
Integrative Systems Biology Investigation of Fabry Disease


Abstract
:
1. Introduction
2. Materials and Methods
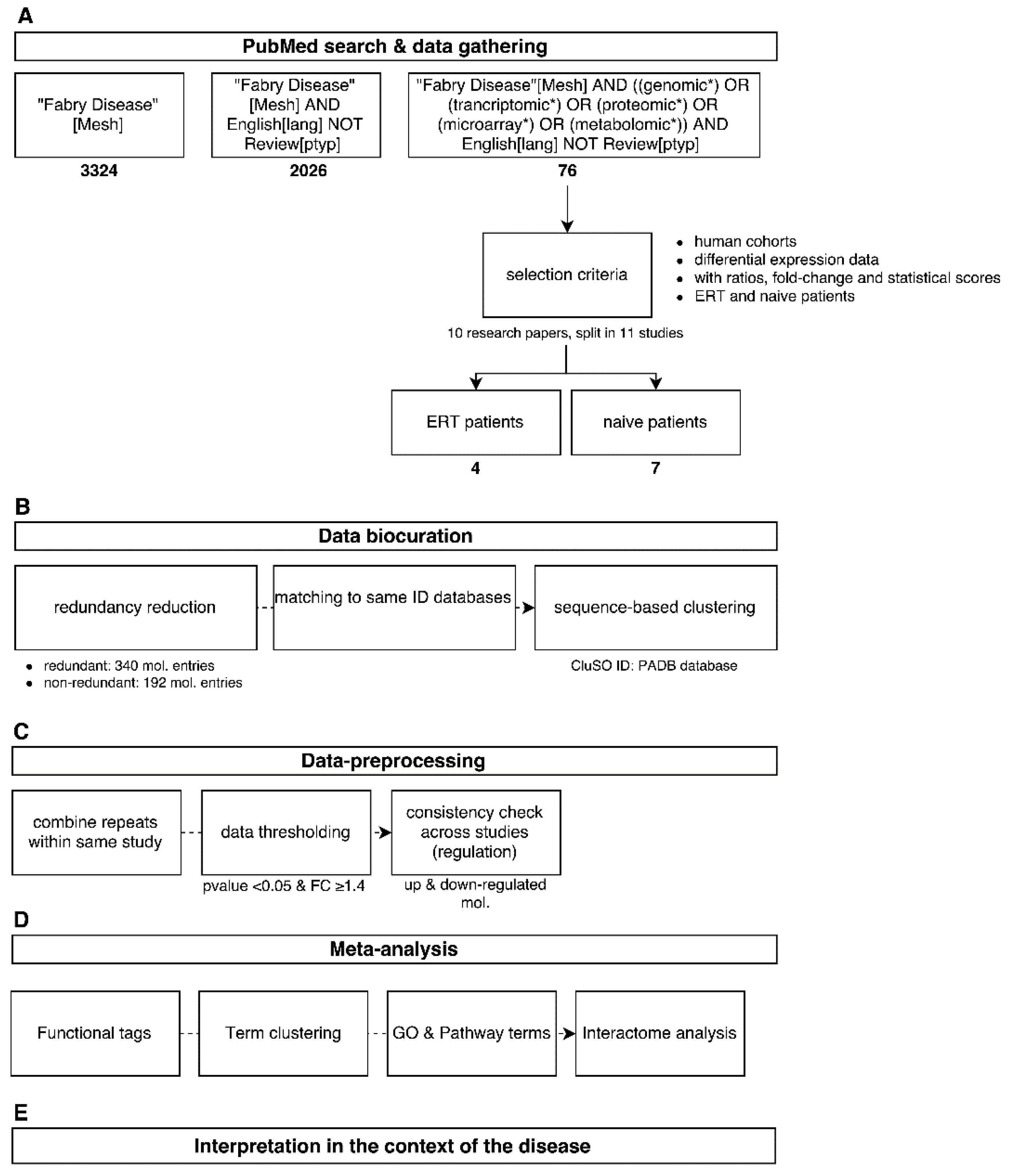
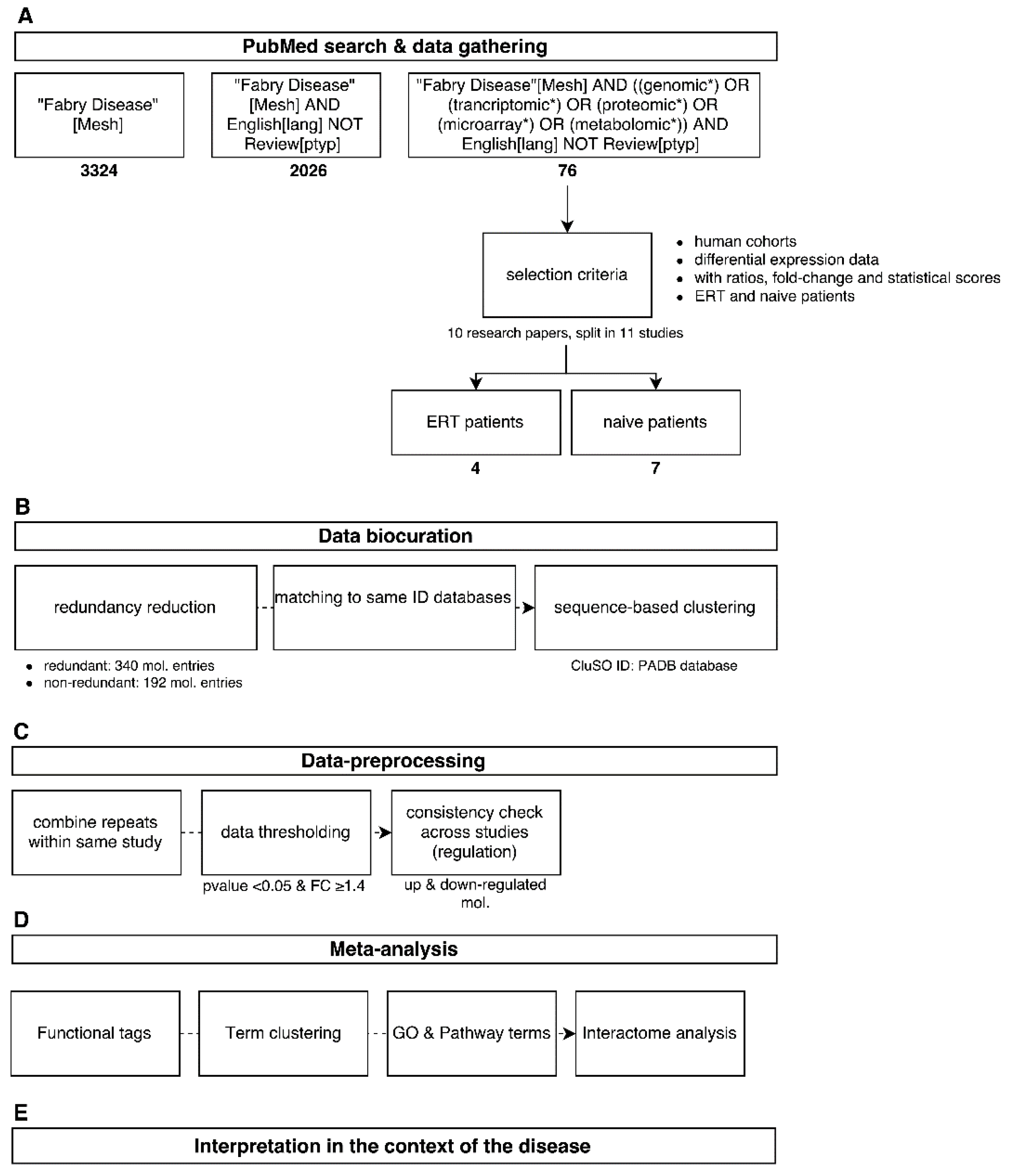
2.1. Data Mining
2.2. Data Biocuration
2.3. Data Pre-Processing
2.4. Meta-Analysis
2.5. Interpretation in the Context of the Disease
3. Results
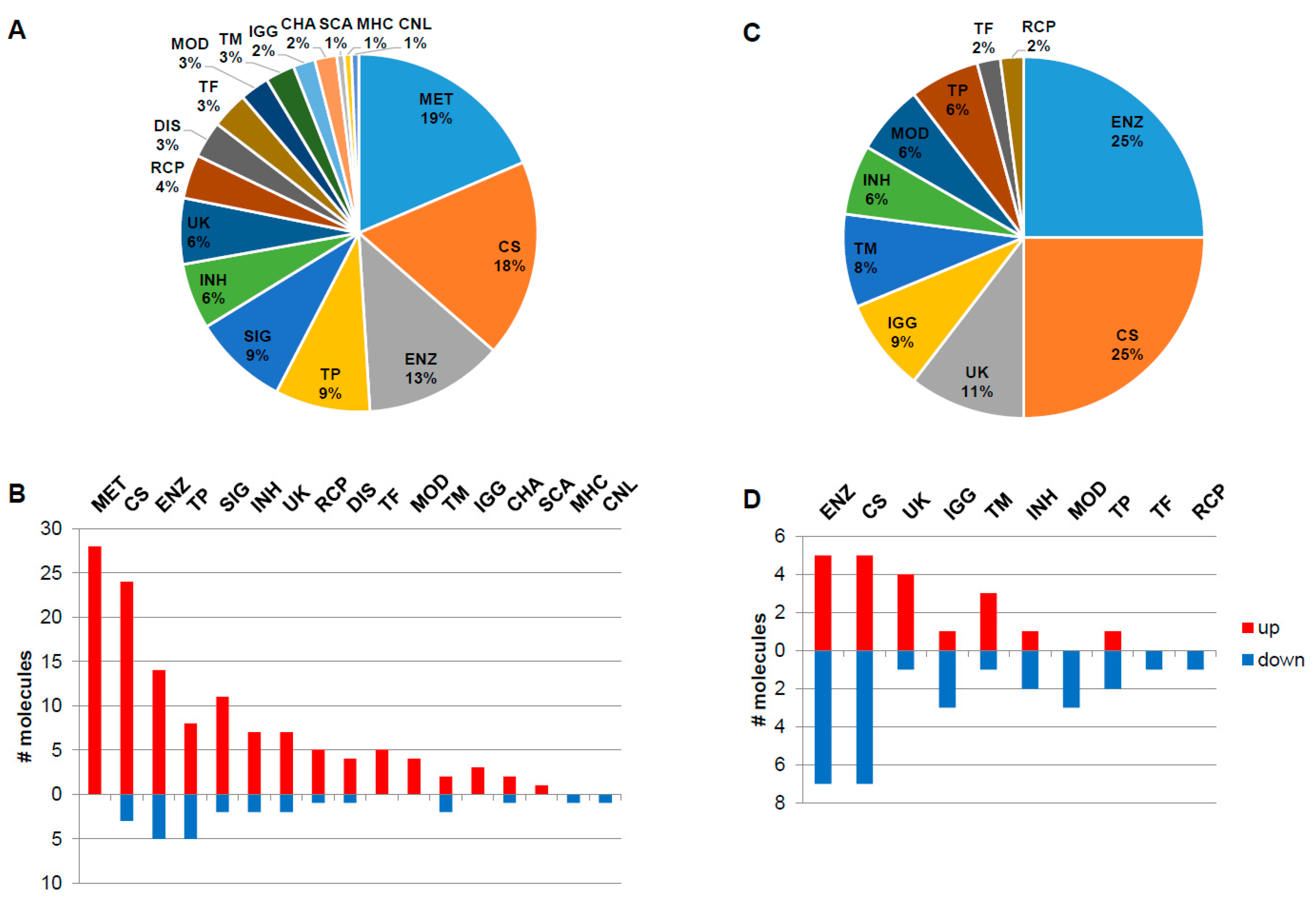
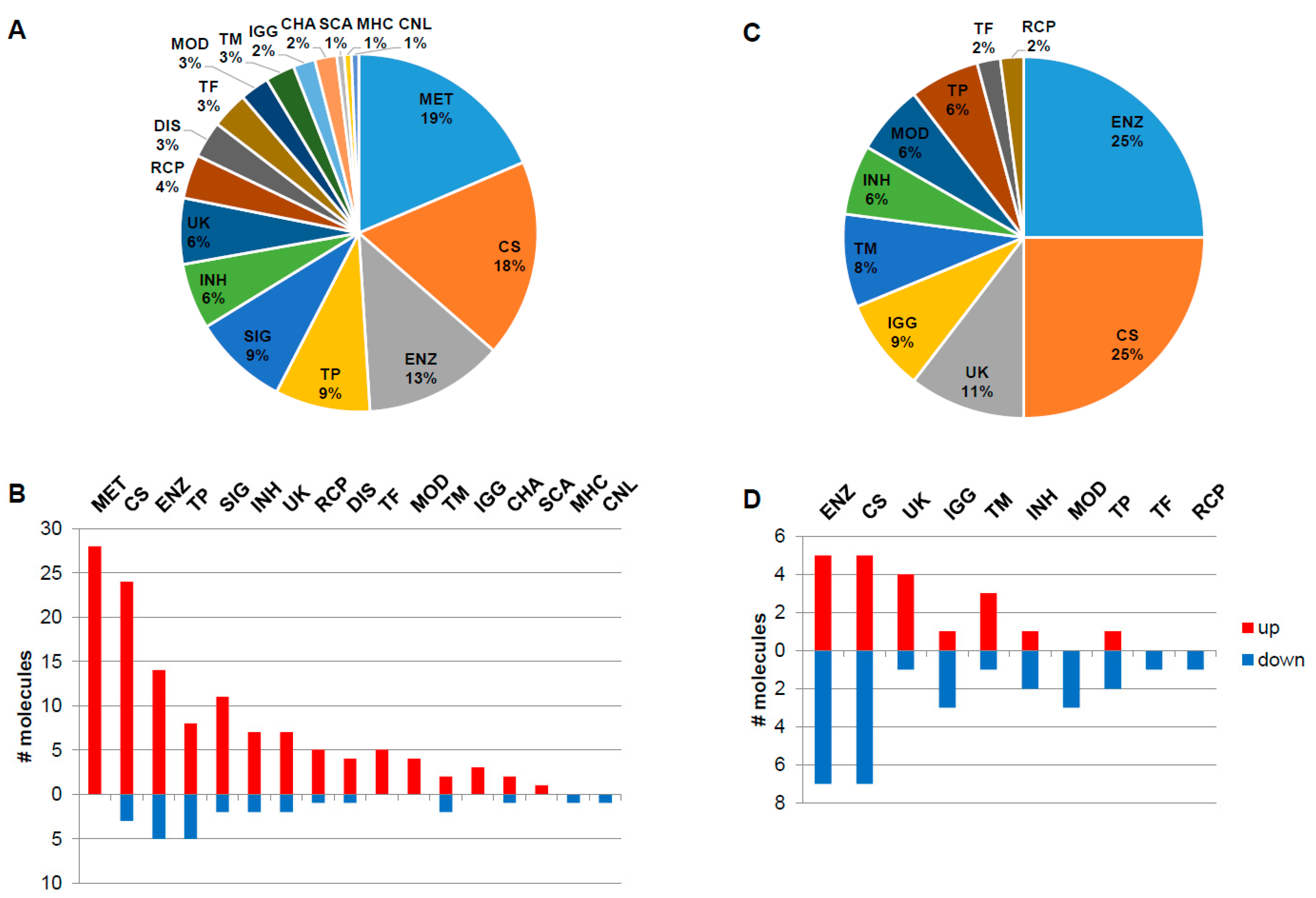
3.1. Dataspace Description
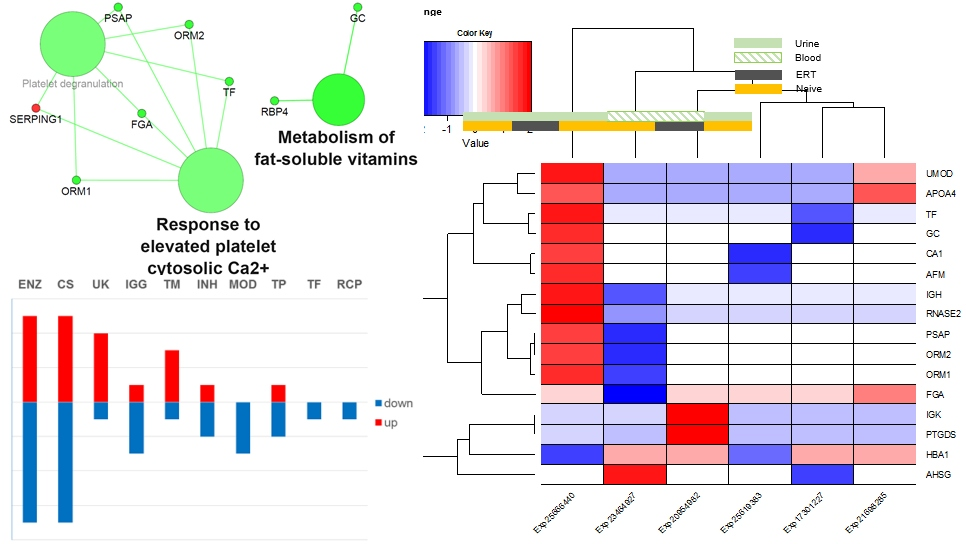
3.2. Functionality Tag Clustering
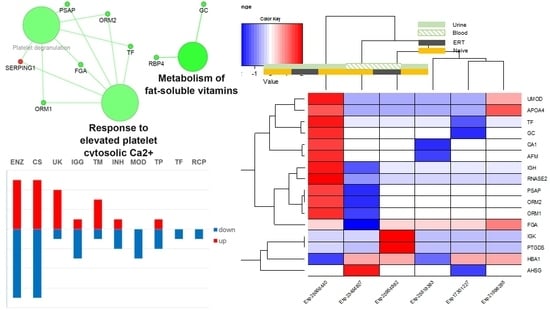
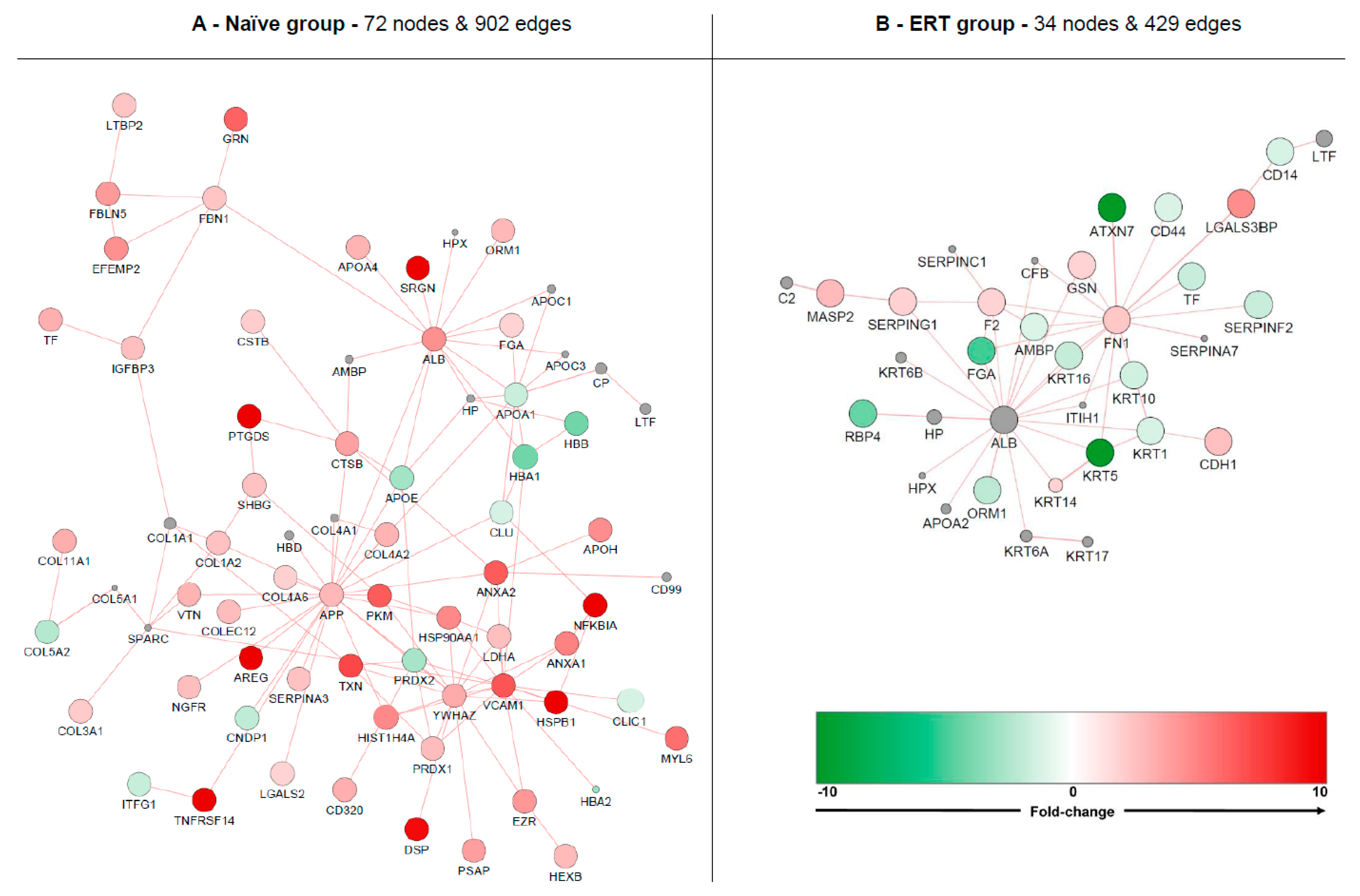
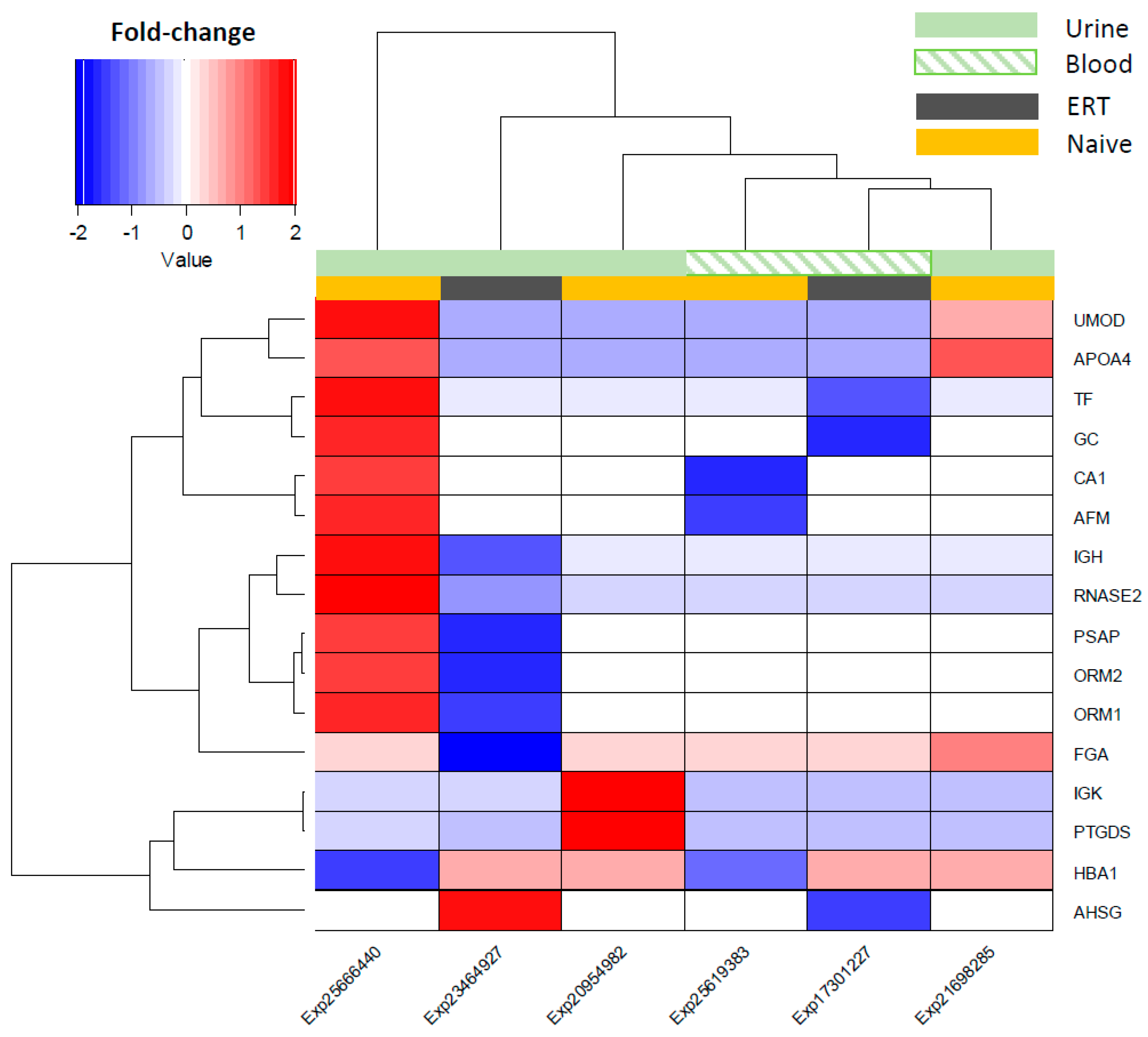
3.3. Expression Correlation Across Fabry Studies and Most Frequent Reported Molecules
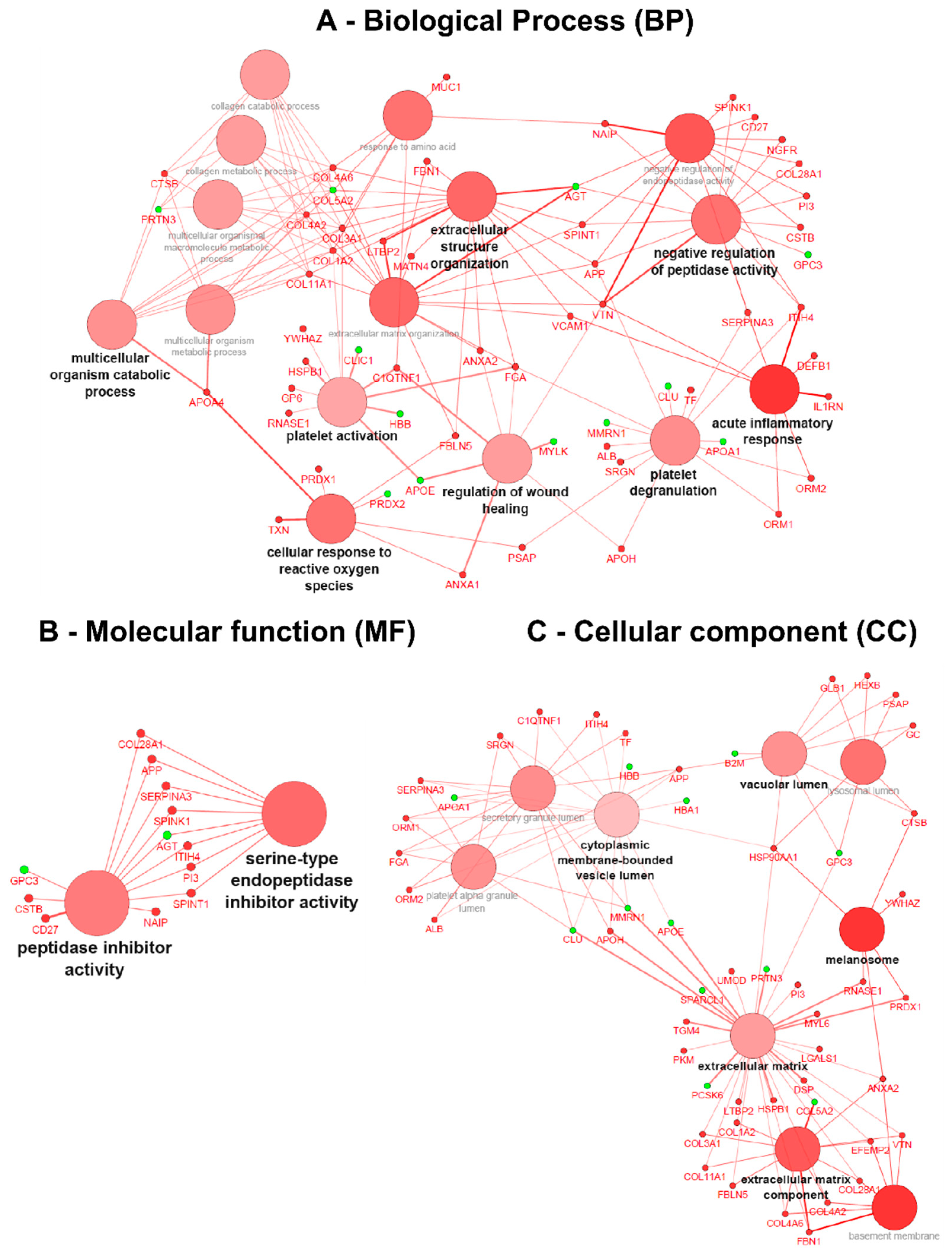
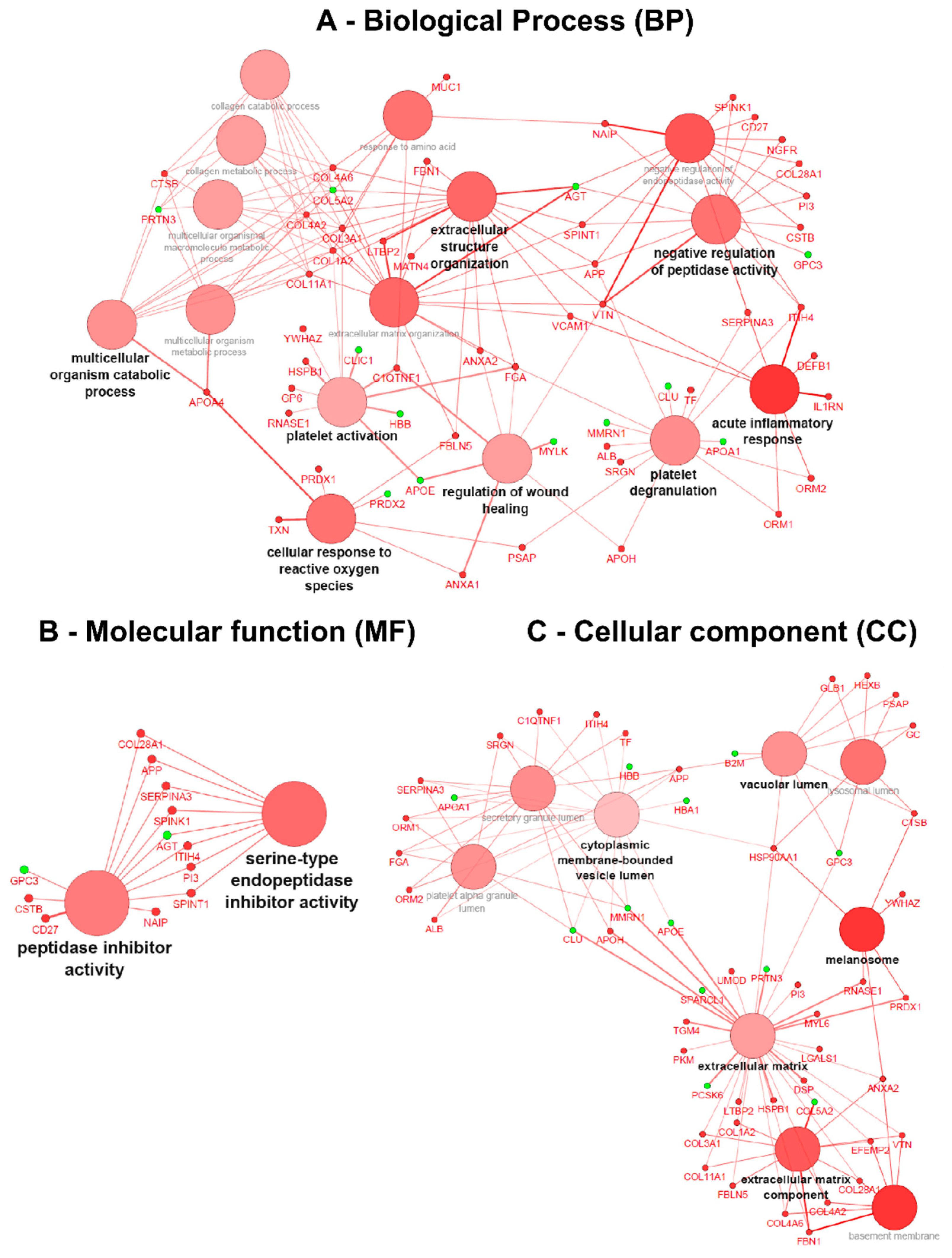
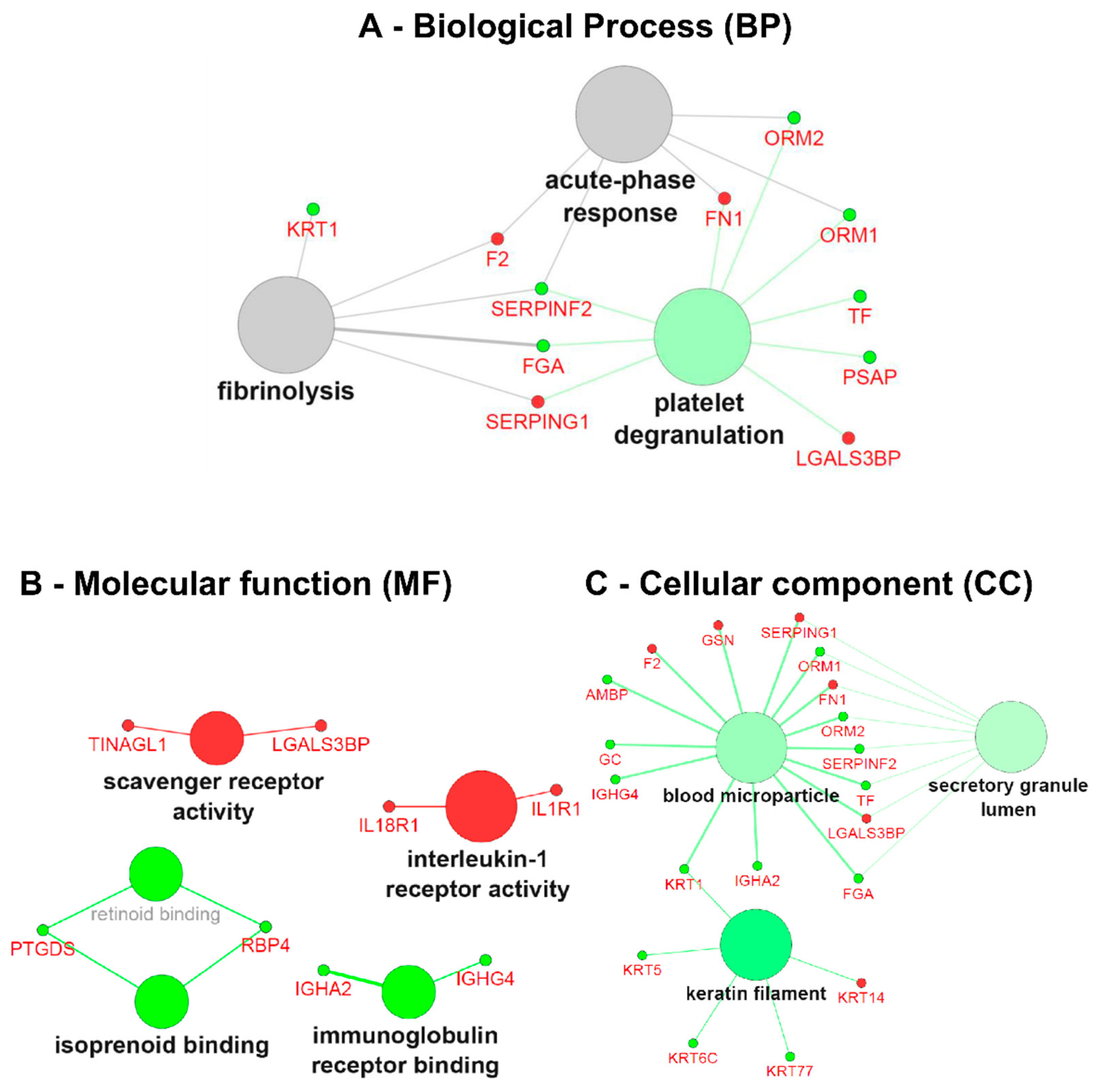
3.4. Gene Ontology (GO) Term Clustering
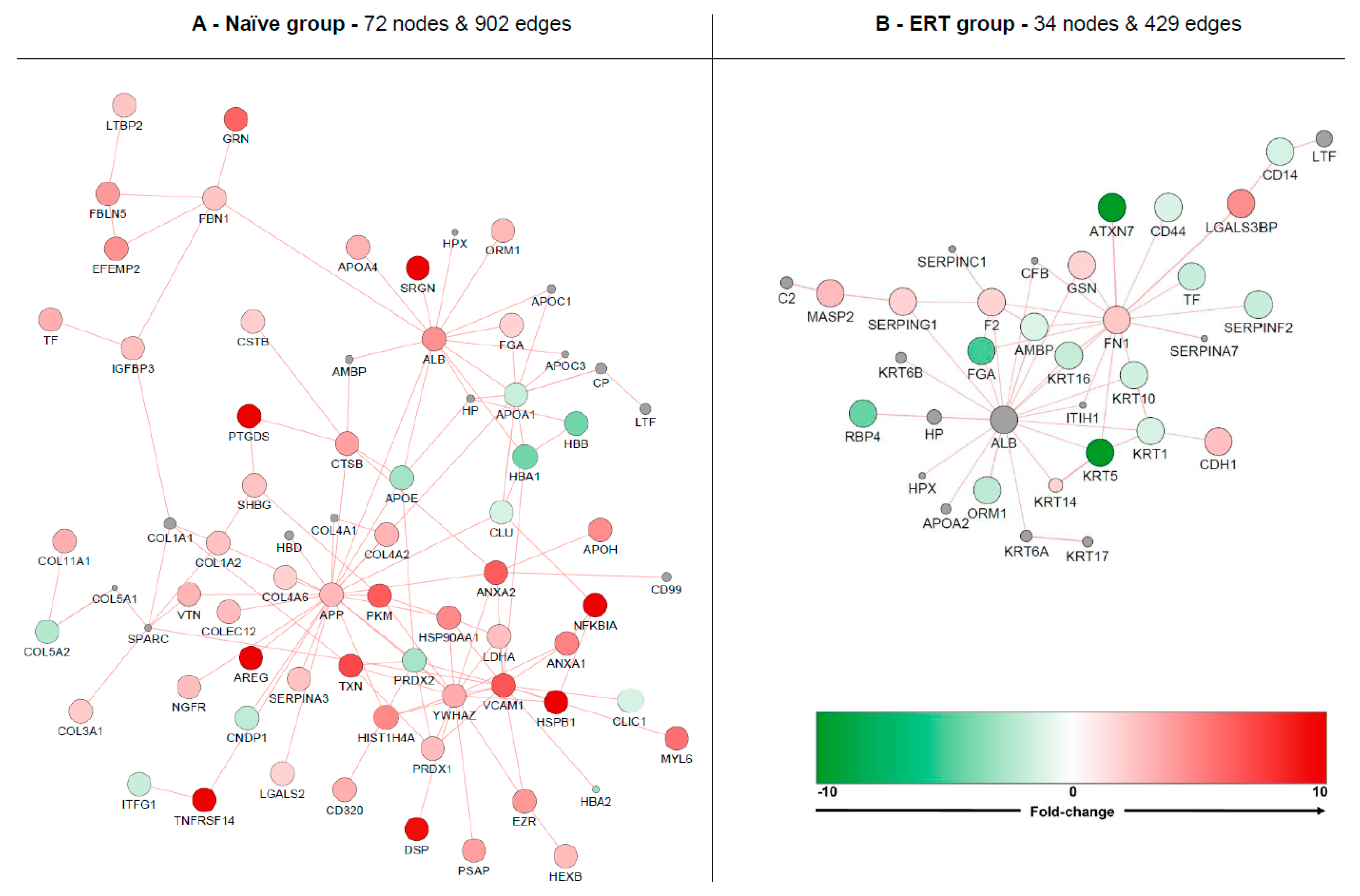
3.5. Interactome Analysis
3.6. Disease Analysis—DisGeNET
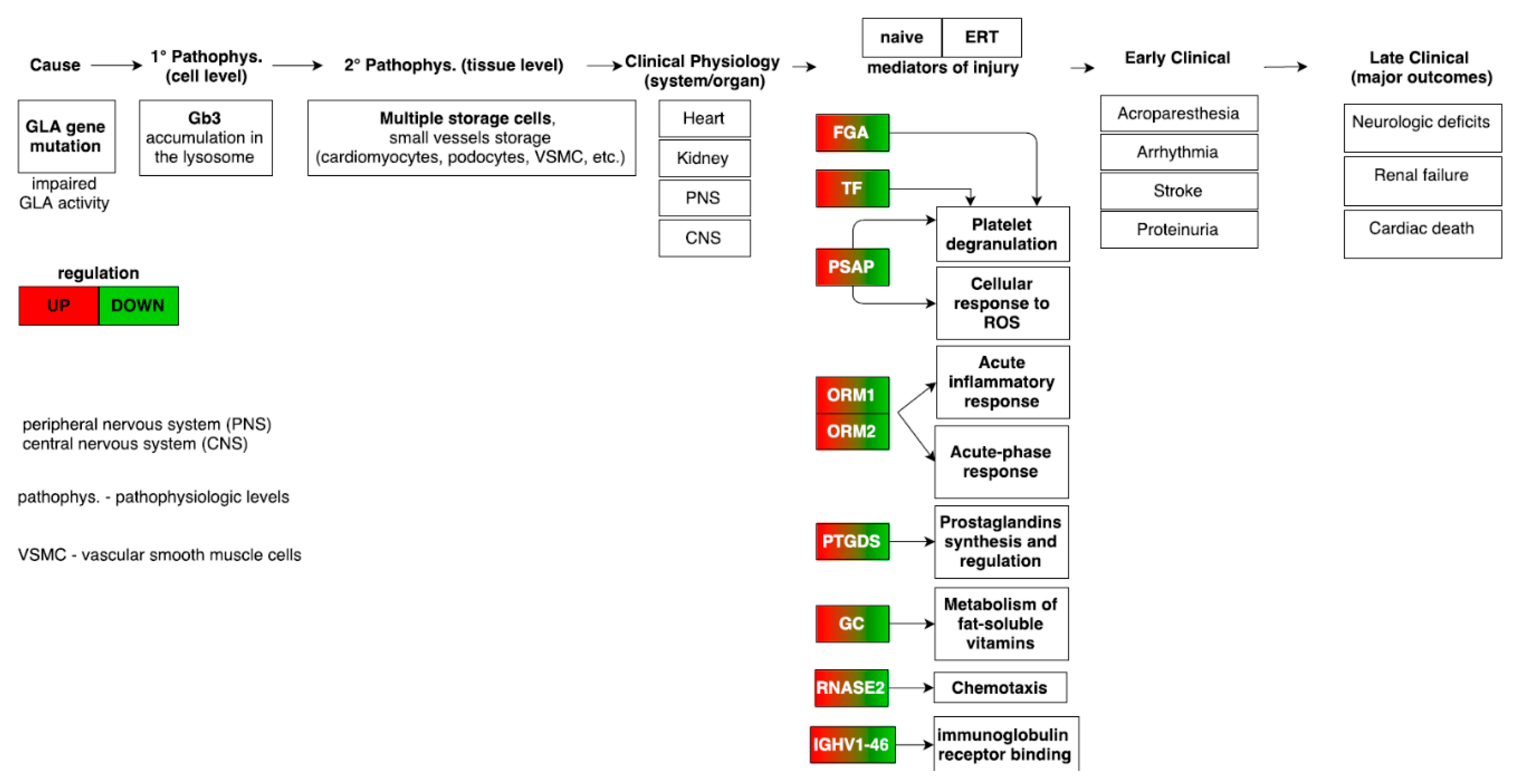
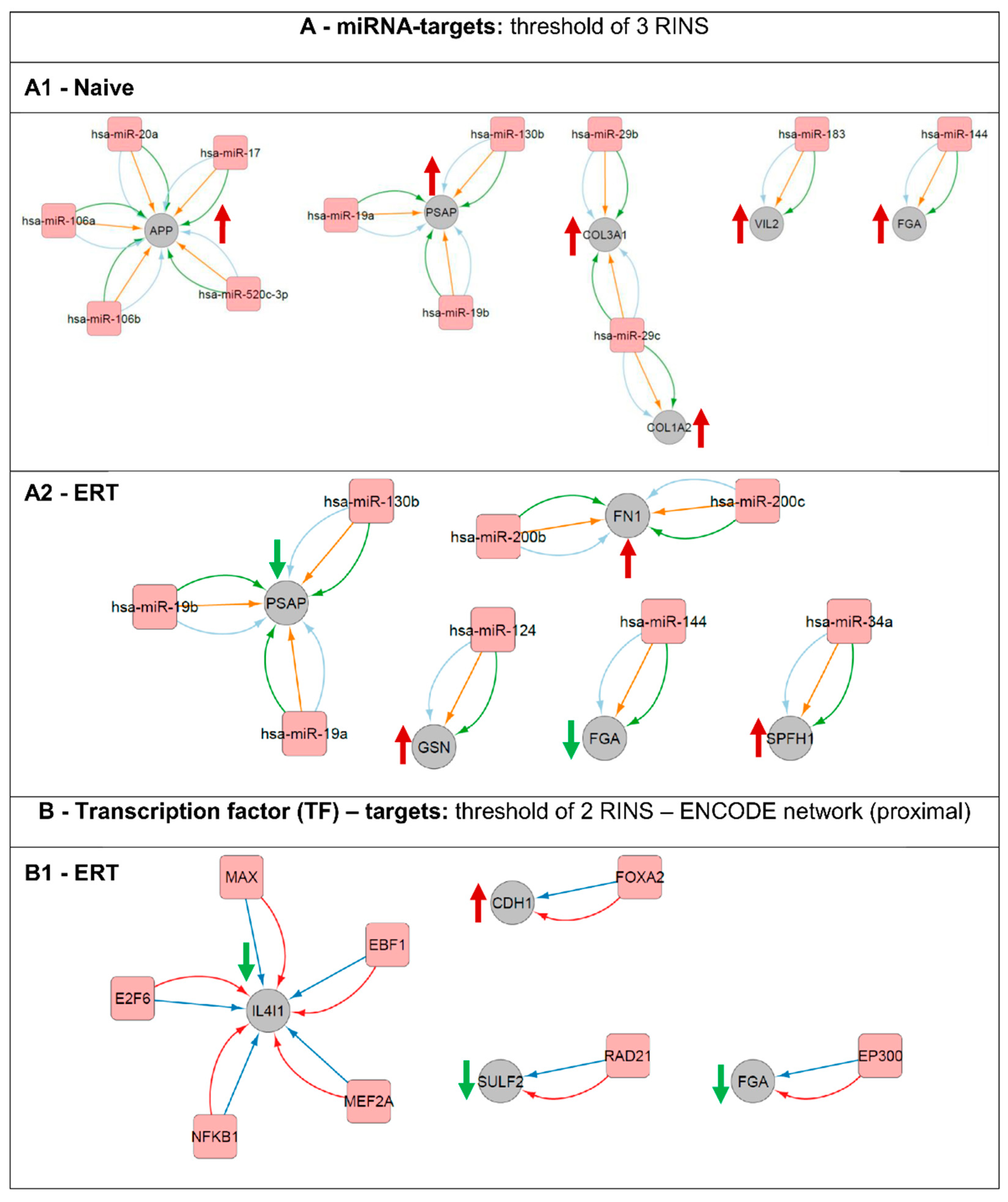
3.7. Regulatory Interactions
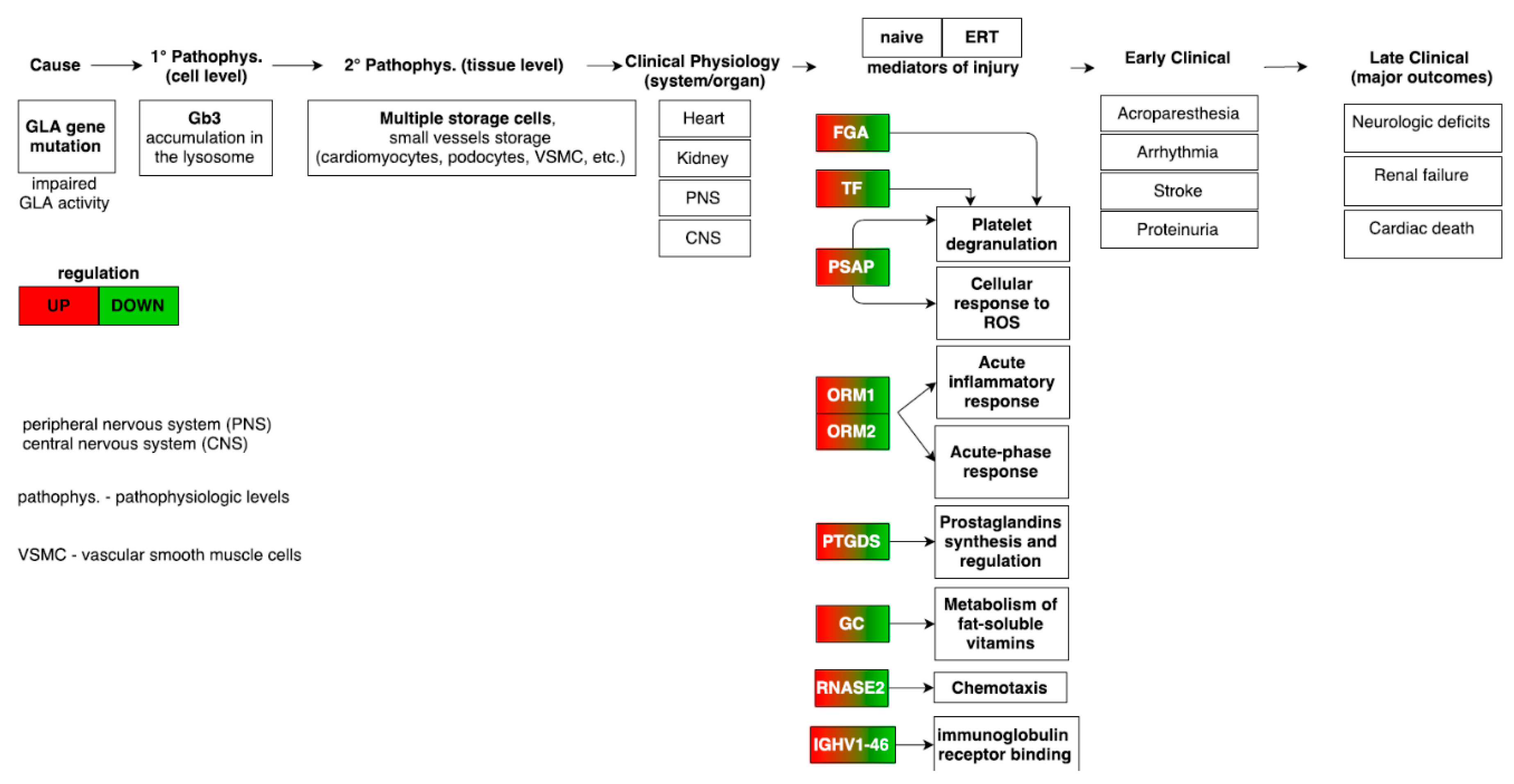
3.8. Merging Results and Overall Interpretation
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schiffmann, R.; Swift, C.; Wang, X.; Blankenship, D.; Ries, M. A prospective 10-year study of individualized, intensified enzyme replacement therapy in advanced Fabry disease. J. Inherit. Metab. Dis. 2015, 38, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Bodamer, O.A.; Watson, M.S.; Wilcox, W.R. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [PubMed]
- DeGraba, T.; Azhar, S.; Dignat-George, F.; Brown, E.; Boutiere, B.; Altarescu, G.; McCarron, R.; Schiffmann, R. Profile of endothelial and leukocyte activation in fabry patients. Ann. Neurol. 2000, 47, 229–233. [Google Scholar] [CrossRef]
- Hilz, M.J.; Kolodny, E.H.; Brys, M.; Stemper, B.; Haendl, T.; Marthol, H. Reduced cerebral blood flow velocity and impaired cerebral autoregulation in patients with Fabry disease. J. Neurol. 2004, 251, 564–570. [Google Scholar] [CrossRef] [PubMed]
- El-Abassi, R.; Singhal, D.; England, J.D. Fabry’s disease. J. Neurol. Sci. 2014, 344, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R. Fabry disease. Pharmacol. Ther. 2009, 122, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M. Is it Fabry disease? Genet. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.; Lee, C.; Moon, M.H.; Hong, G.R.; Cheon, C.K.; Lee, J.S. Unravelling the mechanism of action of enzyme replacement therapy in Fabry disease. J. Hum. Genet. 2016, 61, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Sunder-Plassmann, G. Renal manifestations of Fabry disease. In Fabry Disease: Perspectives from 5 Years of Fos; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Mills, K.; Vellodi, A.; Morris, P.; Cooper, D.; Morris, M.; Young, E.; Winchester, B. Monitoring the clinical and biochemical response to enzyme replacement therapy in three children with Fabry disease. Eur. J. Pediatr. 2004, 163, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Gelderman, M.P.; Ferreira, P.A.; Fuhrmann, S.R.; Yi, H.; Elkahloun, A.; Lix, L.M.; Brady, R.O.; Schiffmann, R.; Goldin, E. Genomic abnormalities of the murine model of Fabry disease after disease-related perturbation, a systems biology approach. Proc. Natl. Acad. Sci. USA 2007, 104, 8065–8070. [Google Scholar] [CrossRef] [PubMed]
- Park, E.S.; Choi, J.O.; Park, J.W.; Lee, M.H.; Park, H.Y.; Jung, S.C. Expression of genes and their responses to enzyme replacement therapy in a Fabry disease mouse model. Int. J. Mol. Med. 2009, 24, 401–407. [Google Scholar] [PubMed]
- Vaquer, G.; Riviere, F.; Mavris, M.; Bignami, F.; Llinares-Garcia, J.; Westermark, K.; Sepodes, B. Animal models for metabolic, neuromuscular and ophthalmological rare diseases. Nat. Rev. Drug Discov. 2013, 12, 287–305. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, T.; Krochmal, M.; Cisek, K.; Fernandes, M.; Husi, H.; Stevens, R.; Bascands, J.L.; Schanstra, J.P.; Klein, J. Omics databases on kidney disease: Where they can be found and how to benefit from them. Clin. Kidney J. 2016, 9, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Ma, L.; Yu, J.; Zhang, Z. Biological databases for human research. Genom. Proteom. Bioinform. 2015, 13, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. Cluego: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. Kegg: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Kelder, T.; van Iersel, M.P.; Hanspers, K.; Kutmon, M.; Conklin, B.R.; Evelo, C.T.; Pico, A.R. Wikipathways: Building research communities on biological pathways. Nucleic Acids Res. 2012, 40, D1301–D1307. [Google Scholar] [CrossRef] [PubMed]
- Montojo, J.; Zuberi, K.; Rodriguez, H.; Kazi, F.; Wright, G.; Donaldson, S.L.; Morris, Q.; Bader, G.D. Genemania cytoscape plugin: Fast gene function predictions on the desktop. Bioinformatics 2010, 26, 2927–2928. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; van Iersel, M.P.; Bohler, A.; Kelder, T.; Nunes, N.; Pico, A.R.; Evelo, C.T. Pathvisio 3: An extendable pathway analysis toolbox. PLoS Comput. Boil. 2015, 11, e1004085. [Google Scholar] [CrossRef] [PubMed]
- Bauer-Mehren, A.; Rautschka, M.; Sanz, F.; Furlong, L.I. Disgenet: A cytoscape plugin to visualize, integrate, search and analyze gene-disease networks. Bioinformatics 2010, 26, 2924–2926. [Google Scholar] [CrossRef] [PubMed]
- Kutmon, M.; Kelder, T.; Mandaviya, P.; Evelo, C.T.; Coort, S.L. Cytargetlinker: A cytoscape app to integrate regulatory interactions in network analysis. PLoS ONE 2013, 8, e82160. [Google Scholar] [CrossRef] [PubMed]
- Matafora, V.; Cuccurullo, M.; Beneduci, A.; Petrazzuolo, O.; Simeone, A.; Anastasio, P.; Mignani, R.; Feriozzi, S.; Pisani, A.; Comotti, C.; et al. Early markers of Fabry disease revealed by proteomics. Mol. BioSyst. 2015, 11, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Krokhin, O.V.; Beavis, R.C.; Ries, M.; Robinson, C.; Goldin, E.; Brady, R.O.; Wilkins, J.A.; Schiffmann, R. Proteomics of specific treatment-related alterations in Fabry disease: A strategy to identify biological abnormalities. Proc. Natl. Acad. Sci. USA 2007, 104, 2873–2878. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Goldin, E.; Gelderman, M.P.; Robinson, C.; Baer, J.; Ries, M.; Elkahloun, A.; Brady, R.O.; Schiffmann, R. Apoptotic abnormalities in differential gene expression in peripheral blood mononuclear cells from children with Fabry disease. Acta Paediatr. 2008, 97, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Vojtova, L.; Zima, T.; Tesar, V.; Michalova, J.; Prikryl, P.; Dostalova, G.; Linhart, A. Study of urinary proteomes in Anderson-Fabry disease. Renal Fail. 2010, 32, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Cigna, D.; D’Anna, C.; Zizzo, C.; Francofonte, D.; Sorrentino, I.; Colomba, P.; Albeggiani, G.; Armini, A.; Bianchi, L.; Bini, L.; et al. Alteration of proteomic profiles in pbmc isolated from patients with Fabry disease: Preliminary findings. Mol. BioSyst. 2013, 9, 1162–1168. [Google Scholar] [CrossRef] [PubMed]
- Manwaring, V.; Heywood, W.E.; Clayton, R.; Lachmann, R.H.; Keutzer, J.; Hindmarsh, P.; Winchester, B.; Heales, S.; Mills, K. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: Evidence of presymptomatic kidney disease in pediatric fabry and type-i diabetic patients. J. Proteome Res. 2013, 12, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Hollander, Z.; Dai, D.L.; Putko, B.N.; Yogasundaram, H.; Wilson-McManus, J.E.; Thompson, R.B.; Khan, A.; West, M.L.; McManus, B.M.; Oudit, G.Y. Gender-specific plasma proteomic biomarkers in patients with anderson-Fabry disease. Eur. J. Heart Fail. 2015, 17, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Kistler, A.D.; Siwy, J.; Breunig, F.; Jeevaratnam, P.; Scherl, A.; Mullen, W.; Warnock, D.G.; Wanner, C.; Hughes, D.A.; Mischak, H.; et al. A distinct urinary biomarker pattern characteristic of female fabry patients that mirrors response to enzyme replacement therapy. PLoS ONE 2011, 6, e20534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, M.; Auray-Blais, C. Metabolomic discovery of novel urinary galabiosylceramide analogs as Fabry disease biomarkers. J. Am. Soc. Mass Spectrom. 2015, 26, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Kakkis, E.D.; O’Donovan, M.; Cox, G.; Hayes, M.; Goodsaid, F.; Tandon, P.K.; Furlong, P.; Boynton, S.; Bozic, M.; Orfali, M.; et al. Recommendations for the development of rare disease drugs using the accelerated approval pathway and for qualifying biomarkers as primary endpoints. Orphanet J. Rare Dis. 2015, 10, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Thon, J.N.; Italiano, J.E. Platelets: Production, morphology and ultrastructure. Handb. Exp. Pharmacol. 2012, 3–22. [Google Scholar] [CrossRef]
- Nording, H.M.; Seizer, P.; Langer, H.F. Platelets in inflammation and atherogenesis. Front. Immunol. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- Herter, J.M.; Rossaint, J.; Zarbock, A. Platelets in inflammation and immunity. J. Thromb. Haemost. JTH 2014, 12, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Weber, C.; Gawaz, M. The platelet—Thrombosis and beyond. Thromb. Haemost. 2013, 110, 857–858. [Google Scholar] [CrossRef] [PubMed]
- Jandrot-Perrus, M.; Busfield, S.; Lagrue, A.H.; Xiong, X.; Debili, N.; Chickering, T.; Le Couedic, J.P.; Goodearl, A.; Dussault, B.; Fraser, C.; et al. Cloning, characterization, and functional studies of human and mouse glycoprotein vi: A platelet-specific collagen receptor from the immunoglobulin superfamily. Blood 2000, 96, 1798–1807. [Google Scholar] [PubMed]
- Zhang, W.; Huang, W.; Jing, F. Contribution of blood platelets to vascular pathology in Alzheimer’s disease. J. Blood Med. 2013, 4, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.S. Platelets and depression in cardiovascular disease: A brief review of the current literature. World J. Psychiatry 2012, 2, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Boil. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Gobbetti, T.; Cooray, S.N. Annexin A1 and resolution of inflammation: Tissue repairing properties and signalling signature. Biol. Chem. 2016, 397, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Campanholle, G.; Ligresti, G.; Gharib, S.A.; Duffield, J.S. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C591–C603. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Sanchez-Nino, M.D.; Politei, J.; Oliveira, J.P.; Wanner, C.; Warnock, D.G.; Ortiz, A. Fibrosis: A key feature of Fabry disease with potential therapeutic implications. Orphanet J. Rare Dis. 2013, 8, 116. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Cray, C.; Zaias, J.; Altman, N.H. Acute phase response in animals: A review. Comp. Med. 2009, 59, 517–526. [Google Scholar] [PubMed]
- Vedder, A.C.; Biro, E.; Aerts, J.M.; Nieuwland, R.; Sturk, G.; Hollak, C.E. Plasma markers of coagulation and endothelial activation in Fabry disease: Impact of renal impairment. Nephrol. Dial. Transplant. 2009, 24, 3074–3081. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EXPREF | Total | N (case) | N (control) | Disease Case | Disease Control | Source | Detection Method | Reference |
|---|---|---|---|---|---|---|---|---|
| Exp25666440 | 23 | 11 | 12 | naïve Fabry patients | ERT | urine | nanoLC-ESI-MS/MS | [25] |
| Exp17301227 | 13 | N/A | N/A | 6 months of ERT | before treatment (baseline) | blood | nanoLC-ESI-MS/MS | [26] |
| Exp18339188a | 13 | N/A | N/A | 6 months of ERT | before treatment (baseline) | blood | microarray | [27] |
| Exp18339188b | 13 | N/A | N/A | naïve Fabry patients | healthy | blood | microarray | [27] |
| Exp20954982 | 30 | 20 | 10 | Fabry | healthy | urine | MALDI-TOF MS | [28] |
| Exp23385635 | 14 | 8 | 6 | Fabry | healthy | blood | MALDI-TOF MS | [29] |
| Exp23464927 | 10 | N/A | N/A | 12 months of ERT | before treatment (baseline) | urine | QTOF MS/MS | [30] |
| Exp25619383 | 46 | 32 | 14 | Fabry | healthy | blood | LC-MS/MS | [31] |
| Exp21698285 | 124 | 35 | 89 | naïve female Fabry | healthy | urine | CE-MS | [32] |
| Exp26490183 | 6 | N/A | N/A | short term of ERT | before treatment (baseline) | blood | Illumina MiSeq instrument | [8] |
| Exp25582508 | 32 | 16 | 16 | untreated Fabry males | healthy | urine | UPLC-ESI-TOF-MS | [33] |
| Disease Name | # Shared Genes | Gene Name |
|---|---|---|
| Malignant neoplasm of breast | 16 | AGT, ALB, APOA1, APOA4, CLU, GC, GRN, IGK, ITIH4, PSAP, PTGDS, RNASE1, SERPINA3, SLURP1, TF, YWHAZ |
| Breast carcinoma | 15 | AGT, ALB, APOA1, APOA4, CLU, GC, GRN, ITIH4, PSAP, PTGDS, RNASE1, SERPINA3, SLURP1, TF, YWHAZ |
| Diabetes mellitus, non-insulin-dependent | 13 | AGT, ALB, APOA1, APOA4, CLU, FGA, GC, GRN, HBA1, PTGDS, RNASE1, SERPINA3, TF |
| Schizophrenia | 13 | APOA1, APOH, CLU, GC, GRN, ITIH4, PSAP, PTGDS, RNASE1, SERPINA3, SHISA5, TF, YWHAZ |
| Hypertensive disease | 12 | AGT, ALB, APOA1, CLU, FGA, GC, GRN, PTGDS, RNASE1, SERPINA3, TF, YWHAZ |
| Liver carcinoma | 12 | AGT, ALB, APOA1, APOA4, APOH, CLU, FGA, GC, GRN, PTGDS, UMOD, YWHAZ |
| Diabetes mellitus | 12 | AGT, ALB, APOA1, APOA4, APOH, CLU, FGA, GC, GRN, PTGDS, UMOD, YWHAZ |
| Diabetes | 12 | AGT, ALB, APOA1, APOA4, APOH, CLU, GC, HBA1, PTGDS, SERPINA3, UMOD, YWHAZ |
| Atherosclerosis | 12 | AGT, ALB, APOA1, APOA4, APOH, CLU, GC, HBA1, PTGDS, SERPINA3, UMOD, YWHAZ |
| Arteriosclerosis | 12 | AGT, ALB, APOA1, APOH, CLU, FGA, GC, HBA1, RNASE1, SERPINA3, UMOD, YWHAZ |
| Alzheimer‘s disease | 12 | AGT, ALB, APOA1, APOA4, APOH, CLU, GC, GRN, RNASE1, SERPINA3, TF, YWHAZ |
| Asthma | 11 | AGT, APOA1, GC, IGHG1, ORM1, PSAP, PTGDS, RNASE2, SERPINA3, TF, YWHAZ |
| Obesity | 11 | AGT, ALB, APOA1, APOA4, APOH, CLU, FGA, HBA1, TF, UMOD, YWHAZ |
| Cardiovascular Diseases | 11 | AGT, ALB, APOA1, APOA4, APOH, FGA, HBA1, RNASE1, RNASE2, SERPINA3, TF |
| Cerebrovascular accident | 11 | AGT, APOA1, CLU, GC, GRN, PSAP, PTGDS, RNASE1, SERPINA3, TF, YWHAZ |
| Malignant neoplasm of prostate | 11 | AGT, ALB, APOA1, APOA4, FGA, GC, GRN, ORM1, PTGDS, TF, YWHAZ |
| Prostate carcinoma | 11 | AGT, APOA1, CLU, GC, GRN, PSAP, PTGDS, RNASE1, SERPINA3, TF, YWHAZ |
| Mammary Neoplasms | 10 | AGT, ALB, CLU, GRN, HBA1, PSAP, PTGDS, RNASE1, SLURP1, YWHAZ |
| Colorectal Cancer | 10 | AGT, ALB, APOA1, CLU, GC, ORM2, PSAP, PTGDS, RNASE1, YWHAZ |
| Neoplasm metastasis | 10 | AGT, CLU, GC, GRN, IGK, PSAP, PTGDS, RNASE1, UMOD, YWHAZ |
| Carcinogenesis | 10 | AGT, ALB, APOA1, CLU, GRN, PSAP, RNASE1, SERPINA3, TF, YWHAZ |
| Disease Name | # Shared Genes | Gene Name |
|---|---|---|
| Asthma | 8 | GC, IGHG1, ORM1, PSAP, PTGDS, RNASE2, SERPING1, TF |
| Obesity | 7 | F2, FGA, GC, PTGDS, RBP4, SERPING1, TF |
| Alzheimer's disease | 7 | AMBP, F2, GC, IGK, PSAP, PTGDS, TF |
| Malignant neoplasm of breast | 7 | F2, FGA, GC, ORM1, PTGDS, RBP4, TF |
| Atherosclerosis | 6 | AMBP, F2, FGA, GC, PTGDS, RBP4 |
| Arteriosclerosis | 6 | AMBP, F2, GC, PSAP, PTGDS, TF |
| Breast carcinoma | 6 | F2, FGA, GC, PTGDS, RBP4, TF |
| Diabetes mellitus, non-insulin-dependent | 5 | AMBP, F2, FGA, GC, PTGDS, RBP4 |
| Malignant neoplasm of prostate | 5 | F2, GC, ORM2, PSAP, PTGDS |
| Diabetes mllitus | 5 | AMBP, F2, FGA, GC, TF |
| Drug-induced liver injury | 5 | F2, GC, RBP4, SERPING1, TF |
| Cardiovascular diseases | 5 | GC, PSAP, PTGDS, RBP4, TF |
| Liver carcinoma | 5 | GC, PSAP, PTGDS, RBP4, TF |
| Colorectal cancer | 4 | AMBP, F2, FGA, RBP4, TF |
| Prostate carcinoma | 4 | AMBP, F2, GC, PTGDS, RBP4 |
| melanoma | 4 | F2, FGA, GC, TF |
| Mammary neoplasms | 4 | F2, FGA, RNASE2, TF |
| Malignant neoplasm of ovary | 4 | F2, GC, PSAP, PTGDS |
| Colorectal carcinoma | 4 | F2, FGA, GC, RBP4 |
| Diabetes | 4 | F2, GC, PTGDS, RBP4 |
| Diabetic nephropathy | 4 | GC, PTGDS, RBP4, TF |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, M.; Husi, H. Integrative Systems Biology Investigation of Fabry Disease. Diseases 2016, 4, 35. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4040035
Fernandes M, Husi H. Integrative Systems Biology Investigation of Fabry Disease. Diseases. 2016; 4(4):35. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4040035
Chicago/Turabian StyleFernandes, Marco, and Holger Husi. 2016. "Integrative Systems Biology Investigation of Fabry Disease" Diseases 4, no. 4: 35. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases4040035