatocellular carcinoma. Cancer Cell 2011, 19, 232–243. [Google Scholar] [CrossRef]
- Filipowicz, W.; Grosshans, H. The liver-specific microRNA miR-122: Biology and therapeutic potential. Prog. Drug Res. 2011, 67, 221–238. [Google Scholar]
- Giordano, S.; Columbano, A. MicroRNAs: New tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology 2013, 57, 840–847. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar]
- Devor, E.J.; Schickling, B.M.; Leslie, K.K. microRNA expression patterns across seven cancers are highly correlated and dominated by evolutionarily ancient families. Biomedical. Rep. 2014, 2, 384–387. [Google Scholar]
- Spearman, C. The proof and measurement of association between two things. Am. J. Psychol. 1904, 15, 72–101. [Google Scholar] [CrossRef]
- Budhu, A.; Jia, H.L.; Forgues, M.; Liu, C.G.; Goldstein, D.; Lam, A.; Zanetti, K.A.; Ye, Q.-H.; Qin, L.-X.; Croce, C.M.; et al. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology 2008, 47, 897–907. [Google Scholar] [CrossRef]
- Kamarswamy, R.; Volkmann, I.; Thum, T. Regulation and function of miR-21 in health and disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting E-cadherin repressors ZEB1 and ZEB2. Genes Develop. 2008, 22, 894–907. [Google Scholar] [CrossRef]
- Takahashi, K.; Yan, I.; Wen, H.-J.; Patel, T. microRNAs in liver disease: From diagnostics to therapeutics. Clin. Biochem. 2013, 46, 946–952. [Google Scholar] [CrossRef]
- Yin, H.; Hu, M.; Zhang, R.; Shen, Z.; Flatow, L.; You, M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J. Biol. Chem. 2012, 287, 9817–9826. [Google Scholar] [CrossRef]
- Meng, Z.; Fu, X.; Chen, X.; Zeng, S.; Tian, Y.; Jove, R.; Xu, R.; Huang, W. miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology 2010, 52, 2148–2157. [Google Scholar] [CrossRef]
- Afanasyeva, E.A.; Mestdagh, P.; Kumps, C.; Vandesompele, J.; Ehemann, V.; Theissen, J.; Fischer, M.; Zapatka, M.; Brors, B.; Savelyeva, L.; et al. MicroRNA miR-885-5p targets CDK2 and MCM5, activates p53 and inhibits proliferation and survival. Cell Death Differ. 2011, 18, 974–984. [Google Scholar] [CrossRef]
- Williams, J.H.; Phillips, T.D.; Jolly, S.E.; Williams, J.H.; Phillips, T.D.; Jolly, P.E.; Stiles, J.K.; Jolly, C.M.; Aggarwal, D. Human aflotoxicosis in developing countries: A review of toxicology, exposure, potential health consequences and interventions. Am. J. Clin. Nutr. 2004, 80, 1106–1122. [Google Scholar]
- Gui, J.; Tian, Y.; Wen, X.; Zhang, W.; Zhang, P.; Gao, J.; Run, W.; Tian, L.; Jia, X.; Gao, Y. Serum microRNA characterization identifies miR-885-5p as a potential marker for detecting liver pathologies. Clin. Sci. 2011, 120, 183–193. [Google Scholar] [CrossRef]
- Wilson, M.S.; Brosens, J.J.; Schwenen, H.D.; Lam, E.W. FOXO and FOXM1 in cancer: The FOXO-FOXM1 axis shapes the outcome of cancer chemotherapy. Curr. Drug Targets 2011, 12, 1256–1266. [Google Scholar] [CrossRef]
- Zhao, F.; Lam, E.W. Role of the forkhead transcription factor FOXO-FOXM1axis in cancer and drug resistance. Front. Med. 2012, 6, 376–380. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, X.X.; He, J.R.; Zhou, C.X.; Guo, M.; He, M.; Li, M.F.; Chen, G.Q.; Zhao, Q. Pathologically decreased miR-26a antagonizes apoptosis and facilitates carcinogenesis by targeting MTDH and EZH2 in breast cancer. Carcinogenesis 2011, 32, 2–9. [Google Scholar] [CrossRef]
- Wierstra, I.; Alves, J. FOXM1c transactivates the human c-myc promoter directly via the two TATA boxes P1 and P2. FEBS J. 2006, 273, 4645–4667. [Google Scholar] [CrossRef]
- Tan, Y.; Raychaudhuri, P.; Costa, R.H. Chk2 mediates stabilization of the FoxM1 transcription factor to stimulate expression of DNA repair genes. Mol. Cell Biol. 2007, 27, 1007–1016. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Meng, X.; Devor, E.J.; Yang, S.; Schickling, B.M.; Leslie, K.K. Role of MTDH, FOXM1 and microRNAs in Drug Resistance in Hepatocellular Carcinoma. Diseases 2014, 2, 209-225. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases2030209
Meng X, Devor EJ, Yang S, Schickling BM, Leslie KK. Role of MTDH, FOXM1 and microRNAs in Drug Resistance in Hepatocellular Carcinoma. Diseases. 2014; 2(3):209-225. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases2030209
Chicago/Turabian StyleMeng, Xiangbing, Eric J. Devor, Shujie Yang, Brandon M. Schickling, and Kimberly K. Leslie. 2014. "Role of MTDH, FOXM1 and microRNAs in Drug Resistance in Hepatocellular Carcinoma" Diseases 2, no. 3: 209-225. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases2030209