
Zebrafish Models of Prader-Willi Syndrome: Fast Track to Pharmacotherapeutics

Abstract
:
1. Introduction
2. Discussion
2.1. Genetics
2.2. PWS-Like Syndromes: 6q16 Deletions
2.3. Non Imprinted PWS Genes
2.4. Mouse Models
2.5. The Zebrafish Model: Conserved Neuroanatomy and Physiology
2.6. Technologies for Phenotypic Analysis: PWS Endophenotypes
2.7. Strategies for Targeted Gene Manipulation
2.8. Approaches for Gene Discovery in Zebrafish
2.9. Pharmacogenomic Methods in Zebrafish
2.10. Fast Track to PWS Pharmacotheraputics
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Invest. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Emerick, J.E.; Vogt, K.S. Endocrine manifestations and management of Prader-Willi syndrome. Int. J. Pediatr. Endocrinol. 2013, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, F.; Hirsch, H.J.; Genstil, L.; Landau, Y.E.; Gross-Tsur, V. Prader-Willi syndrome: Medical prevention and behavioral challenges. Child. Adolesc. Psychiatr. Clin. N. Am. 2007, 16, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Bridges, N. What is the value of growth hormone therapy in Prader willi syndrome? Arch. Dis. Child. 2014, 99, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, A.P. Prader-Willi syndrome: Advances in genetics, pathophysiology and treatment. Trends Endocrinol. MeTable 2004, 15, 12–20. [Google Scholar] [CrossRef]
- Nixon, G.M.; Brouillette, R.T. Sleep and breathing in Prader-Willi syndrome. Pediatr. Pulmonol. 2002, 34, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Lohr, H.; Hammerschmidt, M. Zebrafish in endocrine systems: Recent advances and implications for human disease. Annu. Rev. Physiol. 2011, 73, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Pickart, M.A.; Klee, E.W. Zebrafish approaches enhance the translational research tackle box. Transl. Res. 2014, 163, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Yang, J.H.; Shi, Q.S.; Zheng, L.L.; Liu, J.; Zhou, H.; Zhang, H.; Qu, L.H. Rapid birth-and-death evolution of imprinted snornas in the Prader-Willi syndrome locus: Implications for neural development in euarchontoglires. PLoS ONE 2014, 9, e100329. [Google Scholar] [CrossRef] [PubMed]
- Dunzinger, U.; Haaf, T.; Zechner, U. Conserved synteny of mammalian imprinted genes in chicken, frog, and fish genomes. Cytogenet. Genome Res. 2007, 117, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Bohne, A.; Darras, A.; D'Cotta, H.; Baroiller, J.F.; Galiana-Arnoux, D.; Volff, J.N. The vertebrate makorin ubiquitin ligase gene family has been shaped by large-scale duplication and retroposition from an ancestral gonad-specific, maternal-effect gene. BMC Genomics 2010, 11, 721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.M.; Conery, J.S.; Postlethwait, J.H. Automated identification of conserved synteny after whole-genome duplication. Genome Res. 2009, 19, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.M.; Ekker, M.; Wevrick, R. A MAGE/NDN-like gene in zebrafish. Dev. Dyn. 2003, 228, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Bervini, S.; Herzog, H. Mouse models of Prader-Willi syndrome: A systematic review. Front. Neuroendocrinol. 2013, 34, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Neumann, L.C.; Feiner, N.; Meyer, A.; Buiting, K.; Horsthemke, B. The imprinted npap1 gene in the Prader-Willi syndrome region belongs to a POM121-related family of retrogenes. Genome Biol. Evol. 2014, 6, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Cavaille, J.; Buiting, K.; Kiefmann, M.; Lalande, M.; Brannan, C.I.; Horsthemke, B.; Bachellerie, J.P.; Brosius, J.; Huttenhofer, A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl. Acad. Sci. USA 2000, 97, 14311–14316. [Google Scholar] [CrossRef] [PubMed]
- De los Santos, T.; Schweizer, J.; Rees, C.A.; Francke, U. Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region, which is highly expressed in brain. Am. J. Hum. Genet. 2000, 67, 1067–1082. [Google Scholar] [CrossRef] [PubMed]
- El Khattabi, L.; Guimiot, F.; Pipiras, E.; Andrieux, J.; Baumann, C.; Bouquillon, S.; Delezoide, A.L.; Delobel, B.; Demurger, F.; Dessuant, H.; et al. Incomplete penetrance and phenotypic variability of 6q16 deletions including SIM1. Eur. J. Hum. Genet. 2015, 23, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Raimondo, A.; Stutzmann, F.; Ghoussaini, M.; Ramachandrappa, S.; Bersten, D.C.; Durand, E.; Vatin, V.; Balkau, B.; Lantieri, O.; et al. Loss-of-function mutations in SIM1 contribute to obesity and Prader-Willi-like features. J. Clin. Invest. 2013, 123, 3037–3041. [Google Scholar] [CrossRef] [PubMed]
- Izumi, K.; Housam, R.; Kapadia, C.; Stallings, V.A.; Medne, L.; Shaikh, T.H.; Kublaoui, B.M.; Zackai, E.H.; Grimberg, A. Endocrine phenotype of 6q16.1–q21 deletion involving SIM1 and Prader-Willi syndrome-like features. Am. J. Med. Genet. A. Part. A 2013, 161A, 3137–3143. [Google Scholar] [CrossRef] [PubMed]
- Tolson, K.P.; Gemelli, T.; Gautron, L.; Elmquist, J.K.; Zinn, A.R.; Kublaoui, B.M. Postnatal Sim1 deficiency causes hyperphagic obesity and reduced Mc4r and Oxytocin expression. J. Neurosci. 2010, 30, 3803–3812. [Google Scholar] [CrossRef] [PubMed]
- Tolson, K.P.; Gemelli, T.; Meyer, D.; Yazdani, U.; Kozlitina, J.; Zinn, A.R. Inducible neuronal inactivation of SIM1 in adult mice causes hyperphagic obesity. Endocrinology 2014, 155, 2436–2444. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Avena, N.M.; Baier, L.; Brantley, P.; Bray, G.A.; Burnett, L.C.; Butler, M.G.; Driscoll, D.J.; Egli, D.; Elmquist, J.; et al. Hyperphagia: Current concepts and future directions proceedings of the 2nd international conference on hyperphagia. Obesity 2014, 22 (Suppl. S1), S1–S17. [Google Scholar] [CrossRef] [PubMed]
- Blevins, J.E.; Ho, J.M. Role of oxytocin signaling in the regulation of body weight. Rev. Endocr. Metab. Disord. 2013, 14, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Kasher, P.R.; Schertz, K.E.; Thomas, M.; Jackson, A.; Annunziata, S.; Ballesta-Martinez, M.J.; Campeau, P.M.; Clayton, P.E.; Eaton, J.L.; Granata, T.; et al. Small 6q16.1 deletions encompassing POU3F2 cause susceptibility to obesity and variable developmental delay with intellectual disability. Am. J. Hum. Genet. 2016, 98, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.L.; Glasgow, E. The zebrafish bhlh pas transcriptional regulator, single-minded 1 (SIM1), is required for isotocin cell development. Dev. Dyn. 2006, 235, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Unger, J.L.; Glasgow, E. Expression of isotocin-neurophysin mrna in developing zebratish. Gene Expr. Patterns 2003, 3, 105–108. [Google Scholar] [CrossRef]
- Eaton, J.L.; Holmqvist, B.; Glasgow, E. Ontogeny of vasotocin-expressing cells in zebrafish: Selective requirement for the transcriptional regulators orthopedia and single-minded 1 in the preoptic area. Dev. Dyn. 2008, 237, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Lohr, H.; Ryu, S.; Driever, W. Zebrafish diencephalic A11-related dopaminergic neurons share a conserved transcriptional network with neuroendocrine cell lineages. Development 2009, 136, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.L.; DeRossi, C.; May, N.R.; Holdener, B.C.; Fan, C.-M. ARNT2 acts as the dimerization partner of SIM1 for the development of the hypothalamus. Mech. Dev. 2000, 90, 253–261. [Google Scholar] [CrossRef]
- Michaud, J.L.; Rosenquist, T.; May, N.R.; Fan, C.M. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor SIM1. Genes Dev. 1998, 12, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Nakai, S.; Kawano, H.; Yudate, T.; Nishi, M.; Kuno, J.; Nagata, A.; Jishage, K.; Hamada, H.; Fujii, H.; Kawamura, K. The POU domain transcription factor Brn-2 is required for the determination of specific neuronal lineages in the hypothalamus of the mouse. Genes Dev. 1995, 9, 3109–3121. [Google Scholar] [CrossRef] [PubMed]
- Schonemann, M.D.; Ryan, A.K.; McEvilly, R.J.; O'Connell, S.M.; Arias, C.A.; Kalla, K.A.; Li, P.; Sawchenko, P.E.; Rosenfeld, M.G. Development and survival of the endocrine hypothalamus and posterior pituitary gland requires the neuronal POU domain factor Brn-2. Genes Dev. 1995, 9, 3122–3135. [Google Scholar] [CrossRef] [PubMed]
- Cattanach, B.M.; Barr, J.A.; Evans, E.P.; Burtenshaw, M.; Beechey, C.V.; Leff, S.E.; Brannan, C.I.; Copeland, N.G.; Jenkins, N.A.; Jones, J. A candidate mouse model for Prader-Willi syndrome which shows an absence of snrpn expression. Nat. Genet. 1992, 2, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, J.M.; Merchant, M.; Ohta, T.; Ji, Y.; Caldwell, R.G.; Ramsey, M.J.; Tucker, J.D.; Longnecker, R.; Nicholls, R.D. A transgene insertion creating a heritable chromosome deletion mouse model of Prader-Willi and angelman syndromes. Proc. Natl. Acad. Sci. USA 1999, 96, 9258–9263. [Google Scholar] [CrossRef] [PubMed]
- Dubose, A.J.; Smith, E.Y.; Yang, T.P.; Johnstone, K.A.; Resnick, J.L. A new deletion refines the boundaries of the murine Prader-Willi syndrome imprinting center. Hum. Mol. Gen. 2011, 20, 3461–3466. [Google Scholar] [CrossRef] [PubMed]
- Bressler, J.; Tsai, T.F.; Wu, M.Y.; Tsai, S.F.; Ramirez, M.A.; Armstrong, D.; Beaudet, A.L. The snrpn promoter is not required for genomic imprinting of the Prader-Willi/angelman domain in mice. Nat. Genet. 2001, 28, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.F.; Jiang, Y.H.; Bressler, J.; Armstrong, D.; Beaudet, A.L. Paternal deletion from Snrpn to Ube3a in the mouse causes hypotonia, growth retardation and partial lethality and provides evidence for a gene contributing to Prader-Willi syndrome. Hum. Mol. Gen. 1999, 8, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Adamson, T.E.; Resnick, J.L.; Leff, S.; Wevrick, R.; Francke, U.; Jenkins, N.A.; Copeland, N.G.; Brannan, C.I. A mouse model for Prader-Willi syndrome imprinting-centre mutations. Nat. Genet. 1998, 19, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Bogenpohl, J.W.; Howell, M.P.; Wevrick, R.; Panda, S.; Hogenesch, J.B.; Muglia, L.J.; van Gelder, R.N.; Herzog, E.D.; Stewart, C.L. The imprinted gene Magel2 regulates normal circadian output. Nat. Genet. 2007, 39, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Muscatelli, F.; Abrous, D.N.; Massacrier, A.; Boccaccio, I.; le Moal, M.; Cau, P.; Cremer, H. Disruption of the mouse necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Gen. 2000, 9, 3101–3110. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.E.; Kwolek, E.M.; Bischof, J.M.; van Eede, M.; Henkelman, R.M.; Wevrick, R. Regionally reduced brain volume, altered serotonin neurochemistry, and abnormal behavior in mice null for the circadian rhythm output gene Magel2. Am. J. Med. Genet. B 2009, 150B, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Li, H.H.; Zhang, S.; Solomon, N.M.; Camper, S.A.; Cohen, P.; Francke, U. SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice. PLoS ONE 2008, 3, e1709. [Google Scholar] [CrossRef] [PubMed]
- Bischof, J.M.; Stewart, C.L.; Wevrick, R. Inactivation of the mouse Magel2 gene results in growth abnormalities similar to Prader-Willi syndrome. Hum. Mol. Gen. 2007, 16, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Aycan, Z.; Bas, V.N. Prader-Willi syndrome and growth hormone deficiency. J. Clin. Res. Pediatr. Endocrinol. 2014, 6, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Biran, J.; Tahor, M.; Wircer, E.; Levkowitz, G. Role of developmental factors in hypothalamic function. Front. Neuroanat. 2015, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Markakis, E.A. Development of the neuroendocrine hypothalamus. Front. Neuroendocrinol. 2002, 23, 257–291. [Google Scholar] [CrossRef]
- Shubin, N. Your Inner Fish : A Journey into the 3.5-Billion-Year History of the Human Body, 1st ed.; Vintage Books: New York, NY, USA, 2009; p. 237. [Google Scholar]
- Linden, D.J. The Accidental Mind; Belknap Press of Harvard University Press: Cambridge, MA, USA, 2007; p. 276. [Google Scholar]
- Stewart, A.M.; Kalueff, A.V. Developing better and more valid animal models of brain disorders. Behav. Brain Res. 2015, 276, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Herget, U.; Ryu, S. Coexpression analysis of nine neuropeptides in the neurosecretory preoptic area of larval zebrafish. Front. Neuroanat. 2015, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Herget, U.; Wolf, A.; Wullimann, M.F.; Ryu, S. Molecular neuroanatomy and chemoarchitecture of the neurosecretory preoptic-hypothalamic area in zebrafish larvae. J. Comp. Neurol. 2014, 522, 1542–1564. [Google Scholar] [CrossRef] [PubMed]
- Woods, I.G.; Schoppik, D.; Shi, V.J.; Zimmerman, S.; Coleman, H.A.; Greenwood, J.; Soucy, E.R.; Schier, A.F. Neuropeptidergic signaling partitions arousal behaviors in zebrafish. J. Neurosci. 2014, 34, 3142–3160. [Google Scholar] [CrossRef] [PubMed]
- Bruni, G.; Lakhani, P.; Kokel, D. Discovering novel neuroactive drugs through high-throughput behavior-based chemical screening in the zebrafish. Front. Pharmacol. 2014, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Clift, D.; Richendrfer, H.; Thorn, R.J.; Colwill, R.M.; Creton, R. High-throughput analysis of behavior in zebrafish larvae: Effects of feeding. Zebrafish 2014, 11, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cousin, M.A.; Ebbert, J.O.; Wiinamaki, A.R.; Urban, M.D.; Argue, D.P.; Ekker, S.C.; Klee, E.W. Larval zebrafish model for FDA-approved drug repositioning for tobacco dependence treatment. PLoS ONE 2014, 9, e90467. [Google Scholar] [CrossRef] [PubMed]
- Cross, L.M.; Cook, M.A.; Lin, S.; Chen, J.N.; Rubinstein, A.L. Rapid analysis of angiogenesis drugs in a live fluorescent zebrafish assay. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; McCartin, K.; Liu, T.C.; Peterson, R.T.; Evans, T. A forward chemical screen in zebrafish identifies a retinoic acid derivative with receptor specificity. PLoS ONE 2010, 5, e10004. [Google Scholar] [CrossRef] [PubMed]
- Haesemeyer, M.; Schier, A.F. The study of psychiatric disease genes and drugs in zebrafish. Curr. Opin. Neurobiol. 2015, 30, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, C.K.; White, R.M.; Zon, L. Chemical genetic screening in the zebrafish embryo. Nat. Protoc. 2009, 4, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Pugach, E.K.; Hettmer, S.; Storer, N.Y.; Liu, J.; Wills, A.A.; DiBiase, A.; Chen, E.Y.; Ignatius, M.S.; Poss, K.D.; et al. A novel chemical screening strategy in zebrafish identifies common pathways in embryogenesis and rhabdomyosarcoma development. Development 2013, 140, 2354–2364. [Google Scholar] [CrossRef] [PubMed]
- MacRae, C.A.; Peterson, R.T. Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef] [PubMed]
- McCammon, J.M.; Sive, H. Challenges in understanding psychiatric disorders and developing therapeutics: A role for zebrafish. Dis. Model. Mech. 2015, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Pardo-Martin, C.; Allalou, A.; Medina, J.; Eimon, P.M.; Wahlby, C.; Fatih Yanik, M. High-throughput hyperdimensional vertebrate phenotyping. Nat. Commun. 2013, 4, 1467. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.B.; Westerfield, M. Zebrafish models in translational research: Tipping the scales toward advancements in human health. Dis. Model. Mech. 2014, 7, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Rajpurohit, S.K.; Delaspre, F.; Walker, S.L.; White, D.T.; Ceasrine, A.; Kuruvilla, R.; Li, R.J.; Shim, J.S.; Liu, J.O.; et al. First quantitative high-throughput screen in zebrafish identifies novel pathways for increasing pancreatic β-cell mass. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Weger, B.D.; Weger, M.; Nusser, M.; Brenner-Weiss, G.; Dickmeis, T. A chemical screening system for glucocorticoid stress hormone signaling in an intact vertebrate. ACS Chem. Biol. 2012, 7, 1178–1183. [Google Scholar] [CrossRef] [PubMed]
- Coffey, C.M.; Solleveld, P.A.; Fang, J.; Roberts, A.K.; Hong, S.K.; Dawid, I.B.; Laverriere, C.E.; Glasgow, E. Novel oxytocin gene expression in the hindbrain is induced by alcohol exposure: Transgenic zebrafish enable visualization of sensitive neurons. PLoS ONE 2013, 8, e53991. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.A.; Huang, H.; Yang, Z.; Herzog, W.; Hammerschmidt, M.; Lin, S.; Melmed, S. Pituitary corticotroph ontogeny and regulation in transgenic zebrafish. Mol. Endocrinol. 2003, 17, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xu, W.; He, J.; Yin, Z. In vivo alternative assessment of the chemicals that interfere with anterior pituitary POMC expression and interrenal steroidogenesis in POMC: EGFP transgenic zebrafish. Toxicol. Appl. Pharmacol. 2010, 248, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Golan, M.; Zelinger, E.; Zohar, Y.; Levavi-Sivan, B. Architecture of GnRH-gonadotrope-vasculature reveals a dual mode of gonadotropin regulation in fish. Endocrinology 2015, 156, 4163–4173. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.A.; Liu, Q.; Wawrowsky, K.; Yang, Z.; Lin, S.; Melmed, S. Prolactin receptor signaling mediates the osmotic response of embryonic zebrafish lactotrophs. Mol. Endocrinol. 2006, 20, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.; Oikonomou, G.; Prober, D.A. Norepinephrine is required to promote wakefulness and for hypocretin-induced arousal in zebrafish. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Meguro, S.; Hasumura, T.; Hase, T. Body fat accumulation in zebrafish is induced by a diet rich in fat and reduced by supplementation with green tea extract. PLoS ONE 2015, 10, e0120142. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Gerlai, R. Zebrafish as an emerging model for studying complex brain disorders. Trends Pharmacol. Sci. 2014, 35, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Egan, R.J.; Bergner, C.L.; Hart, P.C.; Cachat, J.M.; Canavello, P.R.; Elegante, M.F.; Elkhayat, S.I.; Bartels, B.K.; Tien, A.K.; Tien, D.H.; et al. Understanding behavioral and physiological phenotypes of stress and anxiety in zebrafish. Behav. Brain Res. 2009, 205, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in scn1a zebrafish mutant identifies clemizole as a potential dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.J.; Groneberg, A.H.; Yeh, C.M.; Castillo Ramirez, L.A.; Ryu, S. Optogenetic elevation of endogenous glucocorticoid level in larval zebrafish. Front. in neural circuits 2013, 7, 82. [Google Scholar] [CrossRef] [PubMed]
- Jordi, J.; Guggiana-Nilo, D.; Soucy, E.; Song, E.Y.; Wee, C.L.; Engert, F. A high-throughput assay for quantifying appetite and digestive dynamics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Wolfe, S.A. Forward and reverse genetic approaches for the analysis of vertebrate development in the zebrafish. Dev. Cell 2011, 21, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Varshney, G.K.; Lu, J.; Gildea, D.E.; Huang, H.; Pei, W.; Yang, Z.; Huang, S.C.; Schoenfeld, D.; Pho, N.H.; Casero, D.; et al. A large-scale zebrafish gene knockout resource for the genome-wide study of gene function. Genome Res. 2013, 23, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Aspatwar, A.; Tolvanen, M.E.; Ojanen, M.J.; Barker, H.R.; Saralahti, A.K.; Bauerlein, C.A.; Ortutay, C.; Pan, P.; Kuuslahti, M.; Parikka, M.; et al. Inactivation of ca10a and ca10b genes leads to abnormal embryonic development and alters movement pattern in zebrafish. PLoS ONE 2015, 10, e0134263. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Hisano, Y.; Kawahara, A.; Higashijima, S. Efficient generation of knock-in transgenic zebrafish carrying reporter/driver genes by CRISPR/Cas9-mediated genome engineering. Sci. Rep. 2014, 4, 6545. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.; Sun, C.; Gao, L.; Zhu, D.; Xu, X.; Zhu, X.; Xiong, J.W.; Xi, J.J. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res. 2013, 23, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef] [PubMed]
- Irion, U.; Krauss, J.; Nusslein-Volhard, C. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 2014, 141, 4827–4830. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Kontarakis, Z.; Gerri, C.; Nolte, H.; Holper, S.; Kruger, M.; Stainier, D.Y. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 2015, 524, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Kok, F.O.; Shin, M.; Ni, C.W.; Gupta, A.; Grosse, A.S.; van Impel, A.; Kirchmaier, B.C.; Peterson-Maduro, J.; Kourkoulis, G.; Male, I.; et al. Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell 2015, 32, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Eisen, J.S.; Smith, J.C. Controlling morpholino experiments: Don't stop making antisense. Development 2008, 135, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.L.; Glasgow, E. Zebrafish orthopedia (otp) is required for isotocin cell development. Dev. Genes Evol. 2007, 217, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Catalina-Rodriguez, O.; Kolukula, V.K.; Tomita, Y.; Preet, A.; Palmieri, F.; Wellstein, A.; Byers, S.; Giaccia, A.J.; Glasgow, E.; Albanese, C.; et al. The mitochondrial citrate transporter, CIC, is essential for mitochondrial homeostasis. Oncotarget 2012, 3, 1220–1235. [Google Scholar] [CrossRef] [PubMed]
- Blechman, J.; Borodovsky, N.; Eisenberg, M.; Nabel-Rosen, H.; Grimm, J.; Levkowitz, G. Specification of hypothalamic neurons by dual regulation of the homeodomain protein orthopedia. Development 2007, 134, 4417–4426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Haffter, P.; Granato, M.; Brand, M.; Mullins, M.C.; Hammerschmidt, M.; Kane, D.A.; Odenthal, J.; van Eeden, F.J.; Jiang, Y.J.; Heisenberg, C.P.; et al. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 1996, 123, 1–36. [Google Scholar] [PubMed]
- Amsterdam, A.; Burgess, S.; Golling, G.; Chen, W.; Sun, Z.; Townsend, K.; Farrington, S.; Haldi, M.; Hopkins, N. A large-scale insertional mutagenesis screen in zebrafish. Genes Dev. 1999, 13, 2713–2724. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Jao, L.E.; Zheng, N.; Dolan, K.; Ivey, J.; Zonies, S.; Wu, X.; Wu, K.; Yang, H.; Meng, Q.; et al. Efficient genome-wide mutagenesis of zebrafish genes by retroviral insertions. Proc. Natl. Acad. Sci. USA 2007, 104, 12428–12433. [Google Scholar] [CrossRef] [PubMed]
- Driever, W.; Solnica-Krezel, L.; Schier, A.F.; Neuhauss, S.C.; Malicki, J.; Stemple, D.L.; Stainier, D.Y.; Zwartkruis, F.; Abdelilah, S.; Rangini, Z.; et al. A genetic screen for mutations affecting embryogenesis in zebrafish. Development 1996, 123, 37–46. [Google Scholar] [PubMed]
- Gaiano, N.; Amsterdam, A.; Kawakami, K.; Allende, M.; Becker, T.; Hopkins, N. Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature 1996, 383, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Takeda, H.; Kawakami, N.; Kobayashi, M.; Matsuda, N.; Mishina, M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell 2004, 7, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Sivasubbu, S.; Balciunas, D.; Amsterdam, A.; Ekker, S.C. Insertional mutagenesis strategies in zebrafish. Genome Bio. 2007, 8 (Suppl. S1). [Google Scholar] [CrossRef] [PubMed]
- Wyatt, C.; Bartoszek, E.M.; Yaksi, E. Methods for studying the zebrafish brain: Past, present and future. Eur. J. Neurosci. 2015, 42, 1746–1763. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.M.; Balciunas, D.; Sivasubbu, S.; Clark, K.J.; Bedell, V.M.; Westcot, S.E.; Myers, S.R.; Moulder, G.L.; Thomas, M.J.; Ekker, S.C. Nicotine response genetics in the zebrafish. Proc. Natl. Acad. Sci. USA 2009, 106, 18662–18667. [Google Scholar] [CrossRef] [PubMed]
- Rennekamp, A.J.; Peterson, R.T. 15 Years of zebrafish chemical screening. Curr. Opin. Chem. Biol. 2015, 24, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Metelo, A.M.; Noonan, H.R.; Li, X.; Jin, Y.; Baker, R.; Kamentsky, L.; Zhang, Y.; van Rooijen, E.; Shin, J.; Carpenter, A.E.; et al. Pharmacological HIF2α inhibition improves VHL disease-associated phenotypes in zebrafish model. J. Clin. Invest. 2015, 125, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.; Reischl, M.; Shah, A.H.; Kronfuss, E.; Mikut, R.; Liebel, U.; Grabher, C. A zebrafish drug-repurposing screen reveals sGC-dependent and sGC-independent pro-inflammatory activities of nitric oxide. PLoS ONE 2015, 10, e0137286. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lahvic, J.L.; Binder, V.; Pugach, E.K.; Riley, E.B.; Tamplin, O.J.; Panigrahy, D.; Bowman, T.V.; Barrett, F.G.; Heffner, G.C.; et al. Epoxyeicosatrienoic acids enhance embryonic haematopoiesis and adult marrow engraftment. Nature 2015, 523, 468–471. [Google Scholar] [CrossRef] [PubMed]
- Evason, K.J.; Francisco, M.T.; Juric, V.; Balakrishnan, S.; Pazmino, M.D.P.L.; Gordan, J.D.; Kakar, S.; Spitsbergen, J.; Goga, A.; Stainier, D.Y. Identification of chemical inhibitors of beta-catenin-driven liver tumorigenesis in zebrafish. PLoS Genet. 2015, 11, e1005305. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, V.E.; Varshney, G.K.; Lee, M.; Bupp, S.; Xu, L.; Shinn, P.; Crawford, N.P.; Inglese, J.; Burgess, S.M. Phenotype-driven chemical screening in zebrafish for compounds that inhibit collective cell migration identifies multiple pathways potentially involved in metastatic invasion. Dis. Model. Mech. 2015, 8, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.H.; Hempel, J.E.; Hao, J.; Frist, A.Y.; Williams, M.M.; Fleming, J.T.; Sulikowski, G.A.; Cooper, M.K.; Chiang, C.; Hong, C.C. An in vivo chemical genetic screen identifies phosphodiesterase 4 as a pharmacological target for hedgehog signaling inhibition. Cell Rep. 2015, 11, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Nath, A.K.; Ryu, J.H.; Jin, Y.N.; Roberts, L.D.; Dejam, A.; Gerszten, R.E.; Peterson, R.T. PTPMT1 inhibition lowers glucose through succinate dehydrogenase phosphorylation. Cell Rep. 2015, 10, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Ninov, N.; Delawary, M.; Osman, S.; Roh, A.S.; Gut, P.; Stainier, D.Y. Whole organism high content screening identifies stimulators of pancreatic β-cell proliferation. PLoS ONE 2014, 9, e104112. [Google Scholar]
- Nishiya, N.; Oku, Y.; Kumagai, Y.; Sato, Y.; Yamaguchi, E.; Sasaki, A.; Shoji, M.; Ohnishi, Y.; Okamoto, H.; Uehara, Y. A zebrafish chemical suppressor screening identifies small molecule inhibitors of the Wnt/β-catenin pathway. Chem. Biol. 2014, 21, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Grimaldi, M.; Curtin, E.; Dougherty, M.; Kaufman, C.; White, R.M.; Zon, L.I.; Liao, E.C. Neural crest development and craniofacial morphogenesis is coordinated by nitric oxide and histone acetylation. Chem. Biol. 2014, 21, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Gebruers, E.; Cordero-Maldonado, M.L.; Gray, A.I.; Clements, C.; Harvey, A.L.; Edrada-Ebel, R.; de Witte, P.A.; Crawford, A.D.; Esguerra, C.V. A phenotypic screen in zebrafish identifies a novel small-molecule inducer of ectopic tail formation suggestive of alterations in non-canonical Wnt/PCP signaling. PLoS ONE 2013, 8, e83293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, J.; Ao, A.; Zhou, L.; Murphy, C.K.; Frist, A.Y.; Keel, J.J.; Thorne, C.A.; Kim, K.; Lee, E.; Hong, C.C. Selective small molecule targeting β-catenin function discovered by in vivo chemical genetic screen. Cell Rep. 2013, 4, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Ridges, S.; Heaton, W.L.; Joshi, D.; Choi, H.; Eiring, A.; Batchelor, L.; Choudhry, P.; Manos, E.J.; Sofla, H.; Sanati, A.; et al. Zebrafish screen identifies novel compound with selective toxicity against leukemia. Blood 2012, 119, 5621–5631. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Pan, L.; Groen, R.W.; Baleydier, F.; Kentsis, A.; Marineau, J.; Grebliunaite, R.; Kozakewich, E.; Reed, C.; Pflumio, F.; et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J. Clin. Invest. 2014, 124, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Kokel, D.; Cheung, C.Y.; Mills, R.; Coutinho-Budd, J.; Huang, L.; Setola, V.; Sprague, J.; Jin, S.; Jin, Y.N.; Huang, X.P.; et al. Photochemical activation of TRPA1 channels in neurons and animals. Nat. Chem. Biol. 2013, 9, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Dinday, M.T.; Baraban, S.C. Large-scale phenotype-based antiepileptic drug screening in a zebrafish model of dravet syndrome(1,2,3). eNeuro 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Gut, P.; Baeza-Raja, B.; Andersson, O.; Hasenkamp, L.; Hsiao, J.; Hesselson, D.; Akassoglou, K.; Verdin, E.; Hirschey, M.D.; Stainier, D.Y. Whole-organism screening for gluconeogenesis identifies activators of fasting metabolism. Nat. Chem. Biol. 2013, 9, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Karin Arndt, A.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a new modulator of the intercalated disc in a zebrafish model of arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Bulut, G.; Hong, S.H.; Chen, K.; Beauchamp, E.M.; Rahim, S.; Kosturko, G.W.; Glasgow, E.; Dakshanamurthy, S.; Lee, H.S.; Daar, I.; et al. Small molecule inhibitors of ezrin inhibit the invasive phenotype of osteosarcoma cells. Oncogene 2012, 31, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Rihel, J.; Prober, D.A.; Arvanites, A.; Lam, K.; Zimmerman, S.; Jang, S.; Haggarty, S.J.; Kokel, D.; Rubin, L.L.; Peterson, R.T.; et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science 2010, 327, 348–351. [Google Scholar] [CrossRef] [PubMed]
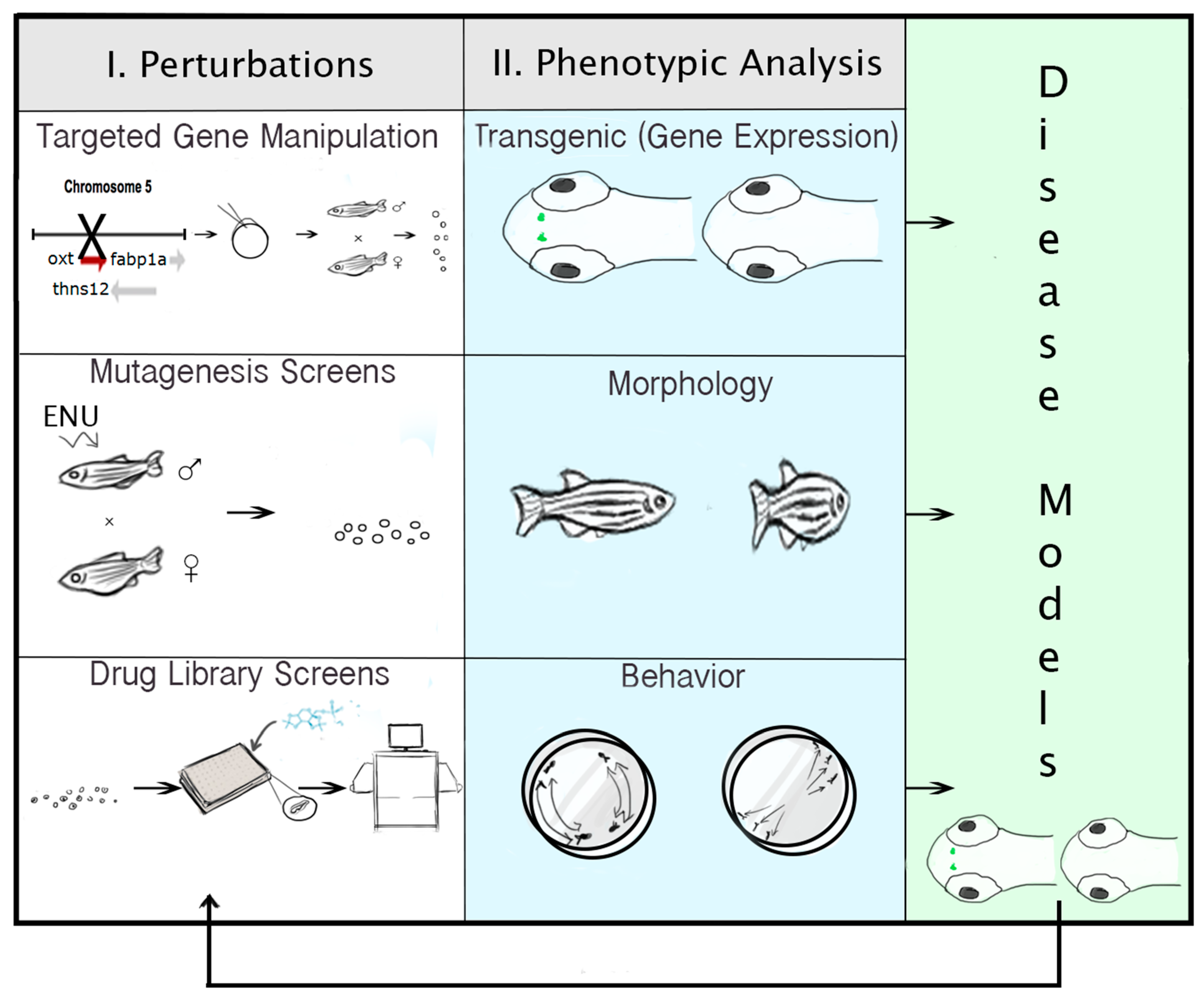

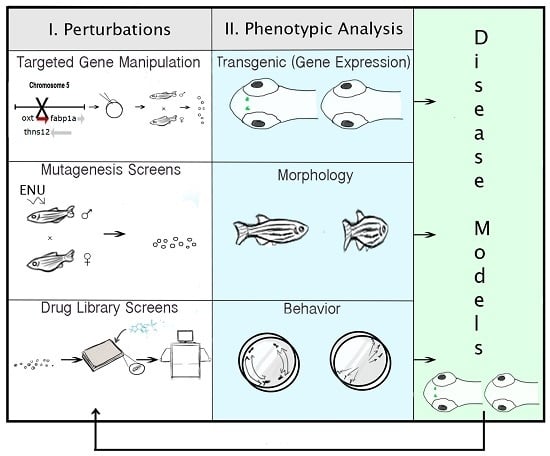
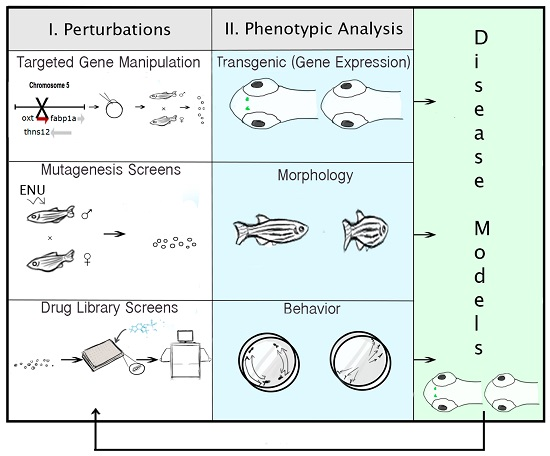{kind=link}
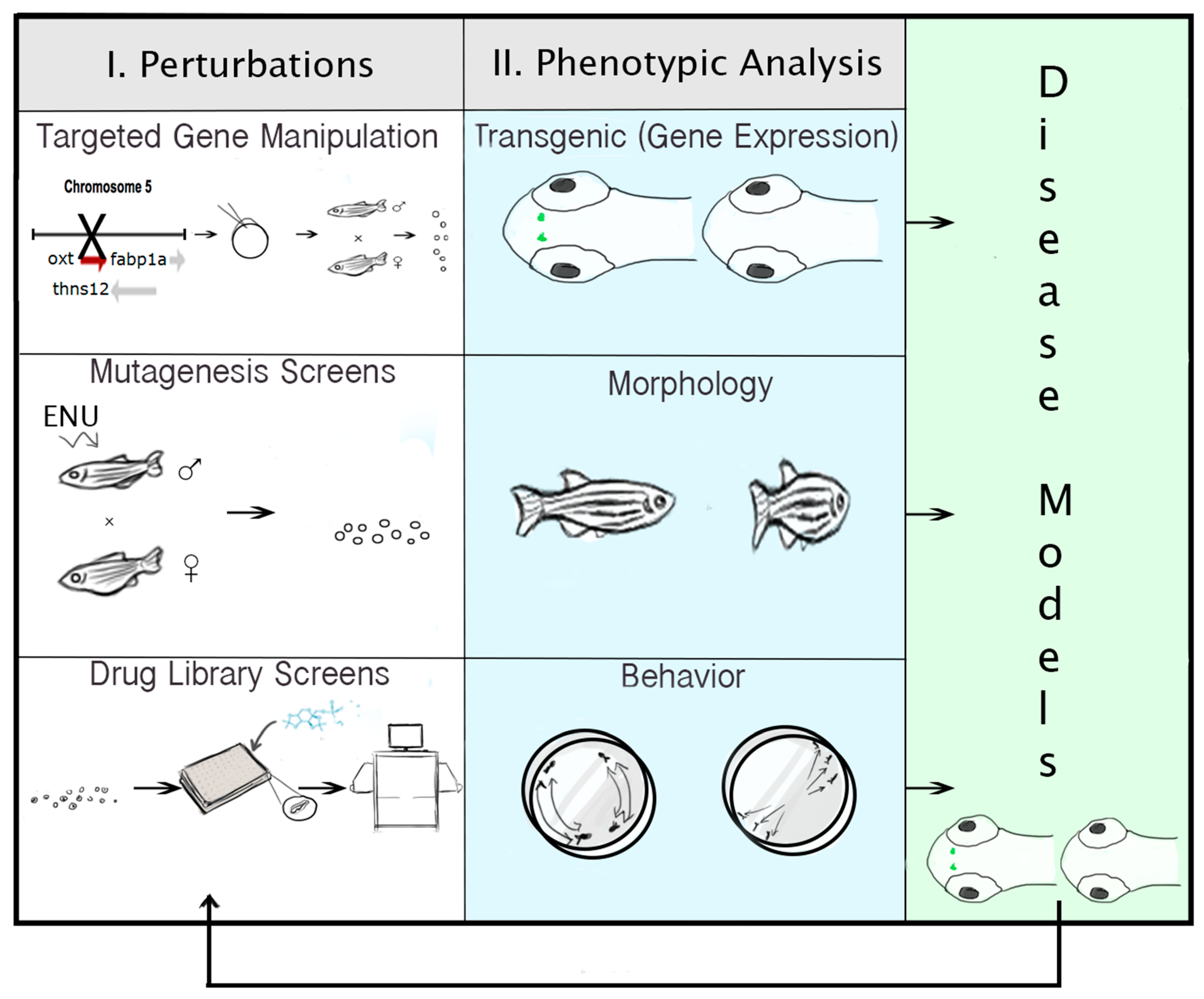{kind=link}
| Medical Symptom | Age of Onset | Treatment Options |
|---|---|---|
| hypotonia and feeding difficulties | 0–9 months | feeding assistance, nasogastric tubes |
| hyperphagia | 4.5–8 years | behavioral therapy |
| short stature | puberty | GH therapy, allows patients to reach full adult height |
| hypoplastic genitalia | birth | hormonal replacement therapy |
| dysmorphic features | birth | none |
| sleep-disordered breathing and daytime hypersomnolence | childhood to adolescence (Nixon and Brouillette) | adenotonsillectomy, nocturnal ventilation, weight control, and behavioral interventions |
| cognitive disability | childhood | none |
| skin picking and obsessive behavior | 5 years | behavioral therapy |
| oppositional behavior and tantrums | 5 years | behavioral therapy and psychiatric drugs |
| Human | Mouse | % aa | Zebrafish | % aa | Chr. |
|---|---|---|---|---|---|
| FRAT1; Chr10 * | Peg12 | 70 | gbp + | 63 | 16 |
| MKRN3 | Mkrn3 | 63 | mkrn1 | 48 | 4 |
| MAGEL2 | Magel2 | 53 | ndnl2 &,+ | 48 | 23 |
| NDN | Ndn | 82 | ndnl2 &,+ | 40 | 23 |
| NPAP1 | Pom121; Chr5 * | 28 | pom121 + | 24 | 10 |
| SNRPN | Snrpn | 100 | snrpb | 93 | 6 |
| SNORD107 | Snord 107 | - | |||
| SNORD64 | Snord 64 | - | |||
| SNORD116@27 | Snord116@27 | - | |||
| SNORD115@41 | Snord115@130 | - | |||
| SNORD108 | - | - | |||
| SNORD109A | - | - | |||
| SNORD109B | - | - |
| Failure to Thrive/Early Fatality | No Observable Abnormal Phenotype |
|---|---|
| Maternal duplication and paternal deletion in PWS region [36] | Deletion of exon 1 of Snrpn [39] |
| 6.8 Mb deletion spanning PWS and AS regions [37] | Deletion of Snrpn exon 2 [40] |
| Deletion spanning from Snrpn to Ube3a [40] | Double deletion of Snrpn exon 2 and Ube3a [40] |
| Deletion of Snrpn exons 1–6 and distal portion of IC [41] | Deletion of Snrpn exon 6, parts of exons 5 and 7 [41] |
| 4.8 kB deletion removing exon 1 of Snrpn and most of the differentially methylated region 1 (DMR1) [39] |
| Endo-Phenotype | Ndn [43] | Magel2 [42,44,46] | Snord116 [45] |
|---|---|---|---|
| Reduced oxytocin expression | Yes | - | - |
| Postnatal growth retardation | X | Yes | Yes |
| Increased anxiety | - | Yes | Yes |
| Respiratory distress | Yes | - | - |
| Skin scraping | Yes | - | - |
| Improved spatial memory and learning | Yes | X | X |
| Altered circadian output | - | Yes | - |
| Impaired fertility | X | Yes | X |
| Increased fat mass | X | Yes | X |
| Hyperphagia | - | X | Yes |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spikol, E.D.; Laverriere, C.E.; Robnett, M.; Carter, G.; Wolfe, E.; Glasgow, E. Zebrafish Models of Prader-Willi Syndrome: Fast Track to Pharmacotherapeutics. Diseases 2016, 4, 13. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4010013
Spikol ED, Laverriere CE, Robnett M, Carter G, Wolfe E, Glasgow E. Zebrafish Models of Prader-Willi Syndrome: Fast Track to Pharmacotherapeutics. Diseases. 2016; 4(1):13. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4010013
Chicago/Turabian StyleSpikol, Emma D., Caroline E. Laverriere, Maya Robnett, Gabriela Carter, Erin Wolfe, and Eric Glasgow. 2016. "Zebrafish Models of Prader-Willi Syndrome: Fast Track to Pharmacotherapeutics" Diseases 4, no. 1: 13. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4010013