Progressive Immunodeficiency with Gradual Depletion of B and CD4+ T Cells in Immunodeficiency, Centromeric Instability and Facial Anomalies Syndrome 2 (ICF2)

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Analysis
2.2. Phenotypic Analyses of Lymphocytes
2.3. Proliferation Assays
2.4. Cytotoxicity Assay
3. Results
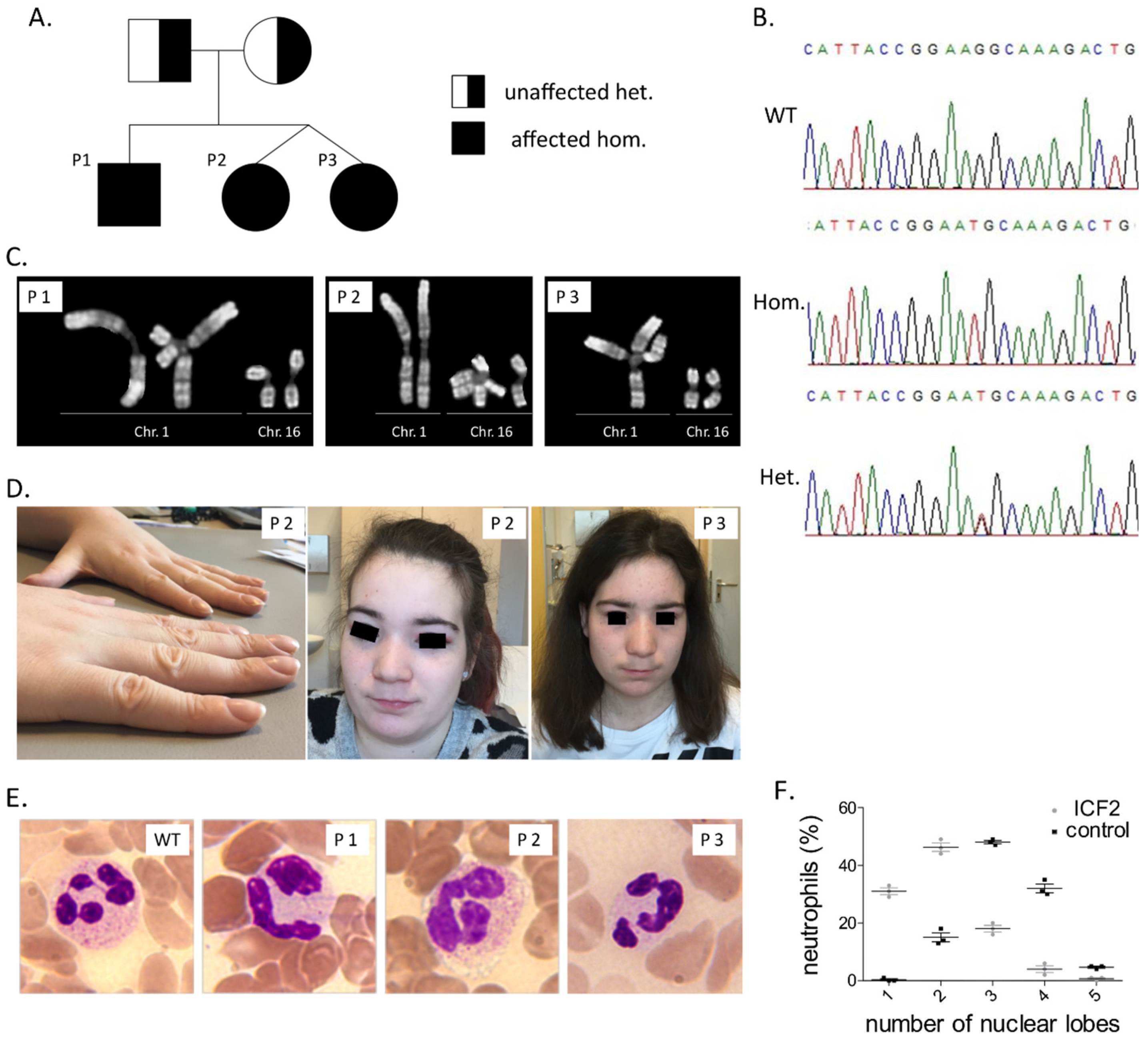
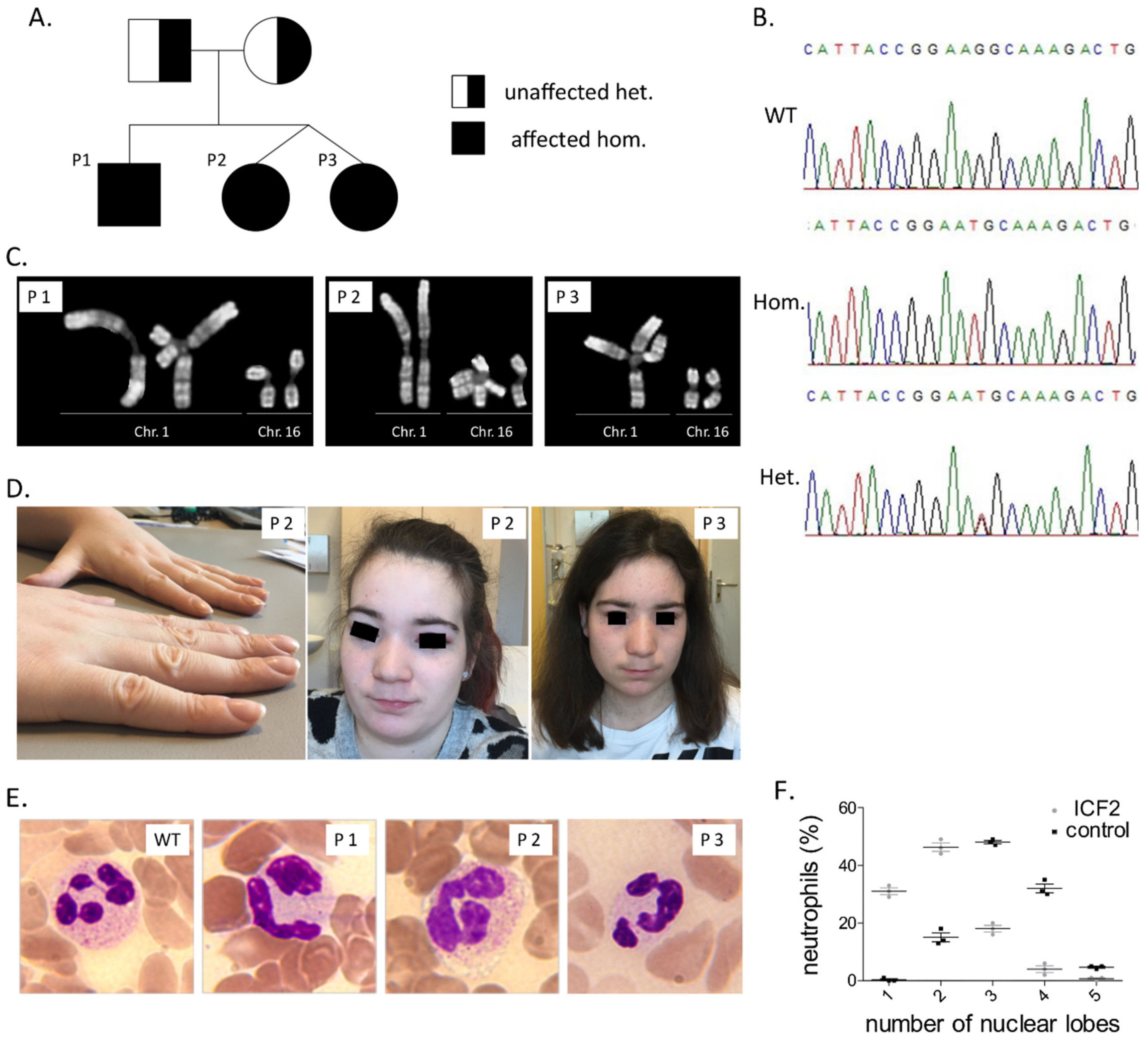
3.1. Clinical Findings
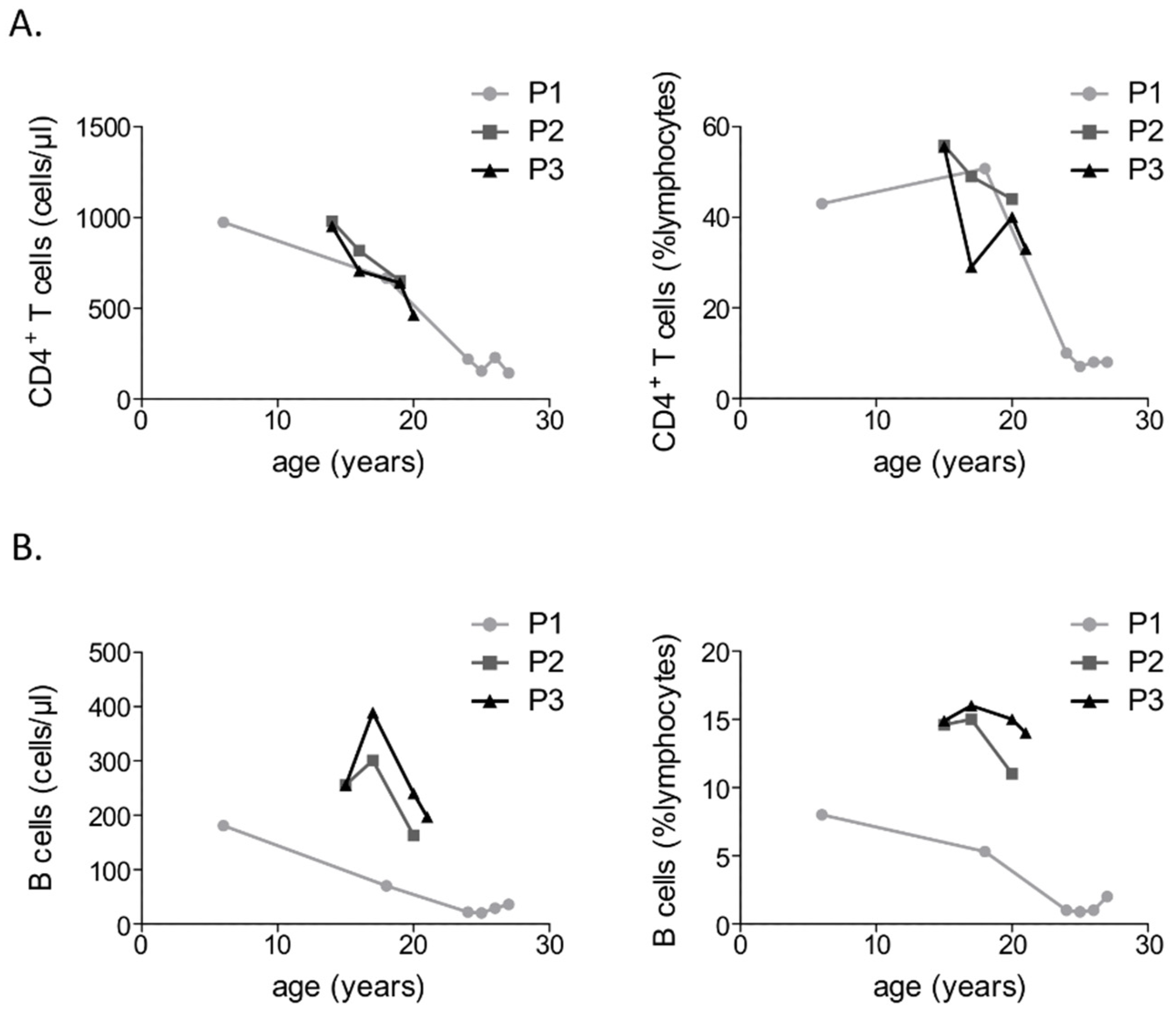
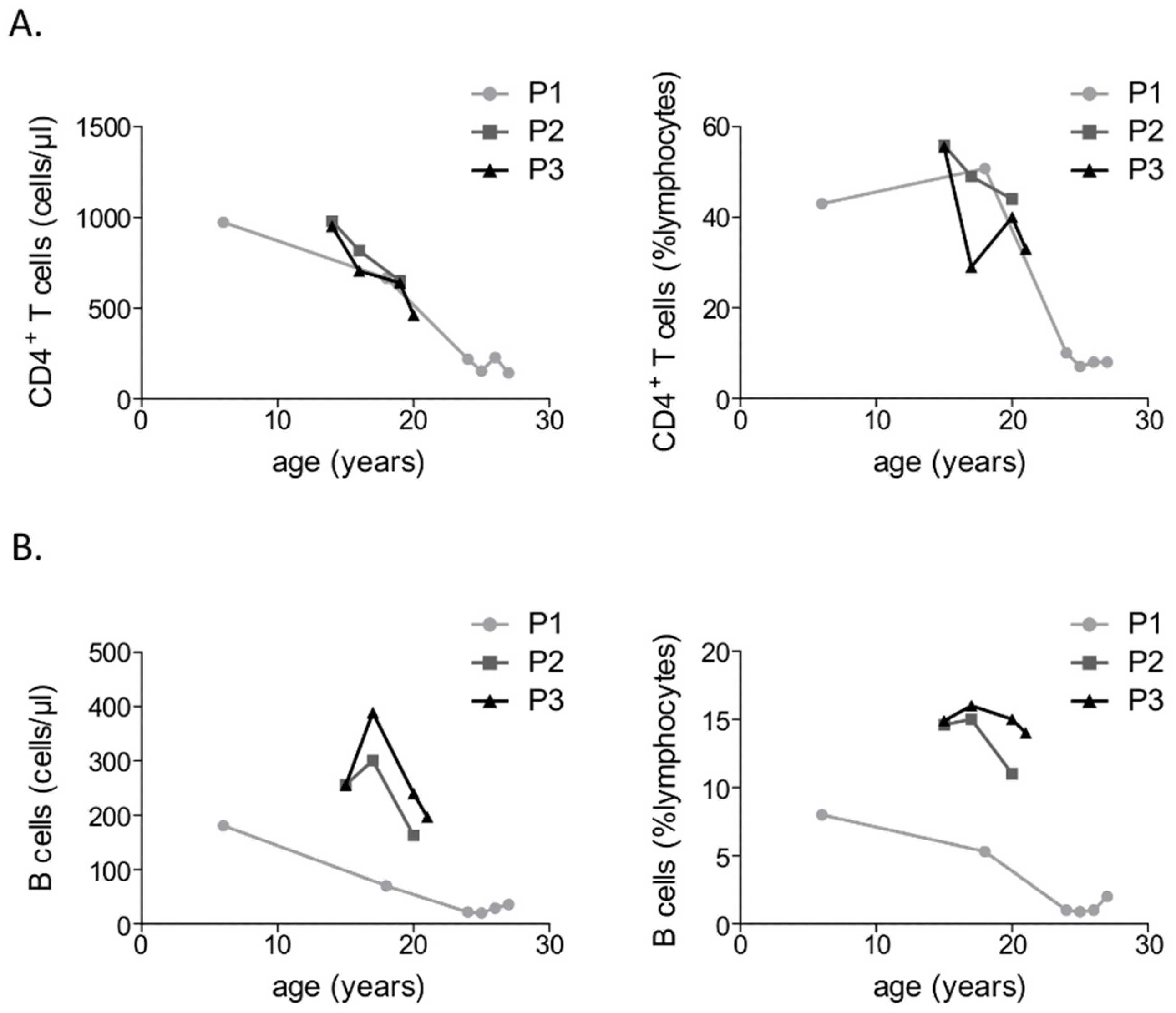
3.2. Immunological Investigations
3.3. Identification of A Pathogenic Mutation in ZBTB24
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ehrlich, M.; Sanchez, C.; Shao, C.; Nishiyama, R.; Kehrl, J.; Kuick, R.; Kubota, T.; Hanash, S.M. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity 2008, 41, 253–271. [Google Scholar] [CrossRef]
- Schuetz, C.; Barbi, G.; Barth, T.F.; Hoenig, M.; Schulz, A.; Möeller, P. ICF syndrome: High variability of the chromosomal phenotype and association with classical Hodgkin lymphoma. Am. J. Med. Genet. A 2007, 143A, 2052–2057. [Google Scholar] [CrossRef]
- Weemaes, C.M.; van Tol, M.J.; Wang, J.; van Ostaijen-ten Dam, M.M.; van Eggermond, M.C.; Thijssen, P.E.; Aytekin, C.; Brunetti-Pierri, N.; van der Burg, M.; Graham Davies, E.; et al. Heterogeneous clinical presentation in ICF syndrome: Correlation with underlying gene defects. Eur. J. Hum. Genet. 2013, 21, 1219–1225. [Google Scholar] [CrossRef]
- Miniou, P.; Bourc’his, D.; Molina Gomes, D.; Jeanpierre, M.; Viegas-Péquignot, E. Undermethylation of Alu sequences in ICF syndrome: Molecular and in situ analysis. Cytogenet. Cell Genet. 1997, 77, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Jeanpierre, M.; Turleau, C.; Aurias, A.; Prieur, M.; Ledeist, F.; Fischer, A.; Viegas-Pequignot, E. An embryonic-like methylation pattern of classical satellite DNA is observed in ICF syndrome. Hum. Mol. Genet. 1993, 2, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Wijmenga, C.; Luo, P.; Stanek, A.M.; Canfield, T.K.; Weemaes, C.M.; Gartler, S.M. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 14412–14417. [Google Scholar] [CrossRef] [PubMed]
- De Greef, J.C.; Wang, J.; Balog, J.; den Dunnen, J.T.; Frants, R.R.; Straasheijm, K.R.; Aytekin, C.; van der Burg, M.; Duprez, L.; Ferster, A.; et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am. J. Hum. Genet. 2011, 88, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, P.E.; Ito, Y.; Grillo, G.; Wang, J.; Velasco, G.; Nitta, H.; Unoki, M.; Yoshihara, M.; Suyama, M.; Sun, Y.; et al. Mutations in CDCA7 and HELLS cause immunodeficiency-centromeric instability-facial anomalies syndrome. Nat. Commun. 2015, 6, 7870. [Google Scholar] [CrossRef]
- Van den Boogaard, M.L.; Thijssen, P.E.; Aytekin, C.; Licciardi, F.; Kıykım, A.A.; Spossito, L.; Dalm, V.A.S.H.; Driessen, G.J.; Kersseboom, R.; de Vries, F.; et al. Expanding the mutation spectrum in ICF syndrome: Evidence for a gender bias in ICF2. Clin. Genet. 2017, 4, 380–387. [Google Scholar] [CrossRef]
- Velasco, G.; Grillo, G.; Touleimat, N.; Ferry, L.; Ivkovic, I.; Ribierre, F.; Deleuze, J.F.; Chantalat, S.; Picard, C.; Francastel, C.; et al. Comparative methylome analysis of ICF patients identifies heterochromatin loci that require ZBTB24, CDCA7 and HELLS for their methylated state. Hum. Mol. Genet. 2018, 27, 2409–2424. [Google Scholar] [CrossRef]
- Liang, J.; Yan, R.; Chen, G.; Feng, J.; Wu, W.W.; Ren, W.; Zhu, C.; Zhao, Y.; Gao, X.M.; Wang, J. Downregulation of ZBTB24 hampers the G0/1- to S-phase cell-cycle transition via upregulating the expression of IRF-4 in human B cells. Genes Immun. 2016, 17, 276–282. [Google Scholar] [CrossRef]
- Chevrier, S.; Corcoran, L.M. BTB-ZF transcription factors, a growing family of regulators of early and late B-cell development. Immunol. Cell Biol. 2014, 92, 481–488. [Google Scholar] [CrossRef]
- Kamae, C.; Imai, K.; Kato, T.; Okano, T.; Honma, K.; Nakagawa, N.; Yeh, T.W.; Noguchi, E.; Ohara, A.; Shigemura, T.; et al. Clinical and Immunological Characterization of ICF Syndrome in Japan. J. Clin. Immunol. 2018, 38, 927–937. [Google Scholar] [CrossRef]
- Sogkas, G.; Fedchenko, M.; Dhingra, A.; Jablonka, A.; Schmidt, R.E.; Atschekzei, F. Primary immunodeficiency disorder caused by phosphoinositide 3-kinase δ deficiency. J. Allergy Clin. Immunol. 2018, 142, 1650–1653. [Google Scholar] [CrossRef]
- Rosenwald, A.; Ott, G.; Katzenberger, T.; Siebert, R.; Kalla, J.; Kuse, R.; Ott, M.M.; Müller-Hermelink, H.K.; Schlegelberger, B. Jumping translocation of 1q as the sole aberration in a case of follicular lymphoma. Cancer Genet. Cytogenet. 1999, 108, 53–56. [Google Scholar] [CrossRef]
- Jacobs, R.; Hintzen, G.; Kemper, A.; Beul, K.; Kempf, S.; Behrens, G.; Sykora, K.W.; Schmidt, R.E. CD56bright cells differ in their KIR repertoire and cytotoxic features from CD56dim NK cells. Eur. J. Immunol. 2001, 31, 3121–3126. [Google Scholar] [CrossRef]
- Chatterjee, D.; Marquardt, N.; Tufa, D.M.; Beauclair, G.; Low, H.Z.; Hatlapatka, T.; Hass, R.; Kasper, C.; von Kaisenberg, C.; Schmidt, R.E.; et al. Role of gamma-secretase in human umbilical-cord derived mesenchymal stem cell mediated suppression of NK cell cytotoxicity. Cell. Commun. Signal. 2014, 12, 63. [Google Scholar] [CrossRef]
- Von Bernuth, H.; Ravindran, E.; Du, H.; Fröhler, S.; Strehl, K.; Krämer, N.; Issa-Jahns, L.; Amulic, B.; Ninnemann, O.; Xiao, M.S.; et al. Combined immunodeficiency develops with age in Immunodeficiency-centromeric instability-facial anomalies syndrome 2 (ICF2). Orphanet J. Rare Dis. 2014, 9, 116. [Google Scholar] [CrossRef]
- Sterlin, D.; Velasco, G.; Moshous, D.; Touzot, F.; Mahlaoui, N.; Fischer, A.; Suarez, F.; Francastel, C.; Picard, C. Genetic, Cellular and Clinical Features of ICF Syndrome: A French National Survey. J. Clin. Immunol. 2016, 36, 149–159. [Google Scholar] [CrossRef]
- Nitta, H.; Unoki, M.; Ichiyanagi, K.; Kosho, T.; Shigemura, T.; Takahashi, H.; Velasco, G.; Francastel, C.; Picard, C.; Kubota, T.; et al. Three novel ZBTB24 mutations identified in Japanese and Cape Verdean type 2 ICF syndrome patients. J. Hum. Genet. 2013, 58, 455–460. [Google Scholar] [CrossRef]
- Licciardi, F.; van den Boogaard, M.; Delle Piane, M.; Tovo, P.A.; Montin, D. EBV-Related Hodgkin Lymphoma in an ICF2 Patient: Is EBV Susceptibility a Hallmark of This ICF Subtype? J. Clin. Immunol. 2019, 1–3. [Google Scholar] [CrossRef]
- Harnisch, E.; Buddingh, E.P.; Thijssen, P.E.; Brooks, A.S.; Driessen, G.L.; Kersseboom, R.; Lankester, A.C. Hematopoietic Stem Cell Transplantation in a Patient with ICF2 Syndrome Presenting With EBV-Induced Hemophagocytic Lymphohystiocytosis. Transplantation 2016, 100, e35–e36. [Google Scholar] [CrossRef]
- Chouery, E.; Abou-Ghoch, J.; Corbani, S.; El Ali, N.; Korban, R.; Salem, N.; Castro, C.; Klayme, S.; Azoury-Abou Rjeily, M.; Khoury-Matar, R.; et al. A novel deletion in ZBTB24 in a Lebanese family with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Clin. Genet. 2012, 82, 489–493. [Google Scholar] [CrossRef]
- Cerbone, M.; Wang, J.; Van der Maarel, S.M.; D’Amico, A.; D’Agostino, A.; Romano, A.; Brunetti-Pierri, N. Immunodeficiency, centromeric instability, facial anomalies (ICF) syndrome, due to ZBTB24 mutations, presenting with large cerebral cyst. Am. J. Med. Genet. A 2012, 158A, 2043–2046. [Google Scholar] [CrossRef]
- Kloeckener-Gruissem, B.; Betts, D.R.; Zankl, A.; Berger, W.; Güngör, T. A new and a reclassified ICF patient without mutations in DNMT3B and its interacting proteins SUMO-1 and UBC9. Am. J. Med. Genet. A 2005, 136, 31–37. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Year of Birth | Age at First Diagnosis of PID/First Immunological Evaluation | Facial Anomalies | Motor Development | Intellectual Development | Infections | Other |
|---|---|---|---|---|---|---|---|---|
| P1 | M | 1992 | 3 | broad flat nasal bridge, micrognathia | normal | Attention deficit hyperactivity disorder, treatment with methylphenidate till the 15th year of life, otherwise normal, work without difficulty | sepsis after birth, three pneumonias (H. influenzae), recurrent upper respiratory tract infections (bronchitis, otitis media, sinusitis) | atopic dermatitis |
| P2 | F | 1996 | 7 | broad flat nasal bridge | normal | Selective mutism in early childhood, otherwise normal, work without difficulty | Atypical mycobacteriosis, recurrent shingles, prolonged fever after vaccination with MMR | atopic dermatitis, scoliosis, nail clubbing, focal bronchial malformation with absence of bronchial glands and cartilage |
| P3 | F | 1996 | 7 | broad flat nasal bridge | normal | Selective mutism in early childhood, otherwise normal, work without difficulty | none | atopic dermatitis, idiopathic epileptic seizures |
| Immunological Test | P1 | P2 | P3 | Normal Range (or Control Value) |
|---|---|---|---|---|
| Year of birth | 1992 | 1996 | 1996 | |
| Serum Ig concentration/year of test | 1997 | 2002 | 2002 | |
| -IgG (g/L) | 2.9 | 10.1 | 9.67 | 5.5–12.3 |
| -IgA (g/L) | 0.73 | 1.81 | 1.16 | 0.23–1.8 |
| -IgM (g/L) | 0.11 | 0.16 | 0.13 | 0.37–1.6 |
| -IgE (IE/mL) | 8.00 | 2 | 2 | 3–75 |
| -IgG1 (g/L) | 2.00 | 8.56 | 8.14 | 4.2–9.9 |
| -IgG2 (g/L) | 0.58 | 1.05 | 1.09 | 0.63–3.5 |
| -IgG3 (g/L) | 0.17 | 0.74 | 0.69 | 0.17–0.88 |
| -IgG4 (g/L) | 0.15 | 0.02 | 0.02 | 0.01–1.2 |
| Lymphocyte subset analysis/year of test | 2017 | 2012 | 2012 | |
| Absolute lymphocyte count (cells/µL) | 2193 | 1755 | 1710 | 1100–4500 |
| Lymphocytes (% leukocytes) | 43 | 27 | 30 | 20–44 |
| CD19 B cells (% lymphocytes) | 0,9 | 14.6 | 14.9 | 4.3–23.1 |
| CD27-IgM+IgD+ naive B cells (% lymphocytes) | NM | 13.8 | 14.1 | 2.6–15.7 |
| CD27+IgM+IgD+ IgM Mem/Marg. Zone B cells (% lymphocytes) | NM | 0.4 | 0.3 | 0.2–12.3 |
| CD27+IgM-IgD-switched mem. Post GC B cells (% lymphocytes) | NM | 0.3 | 0.3 | 1.9–30.4 |
| CD38++IgM++ transitional B cells (% lymphocytes) | NM | 0.3 | 1.1 | 0.6–3.5 |
| CD38+++IgM+/− plasma cells (% lymphocytes) | NM | 0.2 | 0.3 | 0.4–3.6 |
| CD21lowCD38low B cells (% lymphocytes) | NM | 0.1 | 0.1 | 4–26 |
| CD16+CD56+CD3- NK cells (% lymphocytes) | 1.4 | 4.7 | 5.9 | 7–31 |
| CD3+ T cells (% lymphocytes) | 92.4 | 68.8 | 66.6 | 55–83 |
| CD3+CD4+ T helper cells (% lymphocytes) | 7.1 | 55.9 | 55.6 | 27–53 |
| CD3+CD8+ cytotoxic T cells (% lymphocytes) | 84.5 | 11.5 | 9 | 19–34 |
| CD4+CD45RO+ CD4 memory cells (% lymphocytes) | 5.7 | 17 | 19.8 | 11–44 |
| CD4+CD45RA+ naive CD4+ T cells (% lymphocytes) | 1.9 | 40.1 | 37.9 | 21–75 |
| CD4+CD45RA+CD31+ thymus emigrated CD4+ T cells (% lymphocytes) | 1.7 | 36.5 | 35 | 19.4–60.9 |
| CD8+CD27-CD28- late CD8+ effector cells (% lymphocytes) | 5.4 | 1.3 | 0.3 | 2.9–16 |
| CD8+CD27+CD28- CD8+ effector cells (% lymphocytes) | 20 | 1.8 | 1.2 | 2.6–58 |
| Lymphocyte proliferation assay | ||||
| -Medium control (% compared to control) | 126 | 102 | 100 | 100 |
| -Phytohemagglutinin (PHA) (% compared to control) | 71 | 134 | 136 | 100 |
| -Concanavalin A (ConA) (% compared to control) | 80 | 128 | 113 | 100 |
| -Pokeweed mitogen (PWM) (% compared to control) | 80 | 141 | 151 | 100 |
| -Tuberculin purified protein derivative (PPD) (% compared to control) | 120 | 122 | 110 | 100 |
| -Interleukin 2 (IL-2) (% compared to control) | 84 | 129 | 130 | 100 |
| -Anti-CD3-antibody (% compared to control) | 118 | 148 | 154 | 100 |
| NK cell activity | ||||
| -Natural cytotoxicity (1:30) (% compared to control) | 29 | 20 | 47 | 100 |
| -ADCC (1:30) (% compared to control) | 26 | 67 | 67 | 100 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sogkas, G.; Dubrowinskaja, N.; Bergmann, A.K.; Lentes, J.; Ripperger, T.; Fedchenko, M.; Ernst, D.; Jablonka, A.; Geffers, R.; Baumann, U.; et al. Progressive Immunodeficiency with Gradual Depletion of B and CD4+ T Cells in Immunodeficiency, Centromeric Instability and Facial Anomalies Syndrome 2 (ICF2). Diseases 2019, 7, 34. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases7020034
Sogkas G, Dubrowinskaja N, Bergmann AK, Lentes J, Ripperger T, Fedchenko M, Ernst D, Jablonka A, Geffers R, Baumann U, et al. Progressive Immunodeficiency with Gradual Depletion of B and CD4+ T Cells in Immunodeficiency, Centromeric Instability and Facial Anomalies Syndrome 2 (ICF2). Diseases. 2019; 7(2):34. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases7020034
Chicago/Turabian StyleSogkas, Georgios, Natalia Dubrowinskaja, Anke K. Bergmann, Jana Lentes, Tim Ripperger, Mykola Fedchenko, Diana Ernst, Alexandra Jablonka, Robert Geffers, Ulrich Baumann, and et al. 2019. "Progressive Immunodeficiency with Gradual Depletion of B and CD4+ T Cells in Immunodeficiency, Centromeric Instability and Facial Anomalies Syndrome 2 (ICF2)" Diseases 7, no. 2: 34. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases7020034