
Substrate Deprivation Therapy to Reduce Glycosaminoglycan Synthesis Improves Aspects of Neurological and Skeletal Pathology in MPS I Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Maintenance
2.2. Behaviour Testing
2.3. Determination of Tissue GAG
2.4. Determination of Lysosomal Enzyme Activity
2.5. Extraction and Quantification of Brain Monosialogangliosides
2.6. Ex Vivo Micro-CT
2.7. Statistics
3. Results
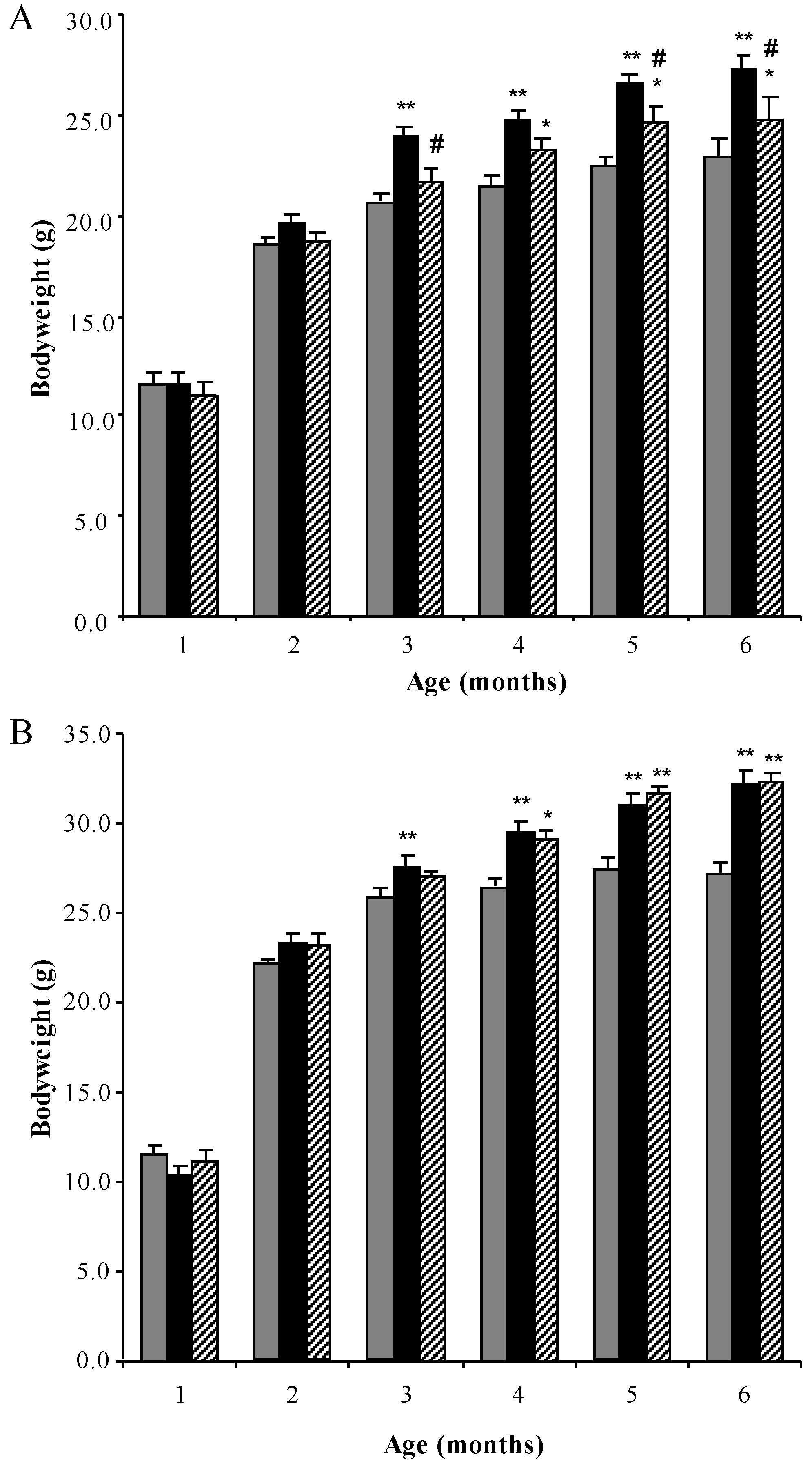
3.1. Bodyweight Profiles
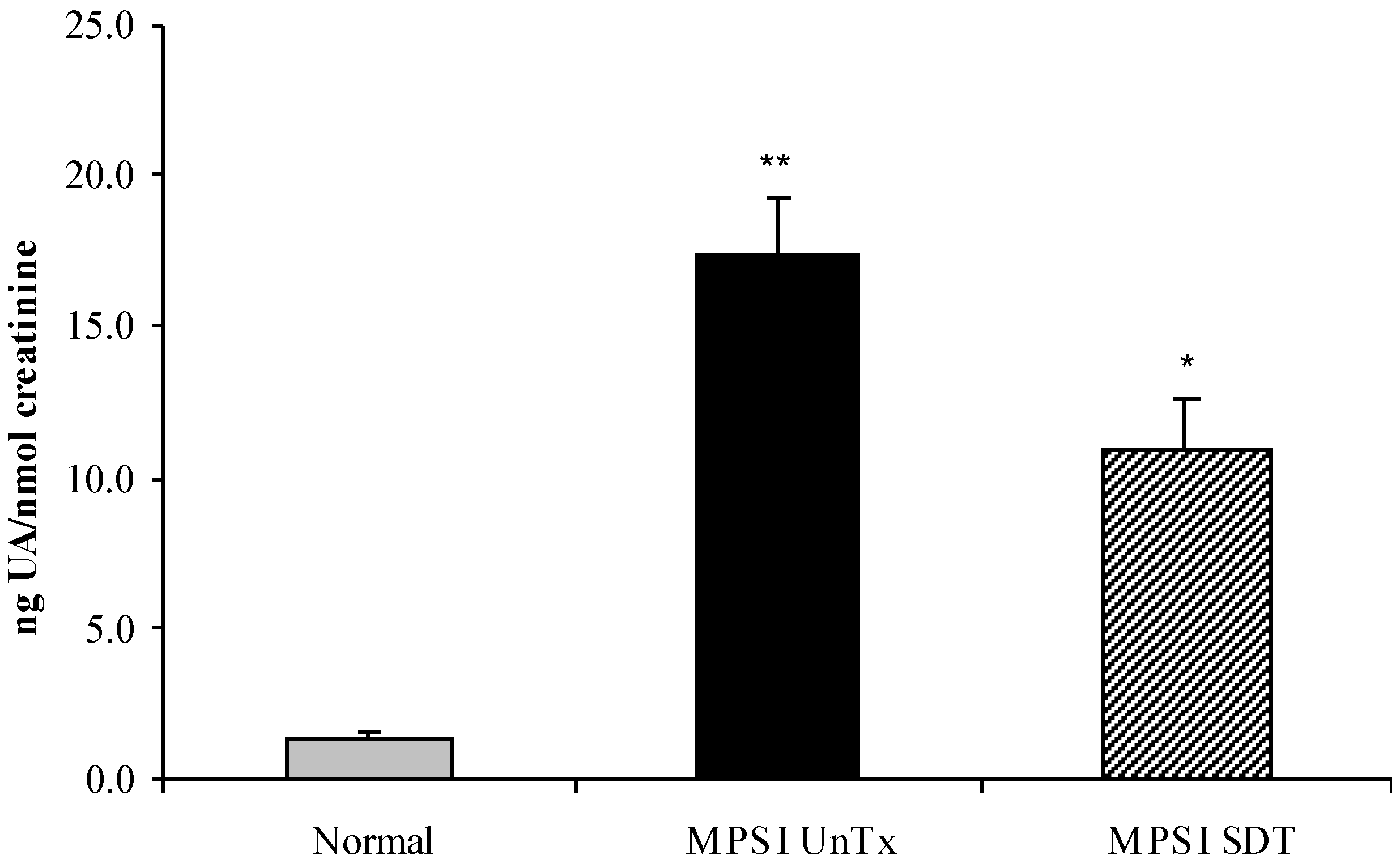
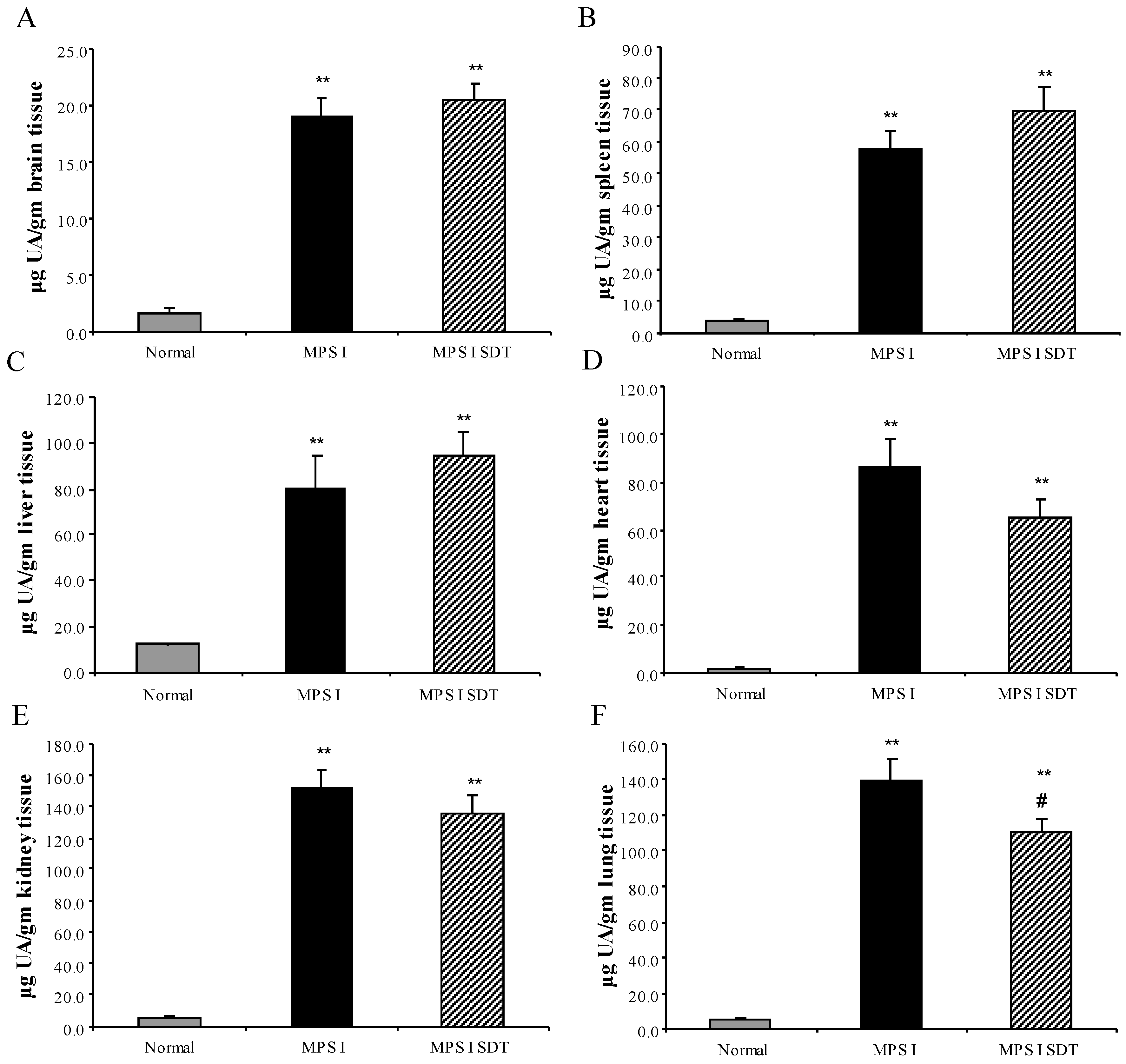
3.2. Biochemical Analysis
3.2.1. Effect of SDT Treatment on Primary and Secondary Storage
3.2.2. Tissue Enzyme Levels
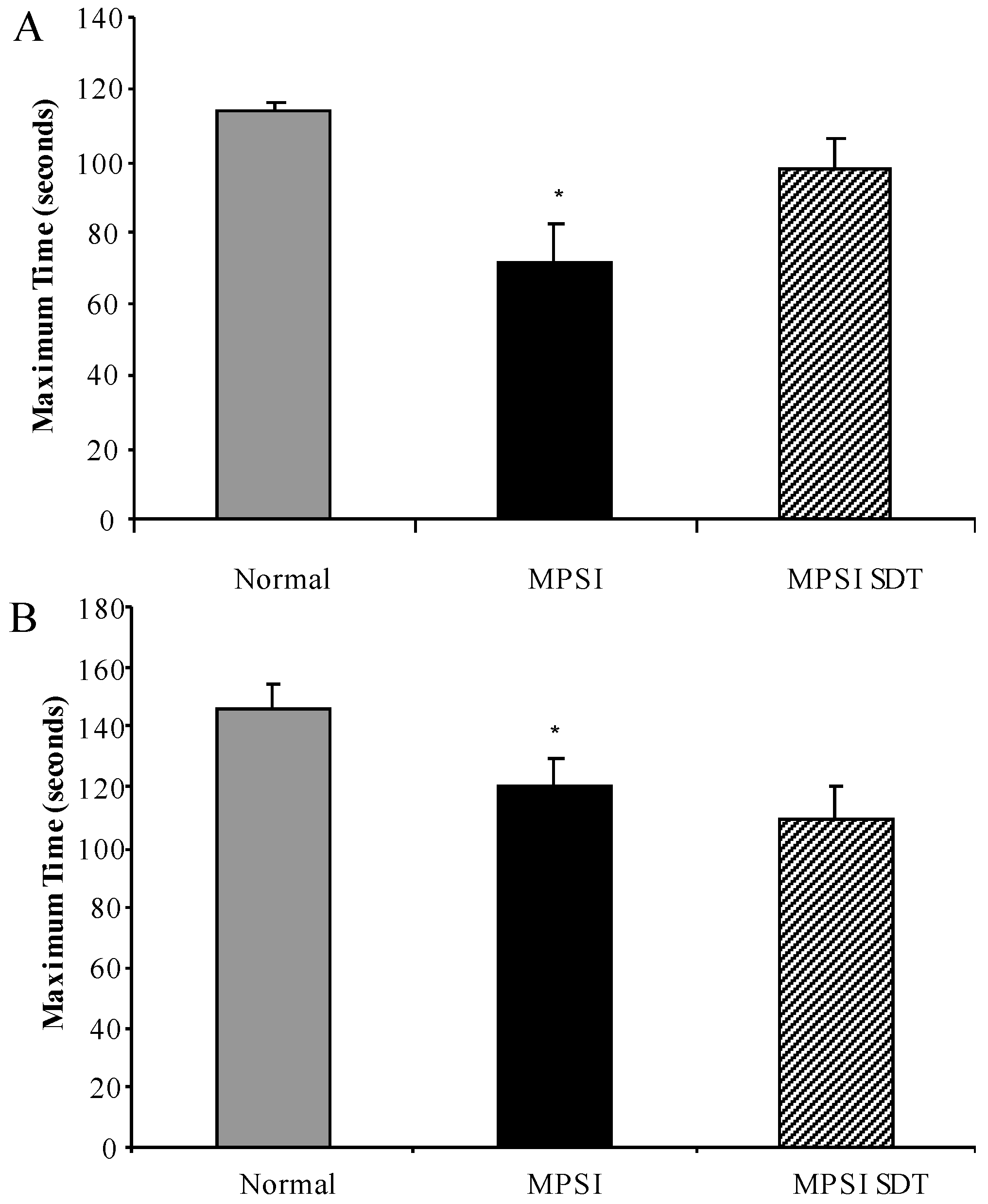
3.3. Effect of SDT Treatment on Behaviour
3.3.1. Inverted Grid
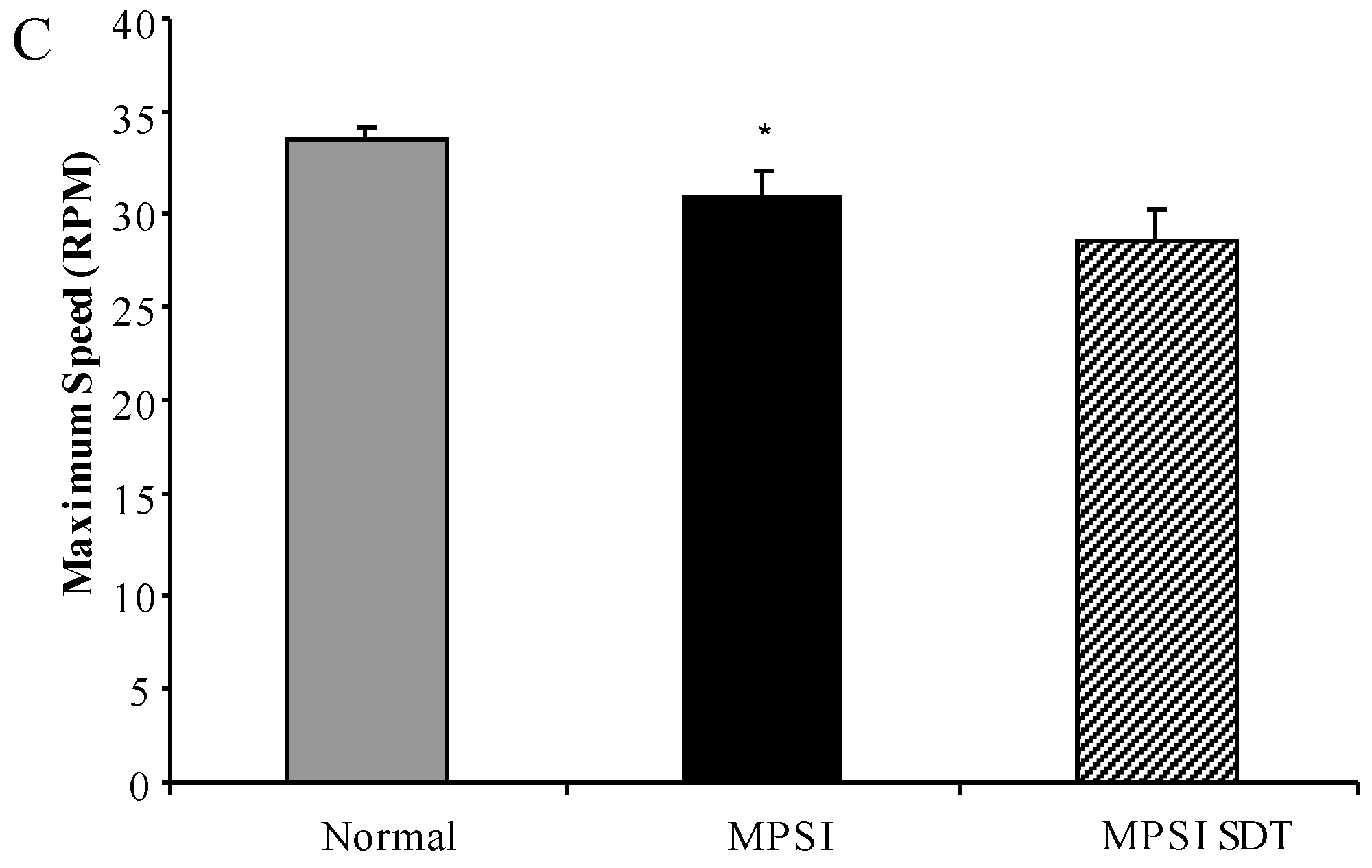
3.3.2. Rotarod
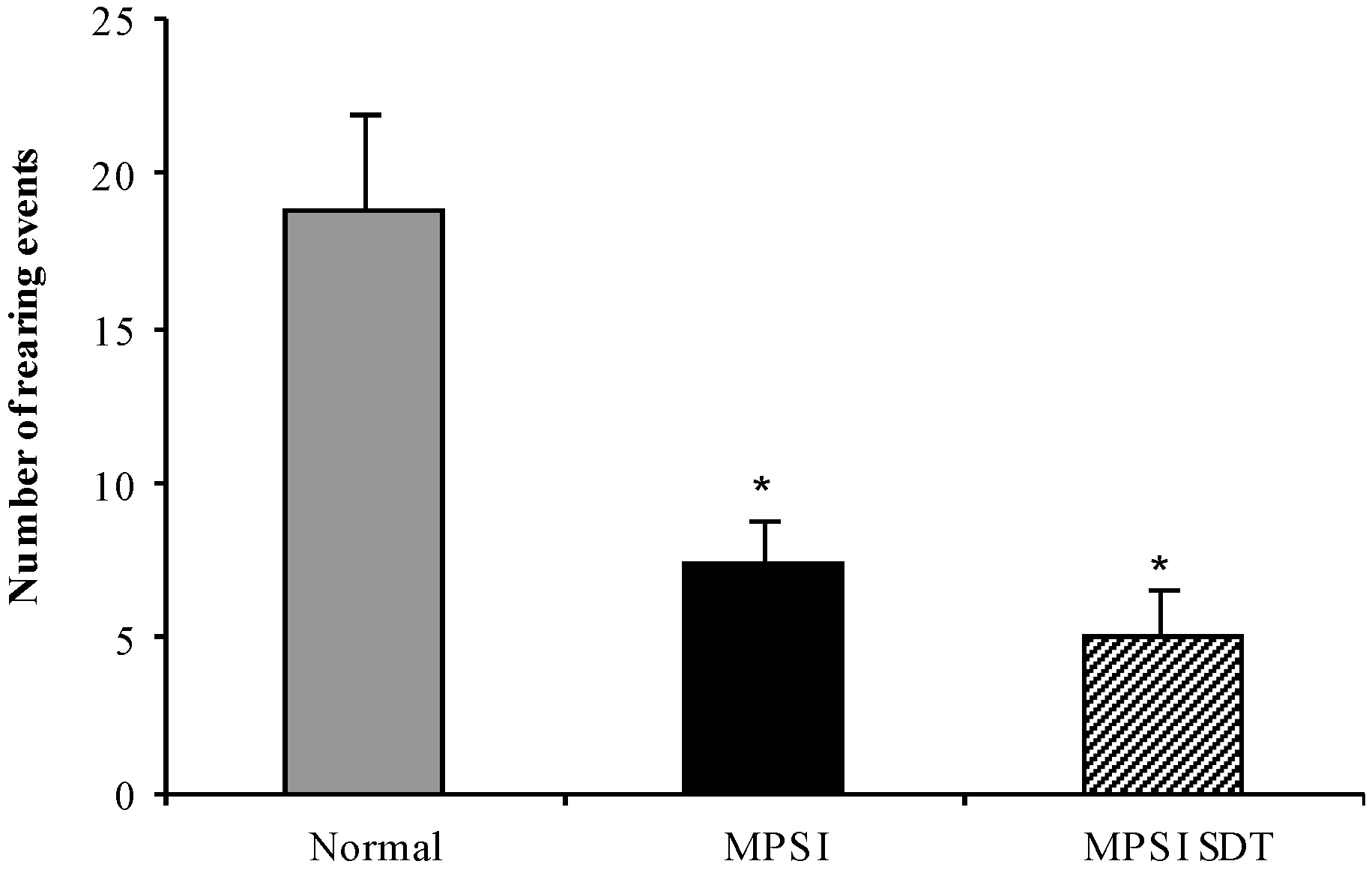
3.3.3. Open Field Exploration
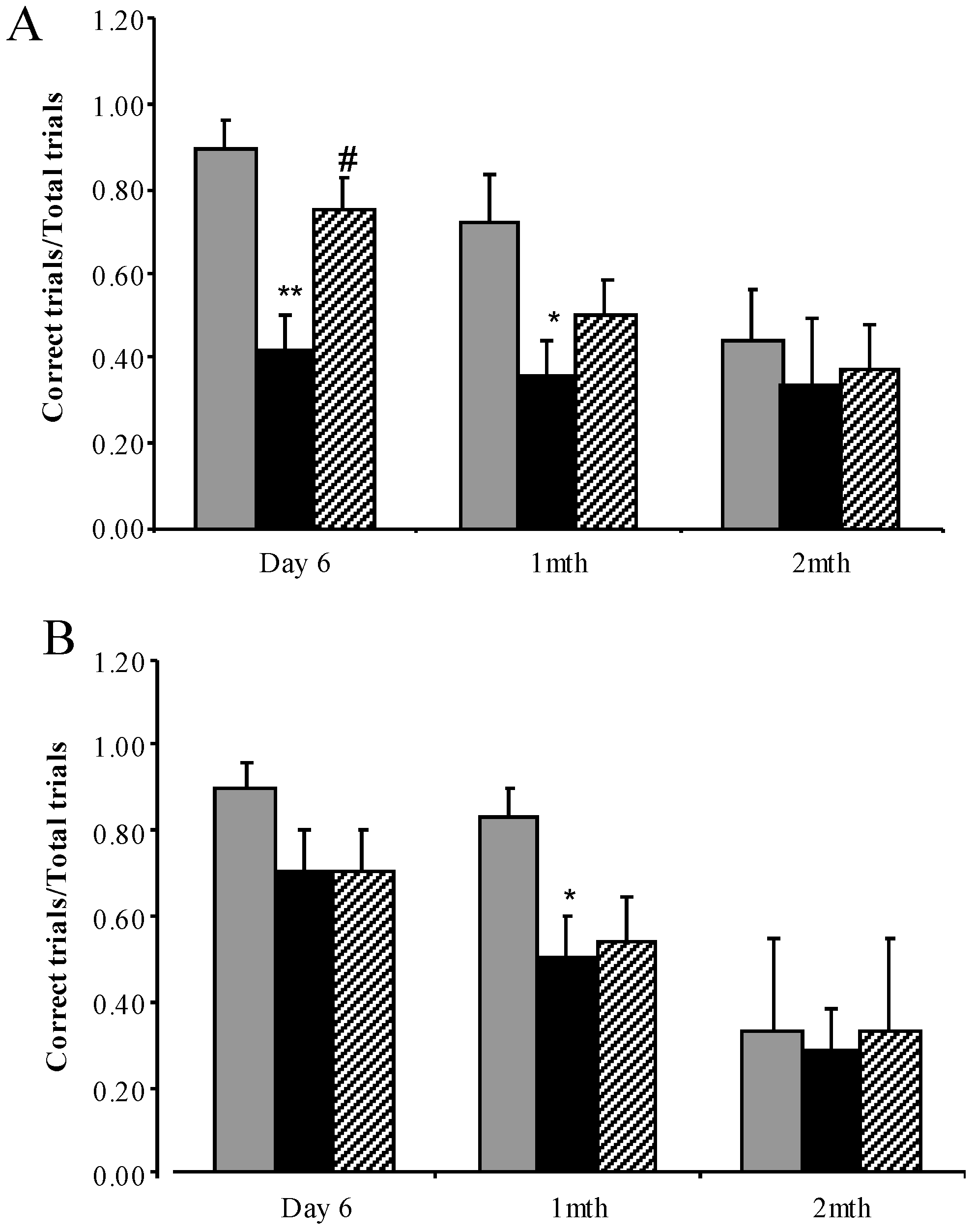
3.3.4. Water Cross Maze
3.4. Effect of SDT Treatment on Skeletal Disease
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill Companies Inc.: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Bunge, S.; Clements, P.R.; Byers, S.; Kleijer, W.J.; Brooks, D.A.; Hopwood, J.J. Genotype-Phenotype Correlations in Mucopolysaccharidosis Type I Using Enzyme Kinetics, Immunoquantification and in Vitro Turnover Studies. Biochim. Biophys. Acta 1998, 1407, 249–256. [Google Scholar] [CrossRef]
- Church, H.; Tylee, K.; Cooper, A.; Thornley, M.; Mercer, J.; Wraith, E.; Carr, T.; O’Meara, A.; Wynn, R.F. Biochemical Monitoring after Haemopoietic Stem Cell Transplant for Hurler Syndrome (Mpsih): Implications for Functional Outcome after Transplant in Metabolic Disease. Bone Marrow Transplant. 2007, 39, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Krivit, W.; Peters, C.; Shapiro, E.G. Bone Marrow Transplantation as Effective Treatment of Central Nervous System Disease in Globoid Cell Leukodystrophy, Metachromatic Leukodystrophy, Adrenoleukodystrophy, Mannosidosis, Fucosidosis, Aspartylglucosaminuria, Hurler, Maroteaux-Lamy, and Sly Syndromes, and Gaucher Disease Type III. Curr. Opin. Neurol. 1999, 12, 167–176. [Google Scholar] [PubMed]
- Field, R.E.; Buchanan, J.A.; Copplemans, M.G.; Aichroth, P.M. Bone-Marrow Transplantation in Hurler's Syndrome. Effect on Skeletal Development. J. Bone Jt. Surg. Br. 1994, 76, 975–981. [Google Scholar] [PubMed]
- Peters, C.; Balthazor, M.; Shapiro, E.G.; King, R.J.; Kollman, C.; Hegland, J.D.; Henslee-Downey, J.; Trigg, M.E.; Cowan, M.J.; Sanders, J.; et al. Outcome of Unrelated Donor Bone Marrow Transplantation in 40 Children with Hurler Syndrome. Blood 1996, 87, 4894–4902. [Google Scholar] [PubMed]
- Kakkis, E.D.; Muenzer, J.; Tiller, G.E.; Waber, L.; Belmont, J.; Passage, M.; Izykowski, B.; Phillips, J.; Doroshow, R.; Walot, I.; et al. Enzyme-Replacement Therapy in Mucopolysaccharidosis I. N Engl. J. Med. 2001, 344, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes, M.; Doroshow, R.; Hoft, R.; Mason, G.; Walot, I.; Diament, M.; Okazaki, S.; Huff, K.; Cox, G.F.; Swiedler, S.J.; et al. A Follow-up Study of Mps I Patients Treated with Laronidase Enzyme Replacement Therapy for 6 Years. Mol. Genet. Metab. 2007, 90, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Barton, R.W.; Neufeld, E.F. The Hurler Corrective Factor. Purification and Some Properties. J. Biol. Chem. 1971, 246, 7773–7779. [Google Scholar] [PubMed]
- Cox-Brinkman, J.; Boelens, J.J.; Wraith, J.E.; O’Meara, A.; Veys, P.; Wijburg, F.A.; Wulffraat, N.; Wynn, R.F. Haematopoietic Cell Transplantation (Hct) in Combination with Enzyme Replacement Therapy (Ert) in Patients with Hurler Syndrome. Bone Marrow Transplant. 2006, 38, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.S.; Wynn, R.; Abdenur, J.E.; Burton, B.K.; Gharib, M.; Haase, C.; Hayashi, R.J.; Shenoy, S.; Sillence, D.; Tiller, G.E.; et al. Safety and Efficacy of Enzyme Replacement Therapy in Combination with Hematopoietic Stem Cell Transplantation in Hurler Syndrome. Genet. Med. 2005, 7, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Friso, A.; Tomanin, R.; Salvalaio, M.; Scarpa, M. Genistein Reduces Glycosaminoglycan Levels in a Mouse Model of Mucopolysaccharidosis Type Ii. Br. J. Pharmacol. 2010, 159, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, M.; Wilkinson, F.L.; Bennett, W.; Langford-Smith, K.J.; O’Leary, H.A.; Jakobkiewicz-Banecka, J.; Wynn, R.; Wraith, J.E.; Wegrzyn, G.; Bigger, B.W. Genistein Reduces Lysosomal Storage in Peripheral Tissues of Mucopolysaccharide Iiib Mice. Mol. Genet. Metab. 2009, 98, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Malinowska, M.; Wilkinson, F.L.; Langford-Smith, K.J.; Langford-Smith, A.; Brown, J.R.; Crawford, B.E.; Vanier, M.T.; Grynkiewicz, G.; Wynn, R.F.; Wraith, J.E.; et al. Genistein Improves Neuropathology and Corrects Behaviour in a Mouse Model of Neurodegenerative Metabolic Disease. PLoS ONE 2010, 5, e14192. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Maryniak, A.; Tylki-Szymanska, A.; Puk, E.; Liberek, A.; Wegrzyn, A.; Czartoryska, B.; Slominska-Wojewodzka, M.; Wegrzyn, G. Two-Year Follow-up of Sanfilippo Disease Patients Treated with a Genistein-Rich Isoflavone Extract: Assessment of Effects on Cognitive Functions and General Status of Patients. Med. Sci. Monit. 2011, 17, CR196–CR202. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, E.; Jakobkiewicz-Banecka, J.; Tylki-Szymanska, A.; Liberek, A.; Maryniak, A.; Malinowska, M.; Czartoryska, B.; Puk, E.; Kloska, A.; Liberek, T.; et al. Genistin-Rich Soy Isoflavone Extract in Substrate Reduction Therapy for Sanfilippo Syndrome: An Open-Label, Pilot Study in 10 Pediatric Patients. Curr. Ther. Res. Clin. Exp. 2008, 69, 166–179. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, J.; Valstar, M.J.; Narajczyk, M.; Wegrzyn, G.; Kulik, W.; Ijlst, L.; Wagemans, T.; van der Wal, W.M.; Wijburg, F.A. Genistein in Sanfilippo Disease: A Randomized Controlled Crossover Trial. Ann. Neurol. 2012, 71, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Malinova, V.; Wegrzyn, G.; Narajczyk, M. The Use of Elevated Doses of Genistein-Rich Soy Extract in the Gene Expression-Targeted Isoflavone Therapy for Sanfilippo Disease Patients. JIMD Rep. 2012, 5, 21–25. [Google Scholar] [PubMed]
- Kim, K.H.; Dodsworth, C.; Paras, A.; Burton, B.K. High Dose Genistein Aglycone Therapy Is Safe in Patients with Mucopolysaccharidoses Involving the Central Nervous System. Mol. Genet. Metab. 2013, 109, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Marucha, J.; Tylki-Szymanska, A.; Jakobkiewicz-Banecka, J.; Piotrowska, E.; Kloska, A.; Czartoryska, B.; Wegrzyn, G. Improvement in the Range of Joint Motion in Seven Patients with Mucopolysaccharidosis Type Ii During Experimental Gene Expression-Targeted Isoflavone Therapy (Get It). Am. J. Med. Genet. A 2011, 155A, 2257–2262. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Fletcher, J.M.; Moore, L.; Byers, S. Trans-Generational Exposure to Low Levels of Rhodamine B Does Not Adversely Affect Litter Size or Liver Function in Murine Mucopolysaccharidosis Type IIIA. Mol. Genet. Metab. 2010, 101, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Rees, M.H.; Klebe, S.; Fletcher, J.M.; Byers, S. Improvement in Behaviour after Substrate Deprivation Therapy with Rhodamine B in a Mouse Model of Mps IIIA. Mol. Genet. Metab. 2007, 92, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.L.; Thomas, B.J.; Wilkinson, A.S.; Fletcher, J.M.; Byers, S. Inhibition of Glycosaminoglycan Synthesis Using Rhodamine B in a Mouse Model of Mucopolysaccharidosis Type Iiia. Pediatr. Res. 2006, 60, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Terlato, N.J.; Cox, G.F. Can Mucopolysaccharidosis Type I Disease Severity Be Predicted Based on a Patient’s Genotype? A Comprehensive Review of the Literature. Genet. Med. 2003, 5, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Valstar, M.J.; Ruijter, G.J.; van Diggelen, O.P.; Poorthuis, B.J.; Wijburg, F.A. Sanfilippo Syndrome: A Mini-Review. J. Inherit. Metab. Dis. 2008, 31, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, G.; Jakobkiewicz-Banecka, J.; Narajczyk, M.; Wisniewski, A.; Piotrowska, E.; Gabig-Ciminska, M.; Kloska, A.; Slominska-Wojewodzka, M.; Korzon-Burakowska, A.; Wegrzyn, A. Why Are Behaviors of Children Suffering from Various Neuronopathic Types of Mucopolysaccharidoses Different? Med. Hypotheses 2010, 75, 605–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskins, M.E.; Jezyk, P.F.; Desnick, R.J.; McDonough, S.K.; Patterson, D.F. Alpha-l-Iduronidase Deficiency in a Cat: A Model of Mucopolysaccharidosis I. Pediatr. Res. 1979, 13, 1294–1297. [Google Scholar] [CrossRef] [PubMed]
- Shull, R.M.; Munger, R.J.; Spellacy, E.; Hall, C.W.; Constantopoulos, G.; Neufeld, E.F. Canine Alpha-l-Iduronidase Deficiency. A Model of Mucopolysaccharidosis I. Am. J. Pathol. 1982, 109, 244–248. [Google Scholar] [PubMed]
- Clarke, L.A.; Russell, C.S.; Pownall, S.; Warrington, C.L.; Borowski, A.; Dimmick, J.E.; Toone, J.; Jirik, F.R. Murine Mucopolysaccharidosis Type I: Targeted Disruption of the Murine Alpha-l-Iduronidase Gene. Hum. Mol. Genet. 1997, 6, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.; Hendson, G.; Jevon, G.; Matlock, T.; Yu, J.; Aklujkar, M.; Ng, K.Y.; Clarke, L.A. Murine Mps I: Insights into the Pathogenesis of Hurler Syndrome. Clin. Genet. 1998, 53, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Sciascia, A., 2nd; Vorhees, C.V.; Williams, M.T. Progression of Multiple Behavioral Deficits with Various Ages of Onset in a Murine Model of Hurler Syndrome. Brain Res. 2008, 1188, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Derrick-Roberts, A.L.; Pyragius, C.E.; Kaidonis, X.M.; Jackson, M.R.; Anson, D.S.; Byers, S. Lentiviral-Mediated Gene Therapy Results in Sustained Expression of Beta-Glucuronidase for up to 12 Months in the Gus(Mps/Mps) and up to 18 Months in the Gus(Tm(L175f)Sly) Mouse Models of Mucopolysaccharidosis Type VII. Hum. Gene Ther. 2014, 25, 798–810. [Google Scholar] [CrossRef] [PubMed]
- Macsai, C.E.; Derrick-Roberts, A.L.; Ding, X.; Zarrinkalam, K.H.; McIntyre, C.; Anderson, P.H.; Anson, D.S.; Byers, S. Skeletal Response to Lentiviral Mediated Gene Therapy in a Mouse Model of Mps VII. Mol. Genet. Metab. 2012, 106, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Blumenkrantz, N.; Asboe-Hansen, G. New Method for Quantitative Determination of Uronic Acids. Anal. Biochem. 1973, 54, 484–489. [Google Scholar] [CrossRef]
- Leaback, D.H.; Walker, P.G. Studies on Glucosaminidase. 4. The Fluorimetric Assay of N-Acetyl-Beta-Glucosaminidase. Biochem. J. 1961, 78, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A Simple Method for the Isolation and Purification of Total Lipides from Animal Tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Williams, M.A.; McCluer, R.H. The Use of Sep-Pak C18 Cartridges During the Isolation of Gangliosides. J. Neurochem. 1980, 35, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Kaidonis, X.; Byers, S.; Ranieri, E.; Sharp, P.; Fletcher, J.; Derrick-Roberts, A. N-Butyldeoxynojirimycin Treatment Restores the Innate Fear Response and Improves Learning in Mucopolysaccharidosis IIIA Mice. Mol. Genet. Metab. 2016, 118, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Derrick-Roberts, A.L.; Panir, K.; Pyragius, C.E.; Zarrinkalam, K.H.; Atkins, G.J.; Byers, S. Reversal of Established Bone Pathology in Mps Vii Mice Following Lentiviral-Mediated Gene Therapy. Mol. Genet. Metab. 2016, 119, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Dire, D.J.; Wilkinson, J.A. Acute Exposure to Rhodamine B. J. Toxicol. Clin. Toxicol. 1987, 25, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Kelner, M.J. Rhodamine B Ingestion as a Cause of Fluorescent Red Urine. West J. Med. 1985, 143, 523–524. [Google Scholar] [PubMed]
- Crawley, A.C.; Gliddon, B.L.; Auclair, D.; Brodie, S.L.; Hirte, C.; King, B.M.; Fuller, M.; Hemsley, K.M.; Hopwood, J.J. Characterization of a C57bl/6 Congenic Mouse Strain of Mucopolysaccharidosis Type IIIA. Brain Res. 2006, 1104, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.Q. A Contradictory Treatment for Lysosomal Storage Disorders: Inhibitors Enhance Mutant Enzyme Activity. Trends Pharmacol. Sci. 2003, 24, 355–360. [Google Scholar] [CrossRef]
- Garcia-Rivera, M.F.; Colvin-Wanshura, L.E.; Nelson, M.S.; Nan, Z.; Khan, S.A.; Rogers, T.B.; Maitra, I.; Low, W.C.; Gupta, P. Characterization of an Immunodeficient Mouse Model of Mucopolysaccharidosis Type I Suitable for Preclinical Testing of Human Stem Cell and Gene Therapy. Brain Res. Bull. 2007, 74, 429–438. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal | MPS I Untreated | MPS I SDT | |
|---|---|---|---|
| GM1 | 14049.98 ± 497.03 | 13154.62 ± 203.63 | 13202.02 ± 198.08 |
| GM2 | 1417.10 ± 56.73 | 4700.51 ± 79.80 ** | 4607.25 ± 53.41 ** |
| GM3 | 1738.95 ± 114.69 | 5175.89 ± 238.03 ** | 4890.26 ± 106.87 ** |
| Normal | MPS I Untreated | MPS I SDT | |
|---|---|---|---|
| Brain | 184.26 ± 13.74 | 1102.85 ± 98.46 ** | 987.06 ± 84.54 ** |
| Liver | 1009.92 ± 103.21 | 9169.05 ± 443.00 ** | 8975.07 ± 488.27 ** |
| Kidney | 859.83 ± 0.58 | 5857.89 ± 11.67 ** | 7239.73 ± 0.001 ** |
| Spleen | 3285.37 ± 380.58 | 5094.60 ± 485.28 * | 4412.64 ± 329.73 |
| Heart | 241.51 ± 23.76 | 5118.18 ± 338.74 ** | 4885.17 ± 414.17 ** |
| Lung | 404.69 ± 54.90 | 6068.17 ± 460.39 ** | 5755.82 ± 453.01 ** |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derrick-Roberts, A.L.K.; Jackson, M.R.; Pyragius, C.E.; Byers, S. Substrate Deprivation Therapy to Reduce Glycosaminoglycan Synthesis Improves Aspects of Neurological and Skeletal Pathology in MPS I Mice. Diseases 2017, 5, 5. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases5010005
Derrick-Roberts ALK, Jackson MR, Pyragius CE, Byers S. Substrate Deprivation Therapy to Reduce Glycosaminoglycan Synthesis Improves Aspects of Neurological and Skeletal Pathology in MPS I Mice. Diseases. 2017; 5(1):5. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases5010005
Chicago/Turabian StyleDerrick-Roberts, Ainslie L. K., Matilda R. Jackson, Carmen E. Pyragius, and Sharon Byers. 2017. "Substrate Deprivation Therapy to Reduce Glycosaminoglycan Synthesis Improves Aspects of Neurological and Skeletal Pathology in MPS I Mice" Diseases 5, no. 1: 5. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases5010005