
Human Coronaviruses: A Review of Virus–Host Interactions

Abstract
:1. Introduction
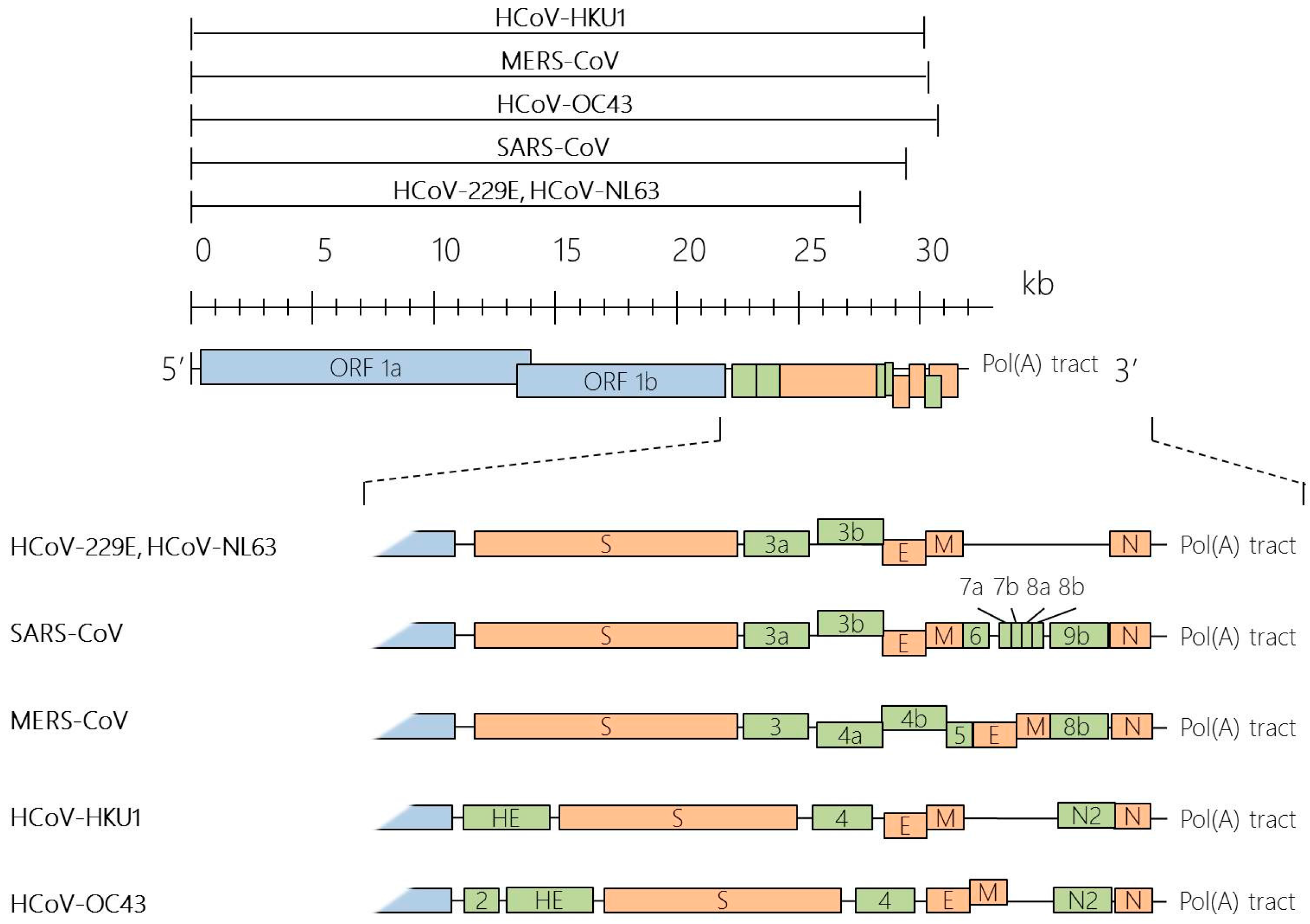
Taxonomy, Genomic Structure and Morphology
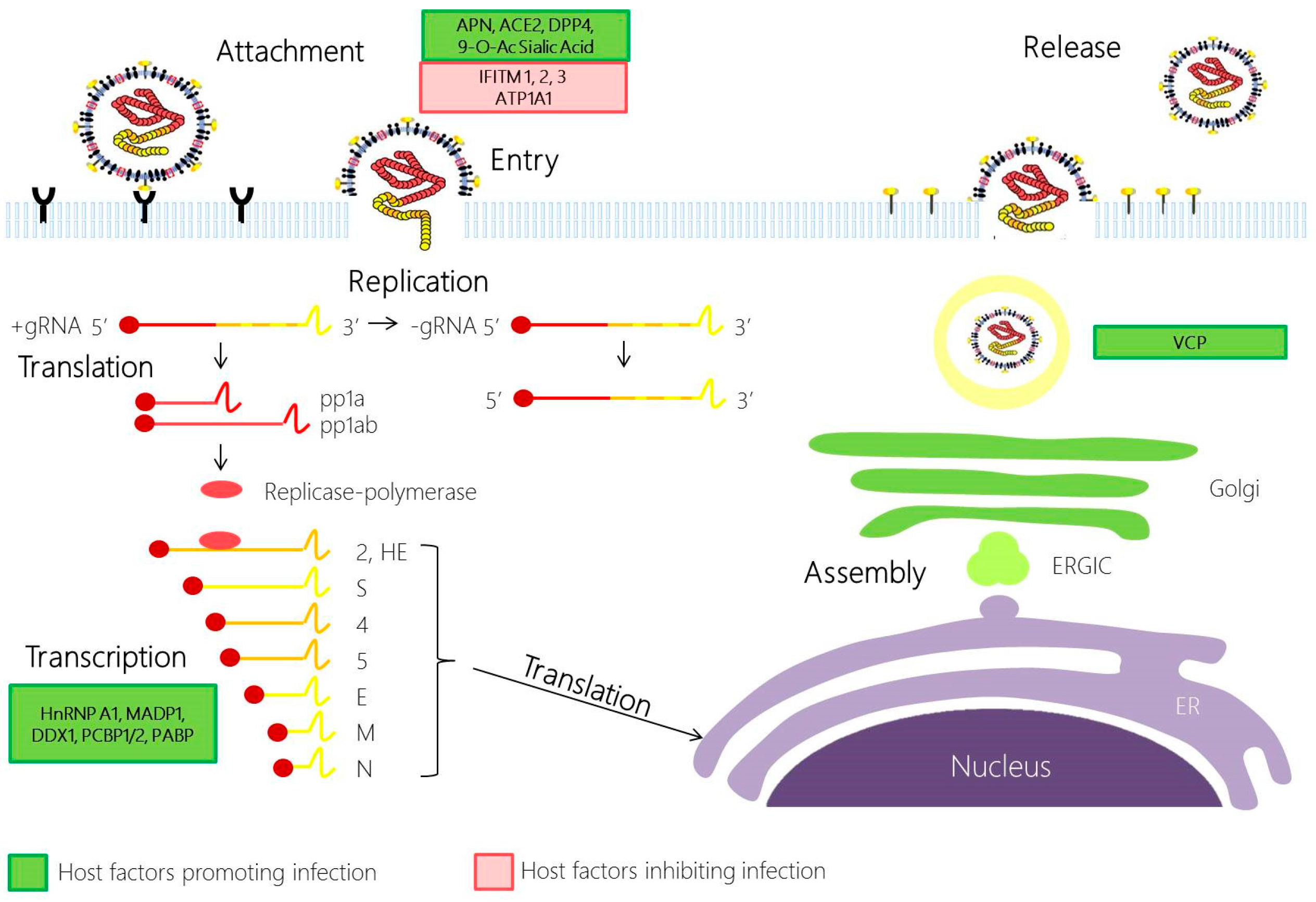
2. Involvement of Host Factors in Viral Replication and Pathogenesis
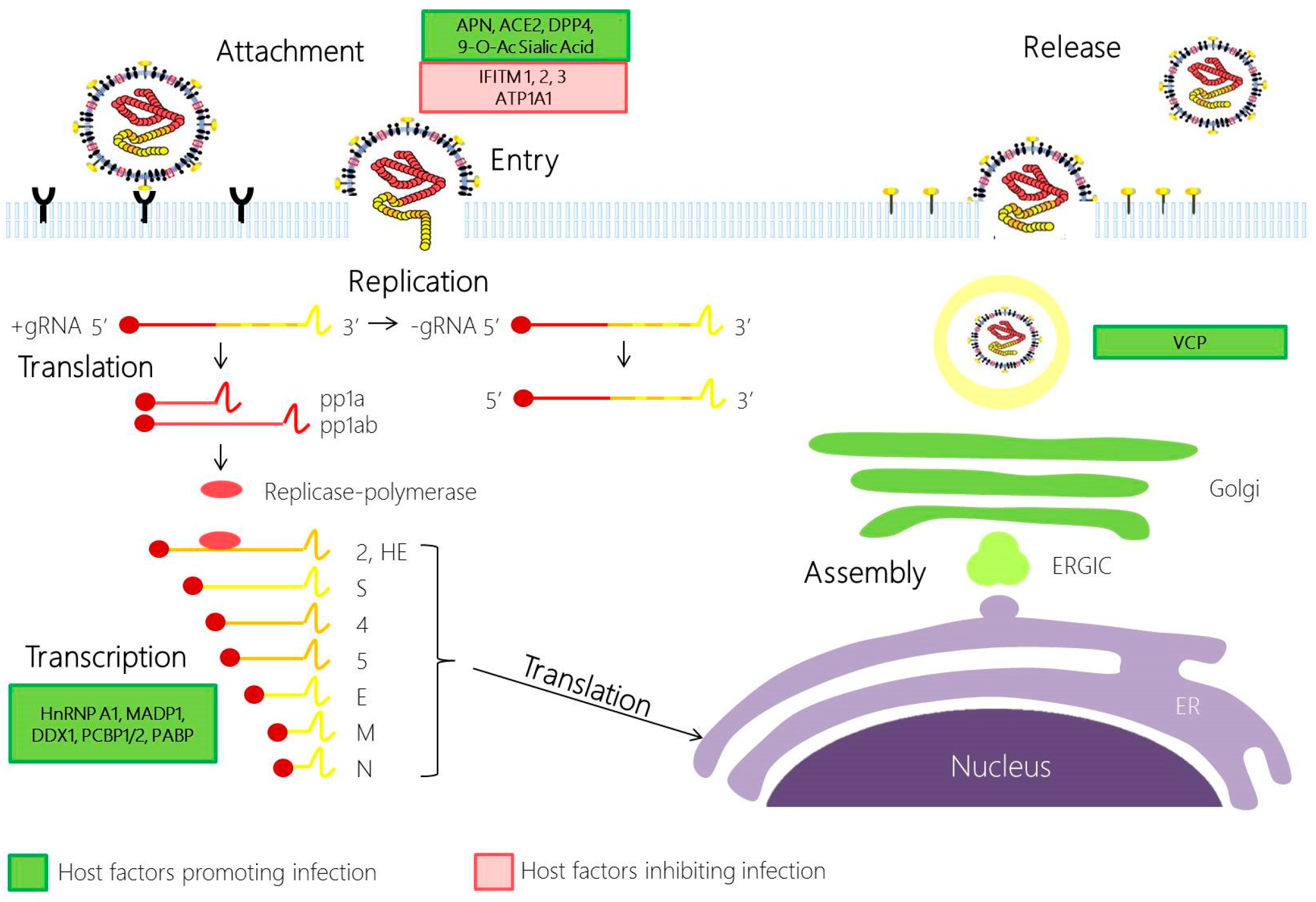
2.1. Coronavirus Attachment and Entry
2.2. Coronavirus Replication
2.3. Coronavirus Assembly and Egress
3. Human Coronavirus Infection and Apoptosis
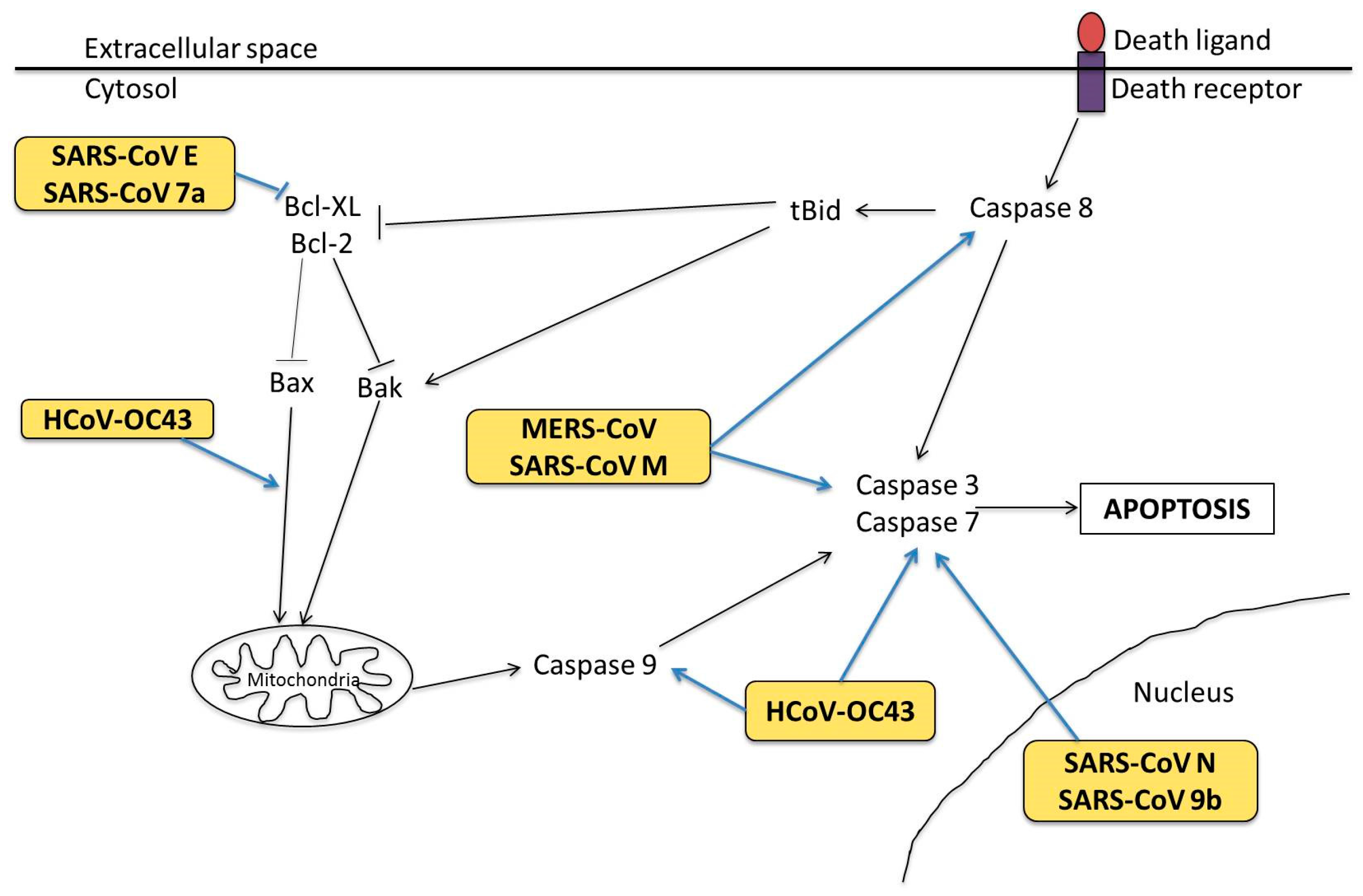
3.1. Cell Tropism and Apoptosis
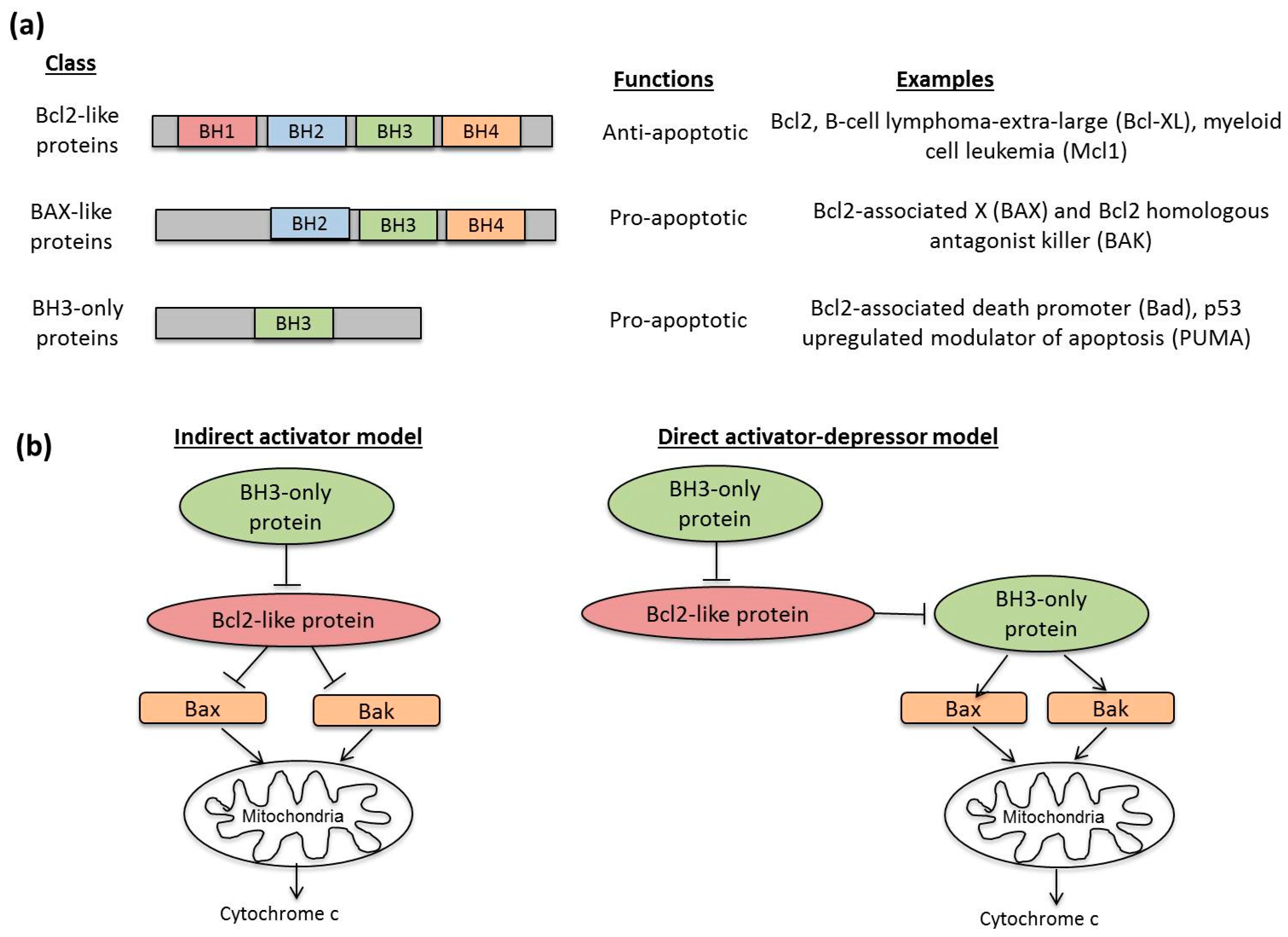
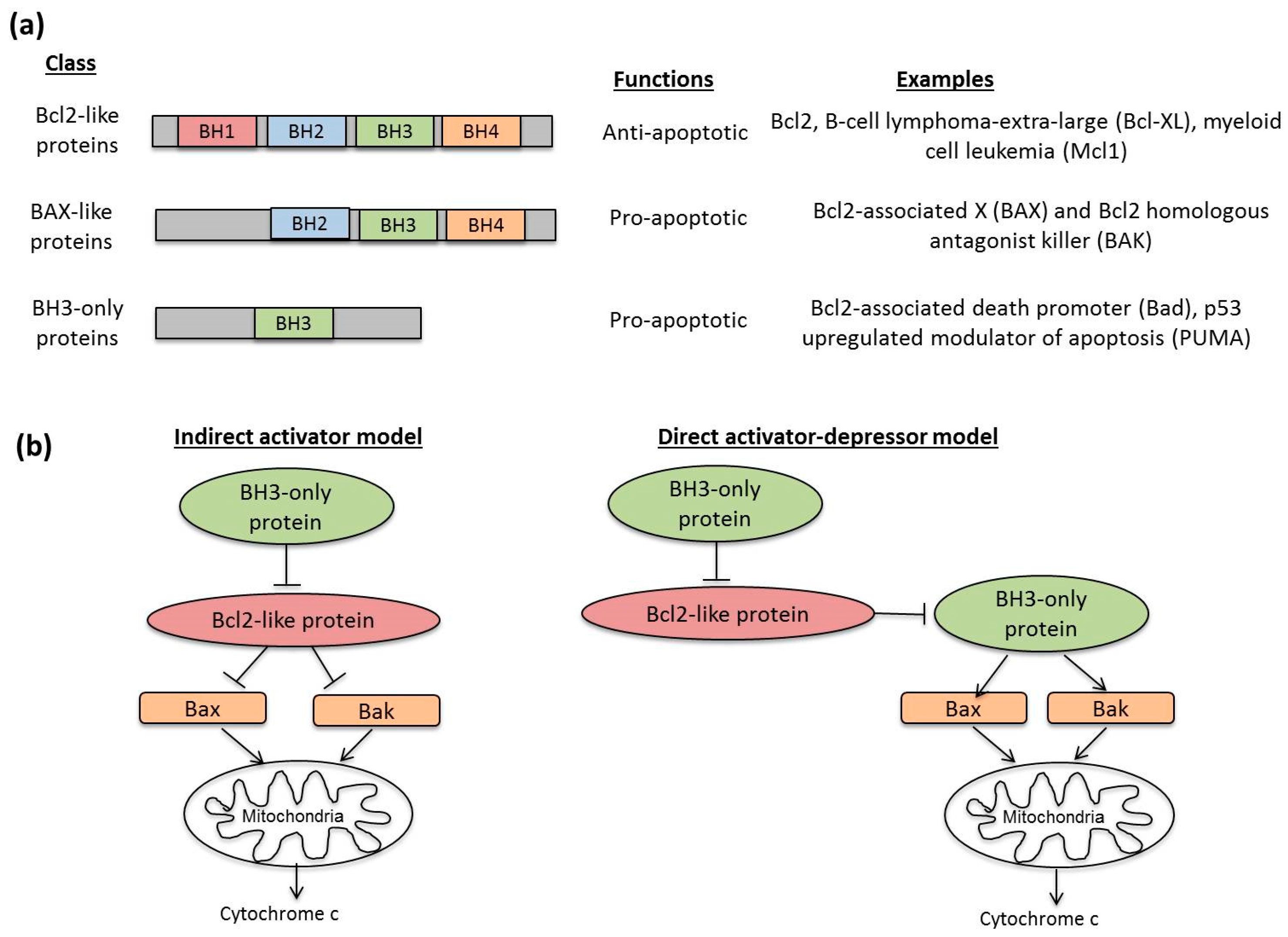
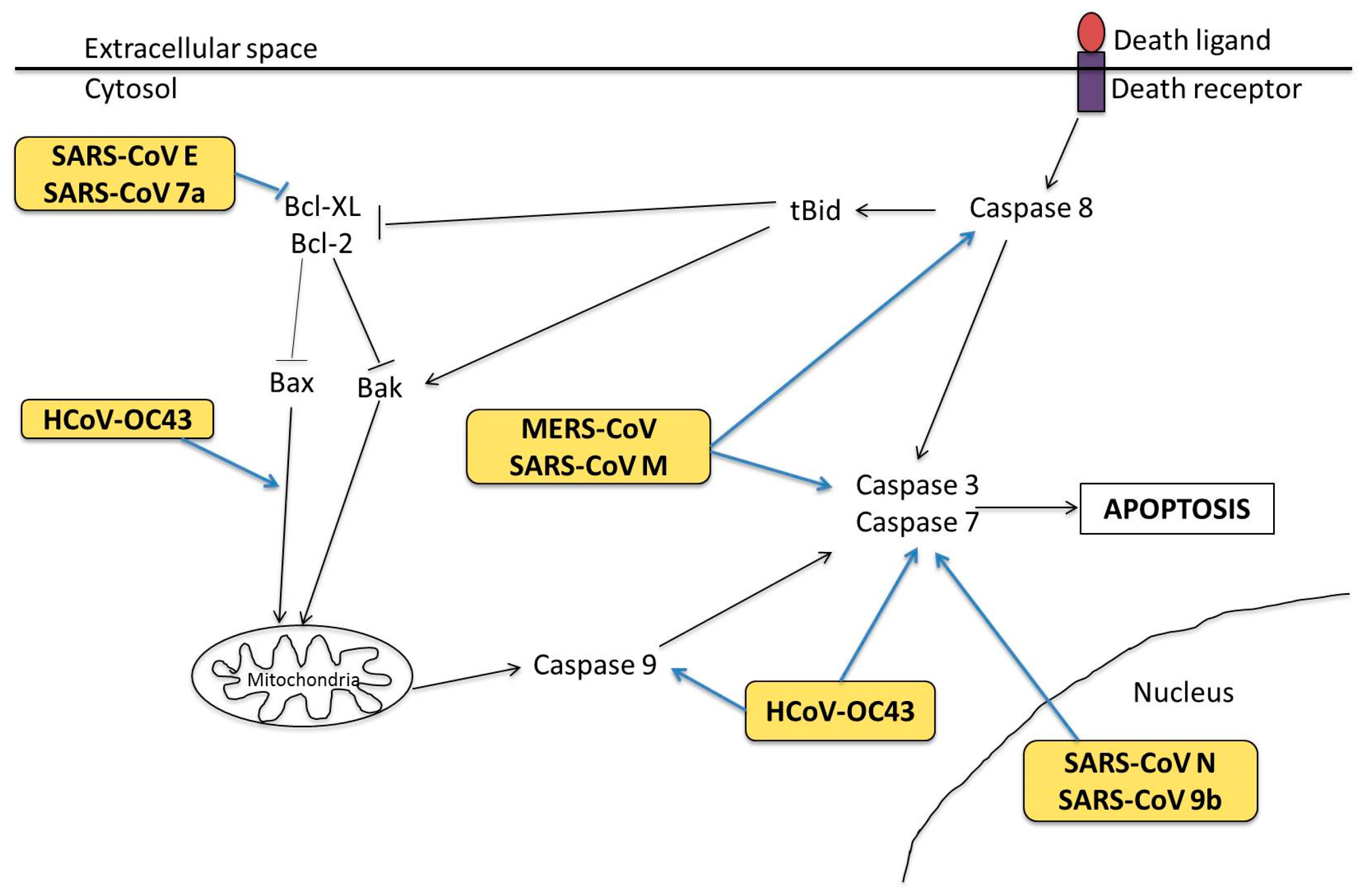
3.2. Molecular Mechanisms in Apoptosis
4. Human Coronavirus Infection and Innate Immunity
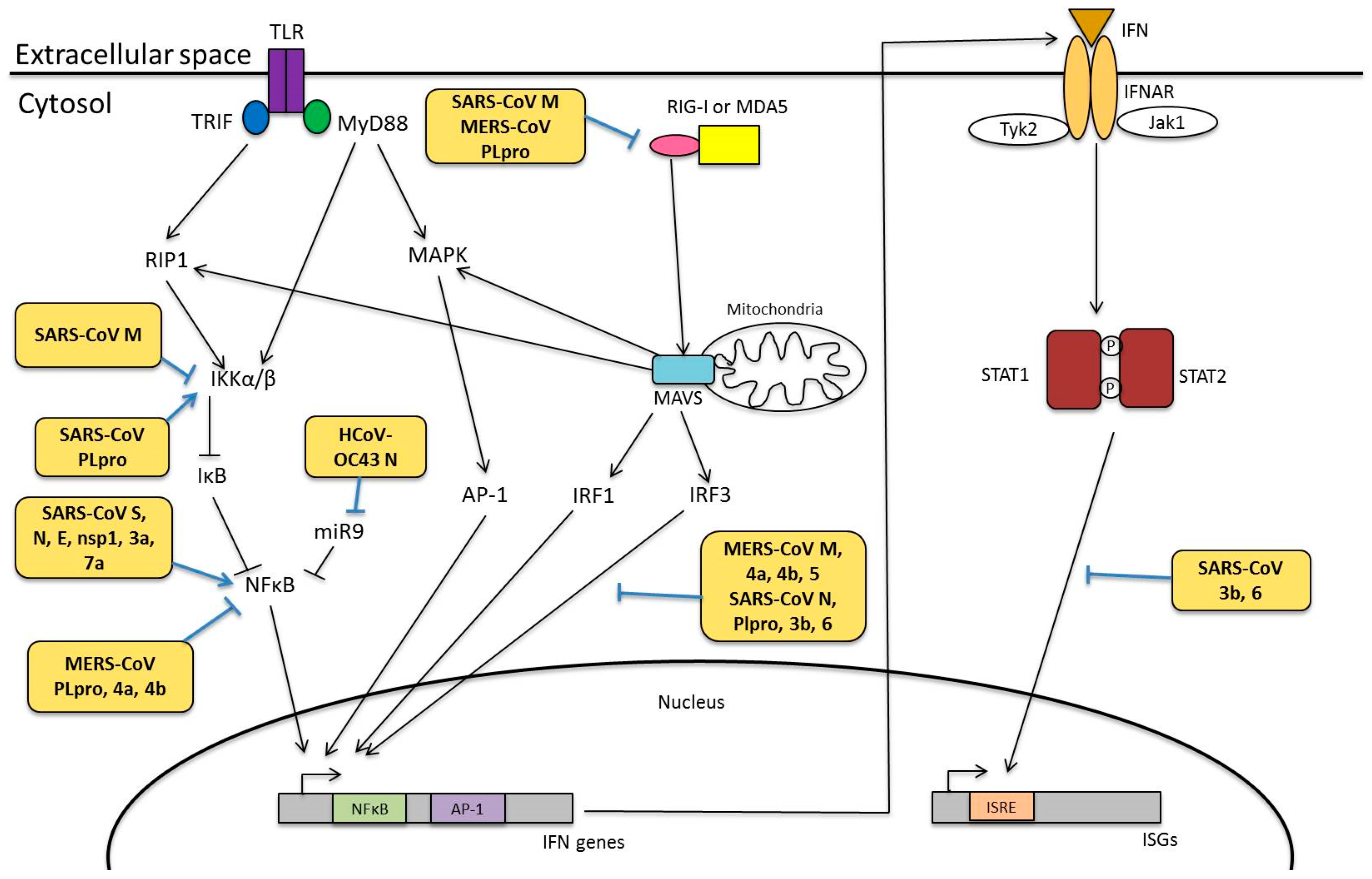
4.1. Pattern Recognition Receptors
4.2. Interferon Responses
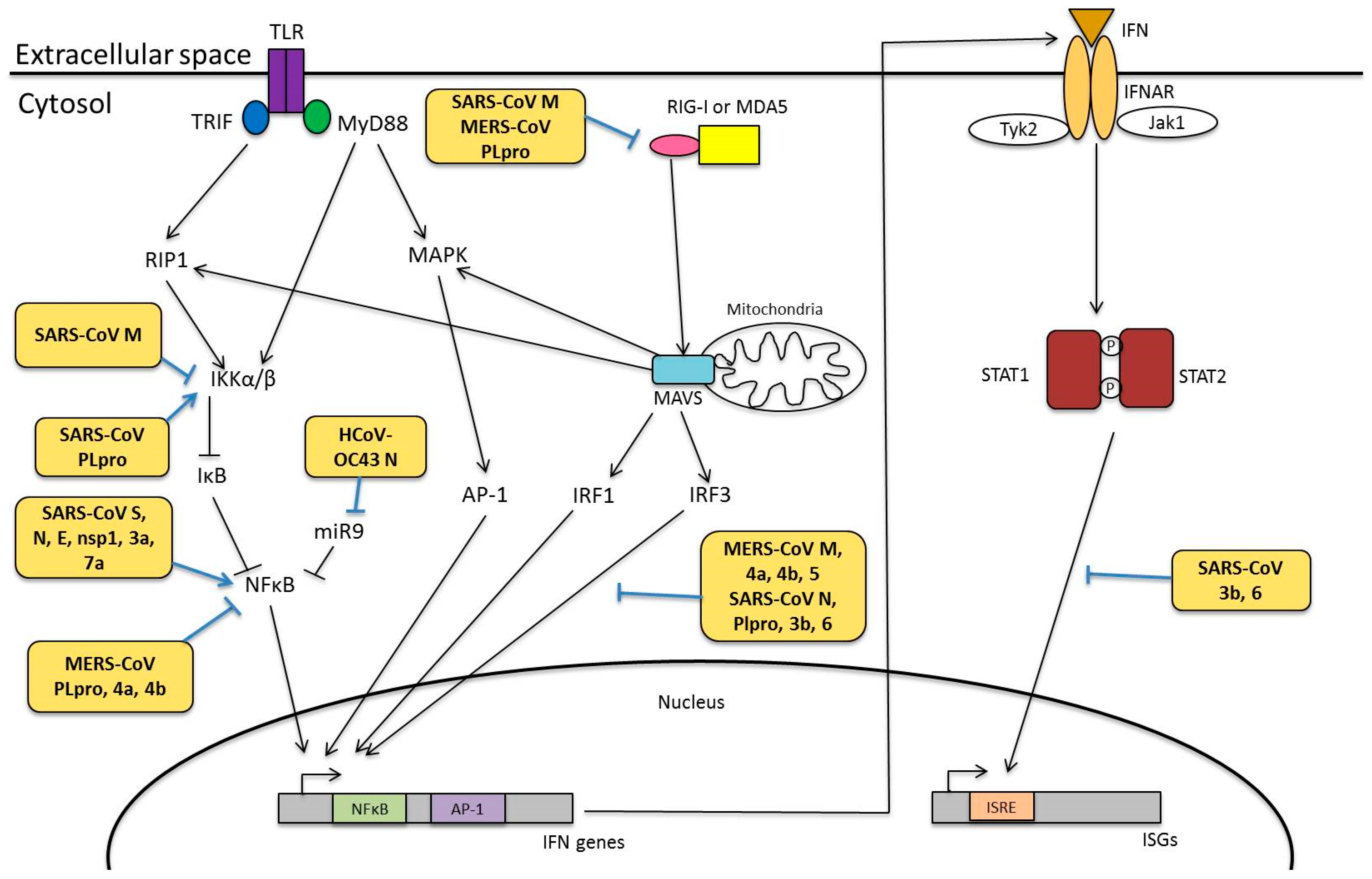
4.3. Modulation of Innate Immunity
4.3.1. Viral Proteins Involved in Innate Immunity
Structural Proteins of HCoVs
Non-Structural and Accessory Proteins of HCoVs
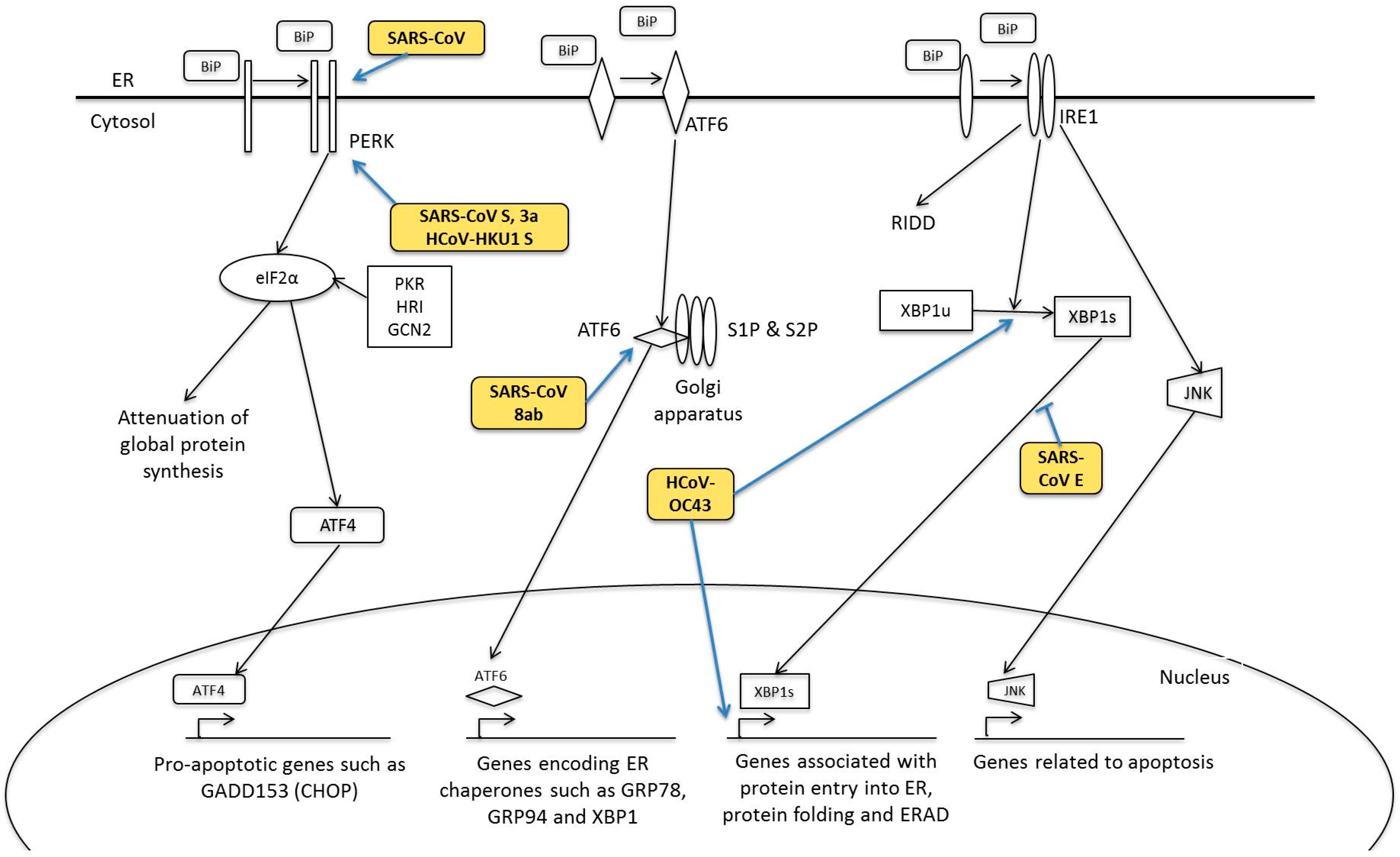
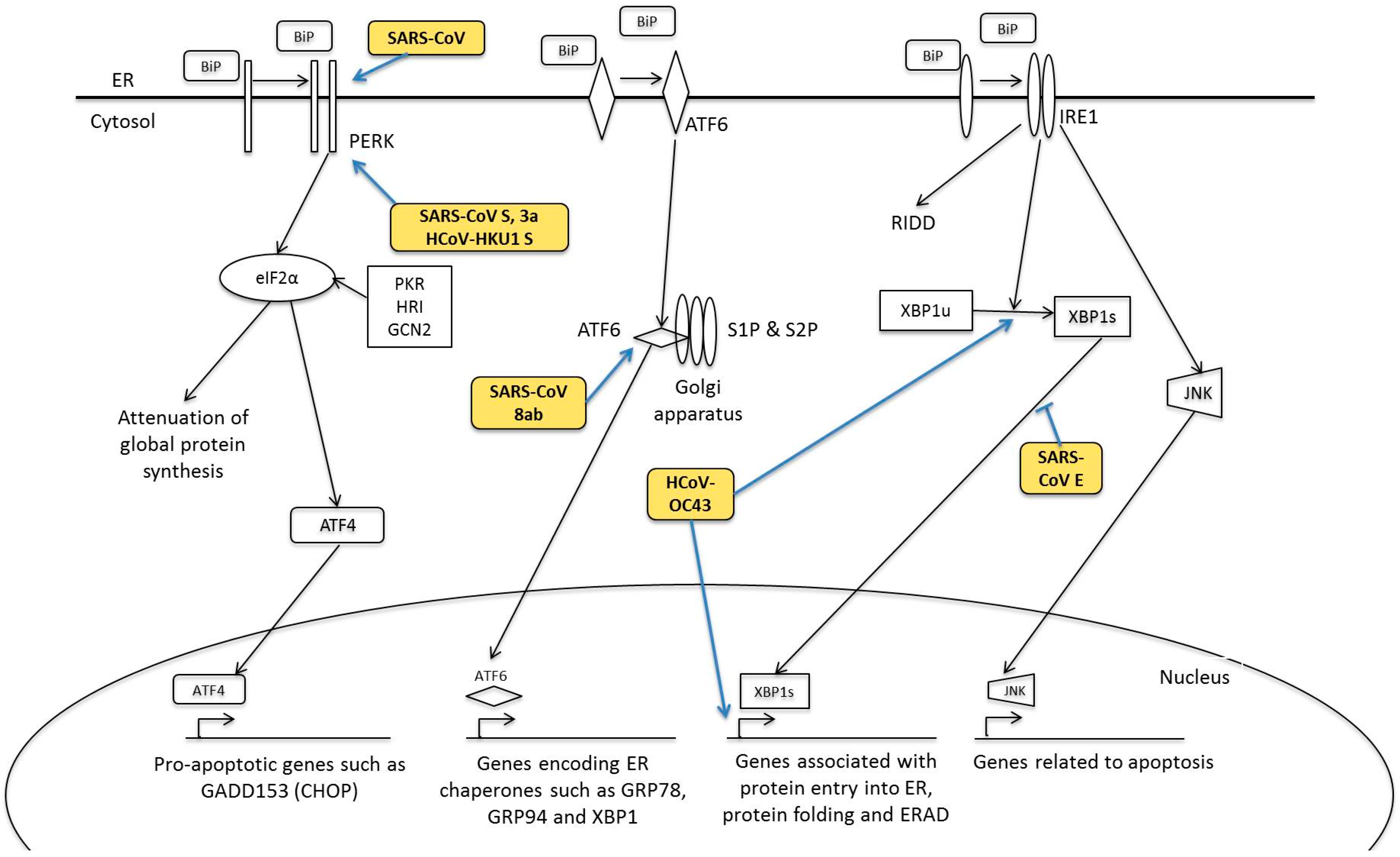
5. Human Coronavirus and ER Stress Response
5.1. PERK Signalling Pathway
5.2. ATF6 Signalling Pathway
5.3. IRE1 Signalling Pathway
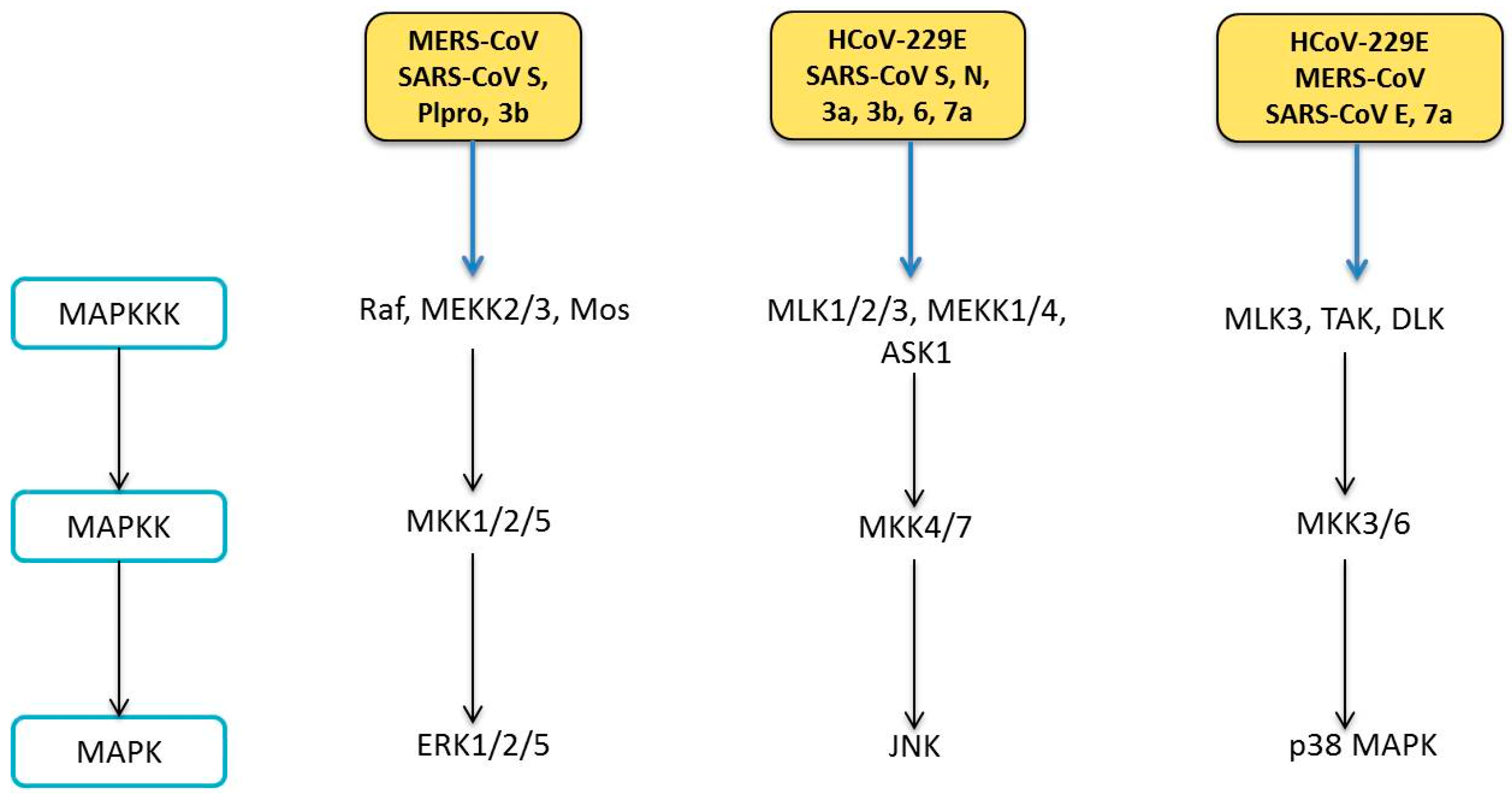
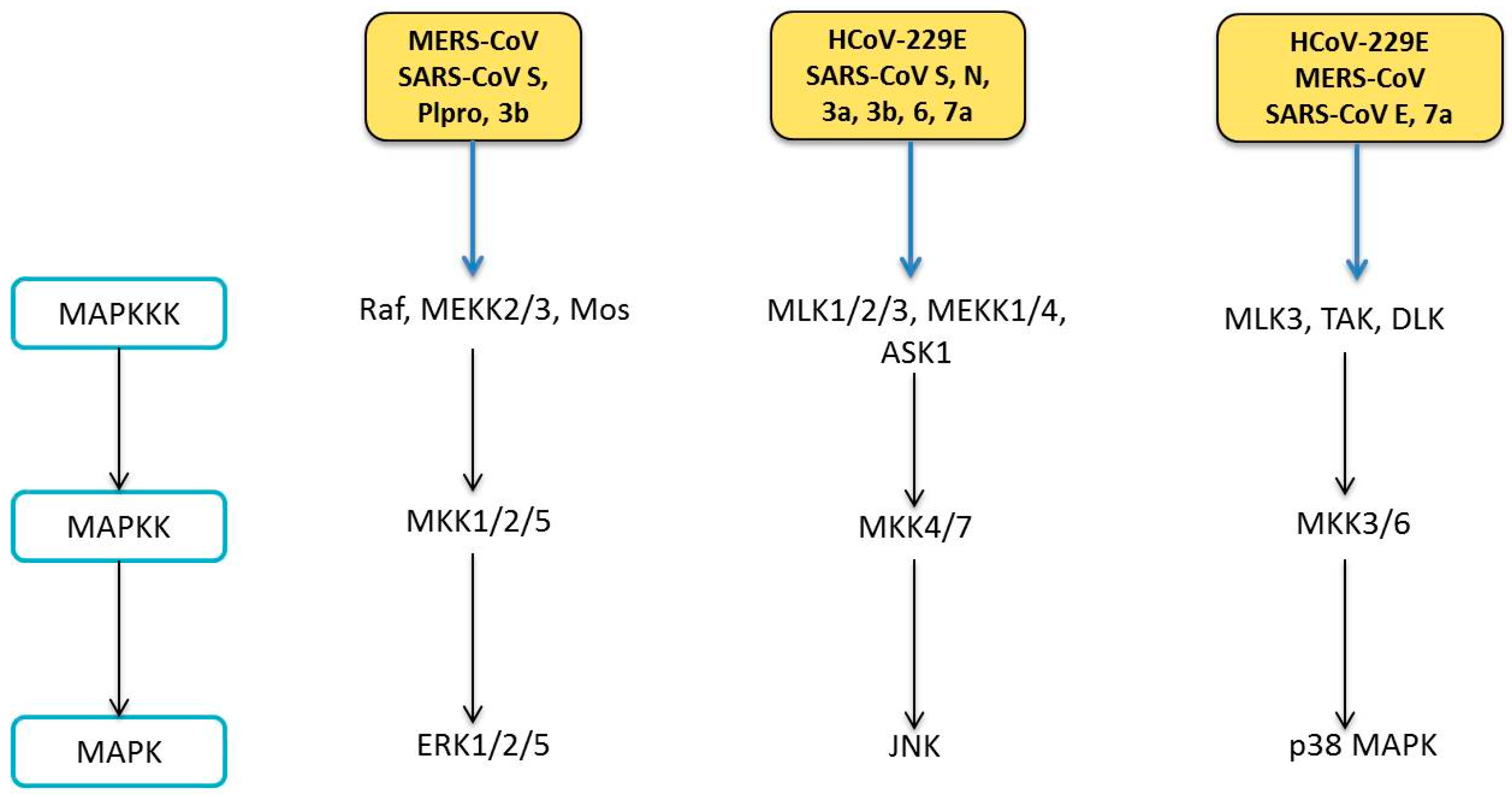
6. Human Coronavirus and MAPK Pathways
6.1. Modulation of MAPK Pathways
6.1.1. ERK Pathway
6.1.2. JNK Pathway
6.1.3. p38 MAPK Pathway
7. Human Coronavirus and NF-κB Pathway
7.1. Modulation of NF-κB Pathway
7.1.1. Structural Proteins
7.1.2. Nonstructural and Accessory Proteins
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pene, F.; Merlat, A.; Vabret, A.; Rozenberg, F.; Buzyn, A.; Dreyfus, F.; Cariou, A.; Freymuth, F.; Lebon, P. Coronavirus 229E-Related Pneumonia in Immunocompromised Patients. Clin. Infect. Dis. 2003, 37, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Maes, P.; Duson, G.; van Ranst, M. Development of One-Step, Real-Time, Quantitative Reverse Transcriptase PCR Assays for Absolute Quantitation of Human Coronaviruses OC43 and 229E. J. Clin. Microbiol. 2005, 43, 5452–5456. [Google Scholar] [CrossRef] [PubMed]
- Jones, B.A.; Grace, D.; Kock, R.; Alonso, S.; Rushton, J.; Said, M.Y.; McKeever, D.; Mutua, F.; Young, J.; McDermott, J.; et al. Zoonosis emergence linked to agricultural intensification and environmental change. Proc. Natl. Acad. Sci. USA 2013, 21, 8399–8340. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoek, L. Human coronaviruses: What do they cause? Antivir. Ther. 2007, 12, 651–658. [Google Scholar] [PubMed]
- Walsh 2007, E.E.; Shin, J.H.; Falsey, A.R. Clinical Impact of Human Coronaviruses 229E and OC43 Infection in Diverse Adult Populations. J. Infect. Dis. 2013, 208, 1634–1642. [Google Scholar] [CrossRef] [PubMed]
- Gorse, G.J.; O’Connor, T.Z.; Hall, S.L.; Vitale, J.N.; Nichol, K.L. Human Coronavirus and Acute Respiratory Illness in Older Adults with Chronic Obstructive Pulmonary Disease. J. Infect. Dis. 2009, 199, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Arbour, N.; Day, R.; Newcombe, J.; Talbot, P.J. Neuroinvasion by Human Respiratory Coronaviruses. J. Virol. 2000, 74, 8913–8921. [Google Scholar] [CrossRef] [PubMed]
- Arbour, N.; Ekandé, S.; Côté, G.; Lachance, C.; Chagnon, F.; Tardieu, M.; Cashman, N.R.; Talbot, P.J. Persistent Infection of Human Oligodendrocytic and Neuroglial Cell Lines by Human Coronavirus 229E. J. Virol. 1999, 73, 3326–3337. [Google Scholar] [PubMed]
- Jacomy, H.; Fragoso, G.; Almazan, G.; Mushynski, W.E.; Talbot, P.J. Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 2006, 349, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Vabret, A.; Mourez, T.; Gouarin, S.; Petitjean, J.; Freymuth, F. An Outbreak of Coronavirus OC43 Respiratory Infection in Normandy, France. Clin. Infect. Dis. 2003, 36, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Smuts, H. Human coronavirus NL63 infections in infants hospitalised with acute respiratory tract infections in South Africa. Influenza Other Respir. Viruses 2008, 2, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Baric, R. Mechanisms of Severe Acute Respiratory Syndrome Pathogenesis and Innate Immunomodulation. Microbiol. Mol. Biol. Rev. MMBR 2008, 72, 672–685. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.M.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yan, M.; Xu, H.; Liang, W.; Kan, B.; Zheng, B.; Chen, H.; Zheng, H.; Xu, Y.; Zhang, E.; et al. SARS-CoV Infection in a Restaurant from Palm Civet. Emerg. Infect. Dis. 2005, 11, 1860–1865. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Ge, X.; Wang, L.-F.; Shi, Z. Bat origin of human coronaviruses. Virol. J. 2015, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cheon, S.; Min, C.-K.; Sohn, K.M.; Kang, Y.J.; Cha, Y.-J.; Kang, J.I.; Han, S.K.; Ha, N.Y.; Kim, G.; et al. Spread of Mutant Middle East Respiratory Syndrome Coronavirus with Reduced Affinity to Human CD26 during the South Korean Outbreak. mBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Oboho, I.K.; Tomczyk, S.M.; Al-Asmari, A.M.; Banjar, A.A.; Al-Mugti, H.; Aloraini, M.S.; Alkhaldi, K.Z.; Almohammadi, E.L.; Alraddadi, B.M.; Gerber, S.I.; et al. 2014 MERS-CoV Outbreak in Jeddah—A Link to Health Care Facilities. N. Engl. J. Med. 2015, 372, 846–854. [Google Scholar] [CrossRef] [PubMed]
- The Korean Society of Infectious Diseases; Korean Society for Healthcare-associated Infection Control and Prevention. An Unexpected Outbreak of Middle East Respiratory Syndrome Coronavirus Infection in the Republic of Korea, 2015. Infect. Chemother. 2015, 47, 120–122. [Google Scholar]
- Masters, P.S. The Molecular Biology of Coronaviruses Advances in Virus Research; Academic Press: Massachusetts, MA, USA, 2006; Volume 66, pp. 193–292. [Google Scholar]
- McBride, R.; Fielding, B.C. The Role of Severe Acute Respiratory Syndrome (SARS)-Coronavirus Accessory Proteins in Virus Pathogenesis. Viruses 2012, 4, 2902–2923. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Snijder, E.J.; Spaan, W.J.M. Severe Acute Respiratory Syndrome Coronavirus Phylogeny: Toward Consensus. J. Virol. 2004, 78, 7863–7866. [Google Scholar] [CrossRef] [PubMed]
- Kolesnikova, L.; Slenczka, W.; Brodt, H.; Klenk, H.; Becker, S. Electron microscopy in diagnostics of SARS case. Microsc. Microanal. 2003, 9, 438–439. [Google Scholar]
- Marsolais, G.; Berthiaume, L.; DiFranco, E.; Marois, P. Rapid Diagnosis by Electron Microscopy of Avian Coronavirus Infection. Can. J. Comp. Med. 1971, 35, 285–288. [Google Scholar] [PubMed]
- Liu, D.X.; Fung, T.S.; Chong, K.K.-L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Yeager, C.L.; Ashmun, R.A.; Williams, R.K.; Cardellichio, C.B.; Shapiro, L.H.; Look, A.T.; Holmes, K.V. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 1992, 357, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Sui, J.; Huang, I.C.; Kuhn, J.H.; Radoshitzky, S.R.; Marasco, W.A.; Choe, H.; Farzan, M. The S proteins of human coronavirus NL63 and severe acute respiratory syndrome coronavirus bind overlapping regions of ACE2. Virology 2007, 367, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef] [PubMed]
- Van Doremalen, N.; Miazgowicz, K.L.; Milne-Price, S.; Bushmaker, T.; Robertson, S.; Scott, D.; Kinne, J.; McLellan, J.S.; Zhu, J.; Munster, V.J. Host Species Restriction of Middle East Respiratory Syndrome Coronavirus through Its Receptor, Dipeptidyl Peptidase 4. J. Virol. 2014, 88, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dong, W.; Milewska, A.; Golda, A.; Qi, Y.; Zhu, Q.K.; Marasco, W.A.; Baric, R.S.; Sims, A.C.; Pyrc, K.; et al. Human Coronavirus HKU1 Spike Protein Uses O-Acetylated Sialic Acid as an Attachment Receptor Determinant and Employs Hemagglutinin-Esterase Protein as a Receptor-Destroying Enzyme. J. Virol. 2015, 89, 7202–7213. [Google Scholar] [CrossRef] [PubMed]
- Butler, N.; Pewe, L.; Trandem, K.; Perlman, S. Murine encephalitis caused by HCoV-OC43, a human coronavirus with broad species specificity, is partly immune-mediated. Virology 2006, 347, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.W.; Azhar, E.I.; Hui, D.C.; Yuen, K. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; Bartelink, W.; Rottier, P.J.M. Cathepsin L Functionally Cleaves the Severe Acute Respiratory Syndrome Coronavirus Class I Fusion Protein Upstream of Rather than Adjacent to the Fusion Peptide. J. Virol. 2008, 82, 8887–8890. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Dominguez, S.R.; Holmes, K.V. Role of the Spike Glycoprotein of Human Middle East Respiratory Syndrome Coronavirus (MERS-CoV) in Virus Entry and Syncytia Formation. PLoS ONE 2013, 8, e76469. [Google Scholar] [CrossRef] [PubMed]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Dijkman, R.; Habjan, M.; Heurich, A.; Gierer, S.; Glowacka, I.; Welsch, K.; Winkler, M.; Schneider, H.; Hofmann-Winkler, H.; et al. TMPRSS2 Activates the Human Coronavirus 229E for Cathepsin-Independent Host Cell Entry and Is Expressed in Viral Target Cells in the Respiratory Epithelium. J. Virol. 2013, 87, 6150–6160. [Google Scholar] [CrossRef] [PubMed]
- Bertram, S.; Glowacka, I.; Müller, M.A.; Lavender, H.; Gnirss, K.; Nehlmeier, I.; Niemeyer, D.; He, Y.; Simmons, G.; Drosten, C.; et al. Cleavage and Activation of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein by Human Airway Trypsin-Like Protease. J. Virol. 2011, 85, 13363–13372. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct Patterns of IFITM-Mediated Restriction of Filoviruses, SARS Coronavirus, and Influenza A Virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Markosyan, R.M.; Zheng, Y.-M.; Golfetto, O.; Bungart, B.; Li, M.; Ding, S.; He, Y.; Liang, C.; Lee, J.C.; et al. IFITM Proteins Restrict Viral Membrane Hemifusion. PLoS Pathog. 2013, 9, e1003124. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Chen, Q.; Chen, J.; Chen, K.; Shen, X.; Jiang, H. The nucleocapsid protein of SARS coronavirus has a high binding affinity to the human cellular heterogeneous nuclear ribonucleoprotein A1. FEBS Lett. 2005, 579, 2623–2628. [Google Scholar] [CrossRef] [PubMed]
- Nanda, S.K.; Leibowitz, J.L. Mitochondrial Aconitase Binds to the 3′ Untranslated Region of the Mouse Hepatitis Virus Genome. J. Virol. 2001, 75, 3352–3362. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-H.; Chen, P.-J.; Yeh, S.-H. Nucleocapsid Phosphorylation and RNA Helicase DDX1 Recruitment Enables Coronavirus Transition from Discontinuous to Continuous Transcription. Cell Host Microb. 2014, 16, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.W.; Hong, W.; Liu, D.X. Binding of the 5′-untranslated region of coronavirus RNA to zinc finger CCHC-type and RNA-binding motif 1 enhances viral replication and transcription. Nucleic Acids Res. 2012, 40, 5065–5077. [Google Scholar] [CrossRef] [PubMed]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wu, D.; Shen, C.; Chen, K.; Shen, X.; Jiang, H. Severe acute respiratory syndrome coronavirus membrane protein interacts with nucleocapsid protein mostly through their carboxyl termini by electrostatic attraction. Int. J. Biochem. Cell Biol. 2006, 38, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Kumar, P.; Tay, F.P.L.; Moreau, D.; Liu, D.X.; Bard, F. Genome-Wide Screen Reveals Valosin-Containing Protein Requirement for Coronavirus Exit from Endosomes. J. Virol. 2015, 89, 11116–11128. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wide-ranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of Poly(ADP-ribose) Polymerase (PARP) Cleavage in Apoptosis: Caspase 3-resistant parp mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, H.; Krammer, P.H. The CD95 (APO-1/Fas) and the TRAIL (APO-2L) Apoptosis Systems. Exp. Cell Res. 2000, 256, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Bender, L.M.; Morgan, M.J.; Thomas, L.R.; Liu, Z.G.; Thorburn, A. The adaptor protein TRADD activates distinct mechanisms of apoptosis from the nucleus and the cytoplasm. Cell Death Differ. 2005, 12, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Jürgensmeier, J.M.; Shin, H.; Deveraux, Q.; Wolf, B.B.; Yang, X.; Zhou, Q.; Ellerby, H.M.; Ellerby, L.M.; Bredesen, D.; et al. Pro-caspase-3 Is a Major Physiologic Target of Caspase-8. J. Biol. Chem. 1998, 273, 27084–27090. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Sebbagh, M.; Renvoize, C.; Hamelin, J.; Riche, N.; Bertoglio, J.; Breard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 2001, 3, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.-J.; Yuan, J. Cleavage of BID by Caspase 8 Mediates the Mitochondrial Damage in the Fas Pathway of Apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Benedict, C.A.; Norris, P.S.; Ware, C.F. To kill or be killed: Viral evasion of apoptosis. Nat. Immunol. 2002, 3, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. Structural biology of the Bcl-2 family and its mimicry by viral proteins. Cell Death Dis. 2013, 4, e909. [Google Scholar] [CrossRef] [PubMed]
- Pearce, A.F.; Lyles, D.S. Vesicular Stomatitis Virus Induces Apoptosis Primarily through Bak Rather than Bax by Inactivating Mcl-1 and Bcl-XL. J. Virol. 2009, 83, 9102–9112. [Google Scholar] [CrossRef] [PubMed]
- Aillet, F.; Masutani, H.; Elbim, C.; Raoul, H.; Chêne, L.; Nugeyre, M.T.; Paya, C.; Barré-Sinoussi, F.; Gougerot-Pocidalo, M.A.; Israël, N. Human immunodeficiency virus induces a dual regulation of Bcl-2, resulting in persistent infection of CD4(+) T- or monocytic cell lines. J. Virol. 1998, 72, 9698–9705. [Google Scholar] [PubMed]
- Tamura, R.; Kanda, T.; Imazeki, F.; Wu, S.; Nakamoto, S.; Tanaka, T.; Arai, M.; Fujiwara, K.; Saito, K.; Roger, T.; et al. Hepatitis C Virus Nonstructural 5A Protein Inhibits Lipopolysaccharide-Mediated Apoptosis of Hepatocytes by Decreasing Expression of Toll-Like Receptor 4. J. Infect. Dis. 2011, 204, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Aweya, J.J.; Sze, C.W.; Bayega, A.; Mohd-Ismail, N.K.; Deng, L.; Hotta, H.; Tan, Y.J. NS5B induces up-regulation of the BH3-only protein, BIK, essential for the hepatitis C virus RNA replication and viral release. Virology 2015, 474, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Lopez, Y.; Villegas-Sepúlveda, N.; Gómez, B. RSV P-protein impairs extrinsic apoptosis pathway in a macrophage-like cell line persistently infected with respiratory syncytial virus. Virus Res. 2015, 204, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Mocarski, E.S.; Upton, J.W.; Kaiser, W.J. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat. Rev. Immunol. 2012, 12, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. Induction of Apoptosis in MRC-5, Diploid Human Fetal Lung Cells after Infection with Human Coronavirus OC43. In The Nidoviruses: Coronaviruses and Arteriviruses; Lavi, E., Weiss, S.R., Hingley, S.T., Eds.; Springer US: Boston, MA, USA, 2001; pp. 677–682. [Google Scholar]
- Pyrc, K.; Berkhout, B.; van der Hoek, L. The Novel Human Coronaviruses NL63 and HKU1. J. Virol. 2007, 81, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Korteweg, C. Pathology and Pathogenesis of Severe Acute Respiratory Syndrome. Am. J. Pathol. 2007, 170, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Hill, T.E.; Morimoto, C.; Peters, C.J.; Ksiazek, T.G.; Tseng, C.-T.K. Bilateral Entry and Release of Middle East Respiratory Syndrome Coronavirus Induces Profound Apoptosis of Human Bronchial Epithelial Cells. J. Virol. 2013, 87, 9953–9958. [Google Scholar] [CrossRef] [PubMed]
- Yeung, M.-L.; Yao, Y.; Jia, L.; Chan, J.F.W.; Chan, K.-H.; Cheung, K.-F.; Chen, H.; Poon, V.K.M.; Tsang, A.K.L.; To, K.K.W.; et al. MERS coronavirus induces apoptosis in kidney and lung by upregulating Smad7 and FGF2. Nat. Microbiol. 2016, 1, 16004. [Google Scholar] [CrossRef]
- Desforges, M.; Coupanec, A.; Brison, É.; Meessen-Pinard, M.; Talbot, P.J. Neuroinvasive and Neurotropic Human Respiratory Coronaviruses: Potential Neurovirulent Agents in Humans. In Proceedings of the Infectious Diseases and Nanomedicine I: First International Conference (ICIDN-2012), Kathmandu, Nepal, 15–18 December 2012; Adhikari, R., Thapa, S., Eds.; Springer: New Delhi, India, 2014; pp. 75–96. [Google Scholar]
- Desforges, M.; Le Coupanec, A.; Stodola, J.K.; Meessen-Pinard, M.; Talbot, P.J. Human coronaviruses: Viral and cellular factors involved in neuroinvasiveness and neuropathogenesis. Virus Res. 2014, 194, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Favreau, D.J.; Meessen-Pinard, M.; Desforges, M.; Talbot, P.J. Human Coronavirus-Induced Neuronal Programmed Cell Death Is Cyclophilin D Dependent and Potentially Caspase Dispensable. J. Virol. 2012, 86, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Krähling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Mühlberger, E. Severe Acute Respiratory Syndrome Coronavirus Triggers Apoptosis via Protein Kinase R but Is Resistant to Its Antiviral Activity. J. Virol. 2009, 83, 2298–2309. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. In Vitro Detection of Apoptosis in Monocytes/Macrophages Infected with Human Coronavirus. Clin. Diagn. Lab. Immunol. 2002, 9, 1392–1395. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xiong, Z.; Zhang, S.; Yan, Y.; Nguyen, J.; Ng, B.; Lu, H.; Brendese, J.; Yang, F.; Wang, H.; et al. Bcl-xL inhibits T-cell apoptosis induced by expression of SARS coronavirus E protein in the absence of growth factors. Biochem. J. 2005, 392, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Ying, T.; Li, W.; Dimitrov, D.S. Discovery of T-Cell Infection and Apoptosis by Middle East Respiratory Syndrome Coronavirus. J. Infect. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Zhou, J.; Wong, B.H.-Y.; Li, C.; Chan, J.F.-W.; Cheng, Z.-S.; Yang, D.; Wang, D.; Lee, A.C.; Li, C.; et al. Middle East Respiratory Syndrome Coronavirus Efficiently Infects Human Primary T Lymphocytes and Activates the Extrinsic and Intrinsic Apoptosis Pathways. J. Infect. Dis. 2016, 213, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Mesel-Lemoine, M.; Millet, J.; Vidalain, P.-O.; Law, H.; Vabret, A.; Lorin, V.; Escriou, N.; Albert, M.L.; Nal, B.; Tangy, F. A Human Coronavirus Responsible for the Common Cold Massively Kills Dendritic Cells but Not Monocytes. J. Virol. 2012, 86, 7577–7587. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, M.; Schneider, K.; Weber, F.; Weidmann, M.; Hufert, F. Interaction of severe acute respiratory syndrome-associated coronavirus with dendritic cells. J. Gen. Virol. 2006, 87, 1953–1960. [Google Scholar] [CrossRef] [PubMed]
- Bordi, L.; Castilletti, C.; Falasca, L.; Ciccosanti, F.; Calcaterra, S.; Rozera, G.; Di Caro, A.; Zaniratti, S.; Rinaldi, A.; Ippolito, G.; et al. Bcl-2 inhibits the caspase-dependent apoptosis induced by SARS-CoV without affecting virus replication kinetics. Arch. Virol. 2005, 151, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Yang, R.; Guo, L.; Qu, J.; Wang, J.; Hung, T. Apoptosis Induced by the SARS-Associated Coronavirus in Vero Cells Is Replication-Dependent and Involves Caspase. DNA Cell Biol. 2005, 24, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.S.F.; Chan, K.-h.; Cheng, V.C.C.; Woo, P.C.Y.; Lau, S.K.P.; Lam, C.C.K.; Chan, T.L.; Wu, A.K.; Hung, I.F.; Leung, S.Y.; et al. Comparative Host Gene Transcription by Microarray Analysis Early after Infection of the Huh7 Cell Line by Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus 229E. J. Virol. 2005, 79, 6180–6193. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.Y.C.; Yeung, Y.S.; Hon, C.C.; Zeng, F.; Law, K.M.; Leung, F.C.C. Adenovirus-mediated expression of the C-terminal domain of SARS-CoV spike protein is sufficient to induce apoptosis in Vero E6 cells. FEBS Lett. 2005, 579, 6699–6704. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Liu, B.; Jameel, S.; Chow, V.T.K.; Lal, S.K. The SARS coronavirus nucleocapsid protein induces actin reorganization and apoptosis in COS-1 cells in the absence of growth factors. Biochem. J. 2004, 383, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-J.; Fielding, B.C.; Goh, P.-Y.; Shen, S.; Tan, T.H.P.; Lim, S.G.; Hong, W. Overexpression of 7a, a Protein Specifically Encoded by the Severe Acute Respiratory Syndrome Coronavirus, Induces Apoptosis via a Caspase-Dependent Pathway. J. Virol. 2004, 78, 14043–14047. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wong, C.K.; Li, P.; Xie, Y. A SARS-CoV protein, ORF-6, induces caspase-3 mediated, ER stress and JNK-dependent apoptosis. Biochim. Biophys. Acta 2008, 1780, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Åkerström, S.; Sharma, A.K.; Chow, V.T.K.; Teow, S.; Abrenica, B.; Booth, S.A.; Booth, T.F.; Mirazimi, A.; Lal, S.K. SARS-CoV 9b Protein Diffuses into Nucleus, Undergoes Active Crm1 Mediated Nucleocytoplasmic Export and Triggers Apoptosis When Retained in the Nucleus. PLoS ONE 2011, 6, e19436. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, H.; Li, L.; Chen, Zhefan S.; Lau, K.-F.; Tsui, S.K.W.; Chan, H.Y.E. The SARS-coronavirus membrane protein induces apoptosis via interfering with PDK1-PKB/Akt signalling. Biochem. J. 2014, 464, 439–447. [Google Scholar] [CrossRef] [PubMed]
- De Diego, M.L.; Nieto-Torres, J.L.; JimÈnez-GuardeÒo, J.M.; Regla-Nava, J.A.; Álvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Regulates Cell Stress Response and Apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar]
- Tan, Y.-X.; Tan, T.H.P.; Lee, M.J.R.; Tham, P.-Y.; Gunalan, V.; Druce, J.; Birch, C.; Catton, M.; Fu, N.Y.; Yu, V.C.; et al. Induction of Apoptosis by the Severe Acute Respiratory Syndrome Coronavirus 7a Protein Is Dependent on Its Interaction with the Bcl-X(L) Protein. J. Virol. 2007, 81, 6346–6355. [Google Scholar] [CrossRef] [PubMed]
- Favreau, D.J.; Desforges, M.; St-Jean, J.R.; Talbot, P.J. A human coronavirus OC43 variant harboring persistence-associated mutations in the S glycoprotein differentially induces the unfolded protein response in human neurons as compared to wild-type virus. Virology 2009, 395, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Diemer, C.; Schneider, M.; Seebach, J.; Quaas, J.; Frösner, G.; Schätzl, H.M.; Gilch, S. Cell Type-Specific Cleavage of Nucleocapsid Protein by Effector Caspases during SARS Coronavirus Infection. J. Mol. Biol. 2008, 376, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Alcami, A.; Koszinowski, U.H. Viral mechanisms of immune evasion. Immunol. Today 2000, 21, 447–455. [Google Scholar] [CrossRef]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Thomsen, A.R. Sensing of RNA Viruses: A Review of Innate Immune Receptors Involved in Recognizing RNA virus Invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.Y.; Kok, K.-H.; Jaume, M.; Cheung, T.K.W.; Yip, T.-F.; Lai, J.C.C.; Guan, Y.; Webster, R.G.; Jin, D.Y.; Peiris, J.S. Toll-like receptor 10 is involved in induction of innate immune responses to influenza virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 3793–3798. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-Stranded RNA Is Produced by Positive-Strand RNA Viruses and DNA Viruses but Not in Detectable Amounts by Negative-Strand RNA Viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [PubMed]
- Krug, A.; Luker, G.D.; Barchet, W.; Leib, D.A.; Akira, S.; Colonna, M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 2003, 103, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.; French, A.R.; Barchet, W.; Fischer, J.A.A.; Dzionek, A.; Pingel, J.T.; Orihuela, M.M.; Akira, S.; Yokoyama, W.M.; Colonna, M. TLR9-Dependent Recognition of MCMV by IPC and DC Generates Coordinated Cytokine Responses that Activate Antiviral NK Cell Function. Immunity 2004, 21, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Zeisel, M.B.; Jilg, N.; Paranhos-Baccalà, G.; Stoll-Keller, F.; Wakita, T.; Hafkemeyer, P.; Blum, H.E.; Barth, H.; Henneke, P.; et al. Toll-like receptor 2 senses hepatitis C virus core protein but not infectious viral particles. J. Innate Immunity 2009, 1, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Lien, E.; Klagge, I.M.; Avota, E.; Schneider-Schaulies, J.; Duprex, W.P.; Wagner, H.; Kirschning, C.J.; Ter Meulen, V.; Schneider-Schaulies, S. Hemagglutinin Protein of Wild-Type Measles Virus Activates Toll-Like Receptor 2 Signaling. J. Virol. 2002, 76, 8729–8736. [Google Scholar] [CrossRef] [PubMed]
- Rallabhandi, P.; Phillips, R.L.; Boukhvalova, M.S.; Pletneva, L.M.; Shirey, K.A.; Gioannini, T.L.; Weiss, J.P.; Chow, J.C.; Hawkins, L.D.; Vogel, S.N.; et al. Respiratory Syncytial Virus Fusion Protein-Induced Toll-Like Receptor 4 (TLR4) Signaling Is Inhibited by the TLR4 Antagonists Rhodobacter sphaeroides Lipopolysaccharide and Eritoran (E5564) and Requires Direct Interaction with MD-2. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Ben Haij, N.; Leghmari, K.; Planès, R.; Thieblemont, N.; Bahraoui, E. HIV-1 Tat protein binds to TLR4-MD2 and signals to induce TNF-α and IL-10. Retrovirology 2013, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-signaling Networks: An Integration of Adaptor Molecules, Kinases, and Cross-talk. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors [mdash] redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and Unique Functions of the DExD/H-Box Helicases RIG-I, MDA5, and LGP2 in Antiviral Innate Immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.R.; Bruns, A.M.; Horvath, C.M. MDA5 and LGP2: Accomplices and Antagonists of Antiviral Signal Transduction. J. Virol. 2014, 88, 8194–8200. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Kato, H.; Kumagai, Y.; Yoneyama, M.; Sato, S.; Matsushita, K.; Tsujimura, T.; Fujita, T.; Akira, S.; Takeuchi, O. LGP2 is a positive regulator of RIG-I- and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. USA 2010, 107, 1512–1517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Zhang, X.; Wang, G.; Zheng, H. The Laboratory of Genetics and Physiology 2: Emerging Insights into the Controversial Functions of This RIG-I-Like Receptor. BioMed Res. Int. 2014, 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, H.-S.; Pyo, H.-M.; Liu, Q.; Zhou, Y. Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J. Virol. 2015, 89, 6067–6079. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U. Mechanisms of RIG-I-like Receptor Activation and Manipulation by Viral Pathogens. J. Virol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.-D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Kindler, E.; Jónsdóttir, H.R.; Muth, D.; Hamming, O.J.; Hartmann, R.; Rodriguez, R.; Geffers, R.; Fouchier, R.A.; Drosten, C.; Müller, M.A.; et al. Efficient Replication of the Novel Human Betacoronavirus EMC on Primary Human Epithelium Highlights Its Zoonotic Potential. mBio 2013, 4, e00611–e00612. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Heise, M.; Baric, R. SARS coronavirus and innate immunity. Virus Res. 2008, 133, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Clementz, M.A.; Chen, Z.; Banach, B.S.; Wang, Y.; Sun, L.; Ratia, K.; Baez-Santos, Y.M.; Wang, J.; Takayama, J.; Ghosh, A.K.; et al. Deubiquitinating and Interferon Antagonism Activities of Coronavirus Papain-Like Proteases. J. Virol. 2010, 84, 4619–4629. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.-H.; Li, C.P.Y.; Chen, H.; Jin, D.-Y.; Chan, J.F.; Woo, P.C.; Yuen, K.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.; Poon, L.L.M.; Ng, I.H.Y.; Luk, W.; Sia, S.-F.; Wu, M.H.S.; Chan, K.H.; Yuen, K.Y.; Gordon, S.; Guan, Y.; et al. Cytokine Responses in Severe Acute Respiratory Syndrome Coronavirus-Infected Macrophages In Vitro: Possible Relevance to Pathogenesis. J. Virol. 2005, 79, 7819–7826. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.J.; Wang, J.; Ito, Y.; Travanty, E.A.; Voelker, D.R.; Holmes, K.V.; Mason, R.J. Infection of human alveolar macrophages by human coronavirus strain 229E. J. Gen. Virol. 2012, 93, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA Replication of Mouse Hepatitis Virus Takes Place at Double-Membrane Vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S.; Netland, J. Coronaviruses post-SARS: Update on replication and pathogenesis. Nature reviews. Microbiology 2009, 7, 439–450. [Google Scholar] [PubMed]
- Siu, K.L.; Kok, K.H.; Ng, M.H.; Poon, V.K.; Yuen, K.Y.; Zheng, B.J.; Jin, D.Y. Severe Acute Respiratory Syndrome Coronavirus M Protein Inhibits Type I Interferon Production by Impeding the Formation of TRAF3·TANK·TBK1/IKKε Complex. J. Biol. Chem. 2009, 284, 16202–16209. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.-L.; Chan, C.-P.; Kok, K.-H.; Chiu-Yat Woo, P.; Jin, D.-Y. Suppression of innate antiviral response by severe acute respiratory syndrome coronavirus M protein is mediated through the first transmembrane domain. Cell. Mol. Immunol. 2014, 11, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, L.; Geng, H.; Deng, Y.; Huang, B.; Guo, Y.; Zhao, Z.; Tan, W. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 2013, 4, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe Acute Respiratory Syndrome Coronavirus Open Reading Frame (ORF) 3b, ORF 6, and Nucleocapsid Proteins Function as Interferon Antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Pan, J.A.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2010, 42, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Lokugamage, K.G.; Rozovics, J.M.; Narayanan, K.; Semler, B.L.; Makino, S. SARS Coronavirus nsp1 Protein Induces Template-Dependent Endonucleolytic Cleavage of mRNAs: Viral mRNAs Are Resistant to nsp1-Induced RNA Cleavage. PLoS Pathog. 2011, 7, e1002433. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Narayanan, K.; Nakagawa, K.; Terasaki, K.; Ramirez, S.I.; Tseng, C.-T.K.; Ramirez, S.I.; Tseng, C.T.; Makino, S. Middle East respiratory syndrome coronavirus nsp1 inhibits host gene expression by selectively targeting nuclear-transcribed mRNAs but spares mRNAs of cytoplasmic origin. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Jauregui, A.R.; Savalia, D.; Lowry, V.K.; Farrell, C.M.; Wathelet, M.G. Identification of Residues of SARS-CoV nsp1 That Differentially Affect Inhibition of Gene Expression and Antiviral Signaling. PLoS ONE 2013, 8, e62416. [Google Scholar] [CrossRef] [PubMed]
- Mielech, A.M.; Kilianski, A.; Baez-Santos, Y.M.; Mesecar, A.D.; Baker, S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 2014, 450–451, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; et al. Regulation of IRF-3 dependent innate immunity by the papain-like protease domain of the sars coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, X.; Bian, G.; Tu, J.; Xing, Y.; Wang, Y.; Chen, Z. Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 2014, 95, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Kuri, T.; Eriksson, K.K.; Putics, A.; Züst, R.; Snijder, E.J.; Davidson, A.D.; Siddell, S.G.; Thiel, V.; Ziebuhr, J.; Weber, F. The ADP-ribose-1′′-monophosphatase domains of severe acute respiratory syndrome coronavirus and human coronavirus 229E mediate resistance to antiviral interferon responses. J. Gen. Virol. 2011, 92, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Stevens, F.J.; Argon, Y. Protein folding in the ER. Semin. Cell Dev. Biol. 1999, 10, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Coronavirus infection, ER stress, apoptosis and innate immunity. Front. Microbiol. 2014, 5, 296. [Google Scholar] [CrossRef] [PubMed]
- Teske, B.F.; Wek, S.A.; Bunpo, P.; Cundiff, J.K.; McClintick, J.N.; Anthony, T.G.; Wek, R.C. The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol. Biol. Cell 2011, 22, 4390–4405. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP//GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2003, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Huang, M.; Liu, D.X. Coronavirus-induced ER stress response and its involvement in regulation of coronavirus-host interactions. Virus Res. 2014, 194, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Padhan, K.; Rani, M.; Khan, N.; Ahmad, F.; Jameel, S. The SARS Coronavirus 3a Protein Causes Endoplasmic Reticulum Stress and Induces Ligand-Independent Downregulation of the Type 1 Interferon Receptor. PLoS ONE 2009, 4, e8342. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.-P.; Siu, K.-L.; Chin, K.-T.; Yuen, K.-Y.; Zheng, B.; Jin, D.-Y. Modulation of the Unfolded Protein Response by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.-L.; Chan, C.-P.; Kok, K.-H.; C-Y Woo, P.; Jin, D.-Y. Comparative analysis of the activation of unfolded protein response by spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus HKU1. Cell Biosci. 2014, 4, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liao, Y.; Yap, P.L.; Png, K.J.; Tam, J.P.; Liu, D.X. Inhibition of Protein Kinase R Activation and Upregulation of GADD34 Expression Play a Synergistic Role in Facilitating Coronavirus Replication by Maintaining De Novo Protein Synthesis in Virus-Infected Cells. J. Virol. 2009, 83, 12462–12472. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Luo, S.; Baumeister, P.; Huang, J.-M.; Gogia, R.K.; Li, M.; Lee, A.S. Underglycosylation of ATF6 as a Novel Sensing Mechanism for Activation of the Unfolded Protein Response. J. Biol. Chem. 2004, 279, 11354–11363. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of Disulfide Bridges Formed in the Luminal Domain of ATF6 in Sensing Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Sung, S.-C.; Chao, C.-Y.; Jeng, K.-S.; Yang, J.-Y.; Lai, M.M.C. The 8ab protein of SARS-CoV is a luminal ER membrane-associated protein and induces the activation of ATF6. Virology 2009, 387, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Oostra, M.; de Haan, C.A.M.; Rottier, P.J.M. The 29-Nucleotide Deletion Present in Human but Not in Animal Severe Acute Respiratory Syndrome Coronaviruses Disrupts the Functional Expression of Open Reading Frame 8. J. Virol. 2007, 81, 13876–13888. [Google Scholar] [CrossRef] [PubMed]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Signalling Pathways in the Unfolded Protein Response: Development from Yeast to Mammals. J. Biochem. 2009, 146, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Credle, J.J.; Finer-Moore, J.S.; Papa, F.R.; Stroud, R.M.; Walter, P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2005, 102, 18773–18784. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded Proteins Are Ire1-Activating Ligands That Directly Induce the Unfolded Protein Response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed]
- Prischi, F.; Nowak, P.R.; Carrara, M.; Ali, M.M.U. Phosphoregulation of Ire1 RNase splicing activity. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Lee, S.-M.; Chen, A.; Zhang, J.; Zhang, D.D.; Kannan, K.; Ortmann, R.A.; Fang, D. Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen-induced arthritis. EMBO Rep. 2008, 9, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 Has Distinct Catalytic Mechanisms for XBP1/HAC1 Splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar]
- Walter, F.; Schmid, J.; Dussmann, H.; Concannon, C.G.; Prehn, J.H.M. Imaging of single cell responses to ER stress indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4 signalling rather than a switch between signalling branches determine cell survival. Cell Death Differ. 2015, 22, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, G.A.; van de Nes, P.S.; Bredenbeek, P.J.; Spaan, W.J.M. The Coronavirus Spike Protein Induces Endoplasmic Reticulum Stress and Upregulation of Intracellular Chemokine mRNA Concentrations. J. Virol. 2007, 81, 10981–10990. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liao, Y.; Liu, D.X. The Endoplasmic Reticulum Stress Sensor IRE1α Protects Cells from Apoptosis Induced by the Coronavirus Infectious Bronchitis Virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 0000, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.J. Signal Transduction to the Nucleus by MAP Kinase. In Signaling Networks and Cell Cycle Control: The Molecular Basis of Cancer and Other Diseases; Gutkind, J.S., Ed.; Humana Press: Totowa, NJ, USA, 2000; pp. 153–164. [Google Scholar]
- Panteva, M.; Korkaya, H.; Jameel, S. Hepatitis viruses and the MAPK pathway: Is this a survival strategy? Virus Res. 2003, 92, 131–140. [Google Scholar] [CrossRef]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Keshet, Y.; Seger, R. The MAP Kinase Signaling Cascades: A System of Hundreds of Components Regulates a Diverse Array of Physiological Functions. In MAP Kinase Signaling Protocols, 2nd ed.; Peger, R., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 3–38. [Google Scholar]
- Mizutani, T.; Fukushi, S.; Murakami, M.; Hirano, T.; Saijo, M.; Kurane, I.; Morikawa, S. Tyrosine dephosphorylation of STAT3 in SARS coronavirus-infected Vero E6 cells. FEBS Lett. 2004, 577, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Phosphorylation of p38 MAPK and its downstream targets in SARS coronavirus-infected cells. Biochem. Biophys. Res. Commun. 2004, 319, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Chang, S.C.; Wu, H.-Y.; Yu, T.-C.; Wei, W.-C.; Lin, S.; Chien, C.L.; Chang, M.F. Upregulation of the Chemokine (C-C Motif) Ligand 2 via a Severe Acute Respiratory Syndrome Coronavirus Spike-ACE2 Signaling Pathway. J. Virol. 2010, 84, 7703–7712. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. Regulation of p90RSK phosphorylation by SARS-CoV infection in Vero E6 cells. FEBS Lett. 2006, 580, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Li, W.; Wu, J.; Germann, U.A.; Su, M.S.; Kuida, K.; Boucher, D.M. Extracellular Signal-Regulated Kinase 2 Is Necessary for Mesoderm Differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 12759–12764. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.T.-C.; Chien, S.-C.; Chen, I.Y.; Lai, C.-T.; Tsay, Y.-G.; Chang, S.C.; Chang, M.F. Surface vimentin is critical for the cell entry of SARS-CoV. J. Biomed.Sci. 2016, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Robidoux, J.; Daniel, K.W.; Guzman, G.; Floering, L.M.; Collins, S. Requirement of Vimentin Filament Assembly for β3-Adrenergic Receptor Activation of ERK MAP Kinase and Lipolysis. J. Biol. Chem. 2007, 282, 9244–9250. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Liu, C.Y.-Y.; Chiang, B.-L.; Chao, Y.-C.; Chen, C.-C. Induction of IL-8 Release in Lung Cells via Activator Protein-1 by Recombinant Baculovirus Displaying Severe Acute Respiratory Syndrome-Coronavirus Spike Proteins: Identification of Two Functional Regions. J. Immunol. 2004, 173, 7602–7614. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-W.; Lai, C.-C.; Ping, J.-F.; Tsai, F.-J.; Wan, L.; Lin, Y.-J.; Kung, S.H.; Lin, C.W. Severe acute respiratory syndrome coronavirus papain-like protease suppressed alpha interferon-induced responses through downregulation of extracellular signal-regulated kinase 1-mediated signalling pathways. J. Gen. Virol. 2011, 92, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Varshney, B.; Lal, S. SARS-CoV Accessory Protein 3b Induces AP-1 Transcriptional Activity through Activation of JNK and ERK Pathways. Biochemistry 2011, 50, 5419–5425. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral Potential of ERK/MAPK and PI3K/AKT/mTOR Signaling Modulation for Middle East Respiratory Syndrome Coronavirus Infection as Identified by Temporal Kinome Analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Tatsumi, K.; Imai, A.M.; Saito, K.; Kuriyama, T.; Shirasawa, H. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: Involvement of p38 MAPK and ERK. Antivir. Res. 2008, 77, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, T.; Fukushi, S.; Saijo, M.; Kurane, I.; Morikawa, S. JNK and PI3k/Akt signaling pathways are required for establishing persistent SARS-CoV infection in Vero E6 cells. Biochim. Biophys. Acta 2005, 1741, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Kanzawa, N.; Nishigaki, K.; Hayashi, T.; Ishii, Y.; Furukawa, S.; Niiro, A.; Yasui, F.; Kohara, M.; Morita, K.; Matsushima, K.; et al. Augmentation of chemokine production by severe acute respiratory syndrome coronavirus 3a/X1 and 7a/X4 proteins through NF-κB activation. FEBS Lett. 2006, 580, 6807–6812. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.; Gu, C.; Yue, Y.; Wu, K.K.; Wu, J.; Zhu, Y. Spike protein of SARS-CoV stimulates cyclooxygenase-2 expression via both calcium-dependent and calcium-independent protein kinase C pathways. FASEB J. 2007, 21, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S. Molecular Characterization of Cellular Stress Responses during Coronavirus Infection. Ph.D. Thesis, Nanyang Technological University, Singapore, Singapore, 2015. [Google Scholar]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a Protein of Severe Acute Respiratory Syndrome Coronavirus Inhibits Cellular Protein Synthesis and Activates p38 Mitogen-Activated Protein Kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.-J.; Teng, E.; Shen, S.; Tan, T.H.P.; Goh, P.-Y.; Fielding, B.C.; Ooi, E.E.; Tan, H.C.; Lim, S.G.; Hong, W. A Novel Severe Acute Respiratory Syndrome Coronavirus Protein, U274, Is Transported to the Cell Surface and Undergoes Endocytosis. J. Virol. 2004, 78, 6723–6734. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Guardeño, J.M.; Nieto-Torres, J.L.; DeDiego, M.L.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Enjuanes, L. The PDZ-Binding Motif of Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Is a Determinant of Viral Pathogenesis. PLoS Pathog. 2014, 10, e1004320. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Chen, R.-F.; Liu, J.-W.; Yeh, W.-T.; Chang, J.-C.; Liu, P.-M.; Eng, H.L.; Lin, M.C.; Yang, K.D. Altered p38 Mitogen-Activated Protein Kinase Expression in Different Leukocytes with Increment of Immunosuppressive Mediators in Patients with Severe Acute Respiratory Syndrome. J. Immunol. 2004, 172, 7841–7847. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.; Rossi, A.; Amici, C. NEW EMBO MEMBER’S REVIEW: NF-κB and virus infection: Who controls whom. EMBO J. 2003, 22, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hiscott, J.; Kwon, H.; Génin, P. Hostile takeovers: Viral appropriation of the NF-κB pathway. J. Clin. Investig. 2001, 107, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- De Diego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeño, J.M.; Fernandez-Delgado, R.; Fett, C.; Castaño-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-κB-Mediated Inflammation in Severe Acute Respiratory Syndrome Coronavirus-Infected Mice Increases Survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS Coronavirus Spike Protein-Induced Innate Immune Response occurs via Activation of the NF-κB pathway in Human Monocyte Macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.-J.; Ye, L.-B.; Timani, K.A.; Zeng, Y.-C.; She, Y.-L.; Ye, L.; Wu, Z.H. Activation of NF-κB by the Full-length Nucleocapsid Protein of the SARS Coronavirus. Acta Biochim. Biophys. Sin. 2005, 37, 607–612. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Leeson, A.; Andonov, A.; Li, Y.; Bastien, N.; Cao, J.; Osiowy, C.; Dobie, F.; Cutts, T.; Ballantine, M.; et al. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 2003, 311, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Gao, J.; Zheng, H.; Li, B.; Kong, L.; Zhang, Y.; Wang, W.; Zeng, Y.; Ye, L. The membrane protein of SARS-CoV suppresses NF-κB activation. J. Med. Virol. 2007, 79, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.W.; Stephenson, K.B.; Mahony, J.; Lichty, B.D. Human Coronavirus OC43 Nucleocapsid Protein Binds MicroRNA 9 and Potentiates NF-κB Activation. J. Virol. 2014, 88, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Law, A.H.Y.; Lee, D.C.W.; Cheung, B.K.W.; Yim, H.C.H.; Lau, A.S.Y. Role for Nonstructural Protein 1 of Severe Acute Respiratory Syndrome Coronavirus in Chemokine Dysregulation. J. Virol. 2007, 81, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe Acute Respiratory Syndrome Coronavirus Papain-Like Protease Ubiquitin-Like Domain and Catalytic Domain Regulate Antagonism of IRF3 and NF-κB Signaling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef] [PubMed]
- Ratia, K.; Kilianski, A.; Baez-Santos, Y.M.; Baker, S.C.; Mesecar, A. Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating Activity of SARS-CoV Papain-Like Protease. PLoS Pathog. 2014, 10, e1004113. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.L.; Coleman, C.M.; van der Meer, Y.; Snijder, E.J.; Frieman, M.B. The ORF4b-encoded accessory proteins of Middle East respiratory syndrome coronavirus and two related bat coronaviruses localize to the nucleus and inhibit innate immune signalling. J. Gen. Virol. 2014, 95, 874–882. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coronaviriniae Genera | Strains | Discovery | Cellular Receptor | Host | References |
|---|---|---|---|---|---|
| Alpha-coronavirus | HCoV-229E | 1966 | Human Aminopeptidase N (CD13) | Bats | [1,2,21] |
| HCoV-NL63 | 2004 | ACE2 | Palm Civets, Bats | [3,21] | |
| Beta-coronavirus | HCoV-OC43 | 1967 | 9-O-Acetylated sialic acid | Cattle | [4,5] |
| HcoV-HKU1 | 2005 | 9-O-Acetylated sialic acid | Mice | [6,7] | |
| SARS-CoV | 2003 | ACE2 | Palm Civets, Bats | [8,19,21] | |
| MERS-CoV | 2012 | DPP4 | Bats, Camels | [9] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, Y.X.; Ng, Y.L.; Tam, J.P.; Liu, D.X. Human Coronaviruses: A Review of Virus–Host Interactions. Diseases 2016, 4, 26. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4030026
Lim YX, Ng YL, Tam JP, Liu DX. Human Coronaviruses: A Review of Virus–Host Interactions. Diseases. 2016; 4(3):26. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4030026
Chicago/Turabian StyleLim, Yvonne Xinyi, Yan Ling Ng, James P. Tam, and Ding Xiang Liu. 2016. "Human Coronaviruses: A Review of Virus–Host Interactions" Diseases 4, no. 3: 26. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4030026