Outcomes of Kidney Transplantation in Fabry Disease: A Meta-Analysis

 , , , , , , and
, , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Search Strategies
2.2. Study Selection
2.3. Data Extraction
2.4. Data Synthesis and Statistical Analysis
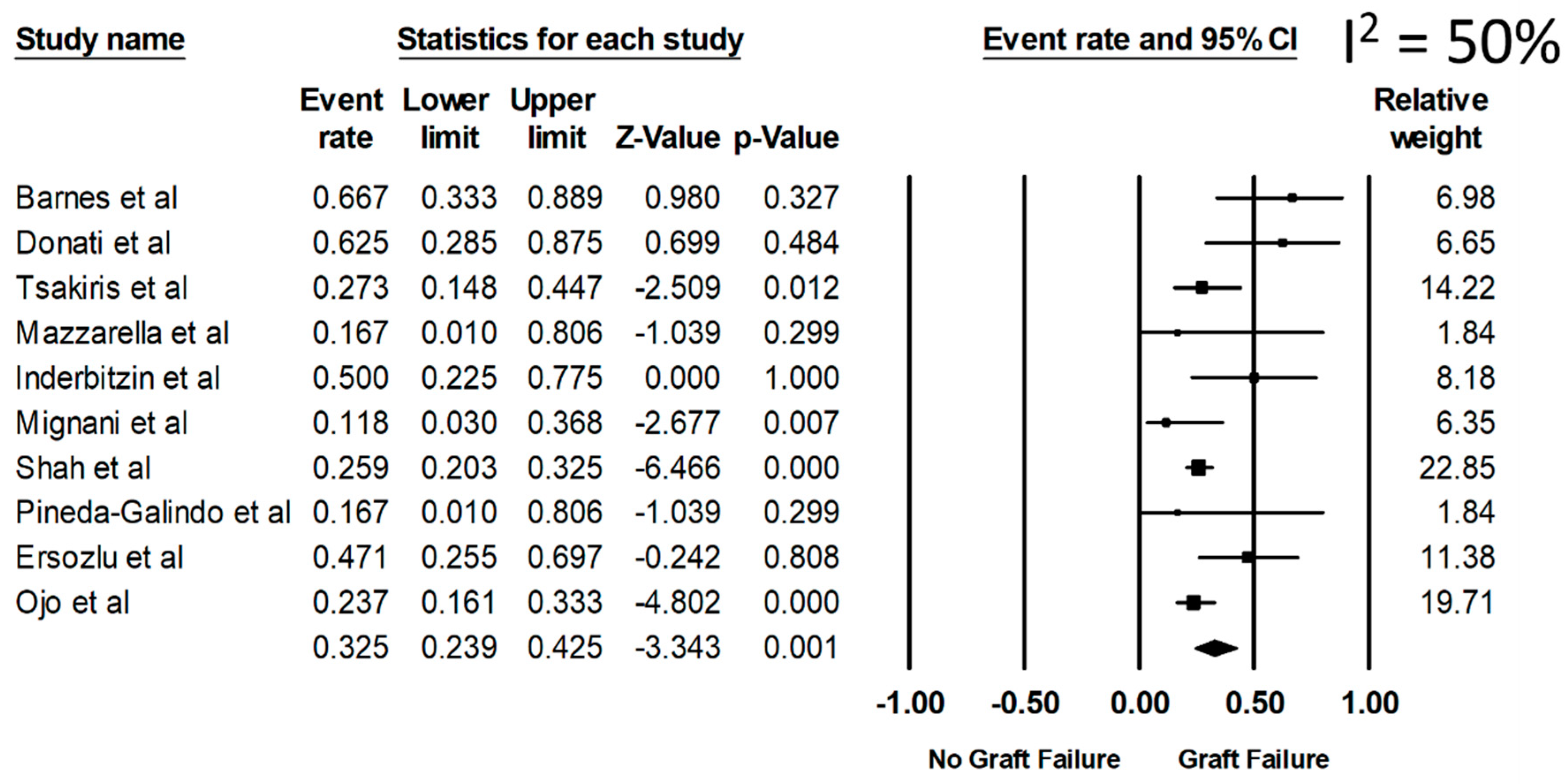
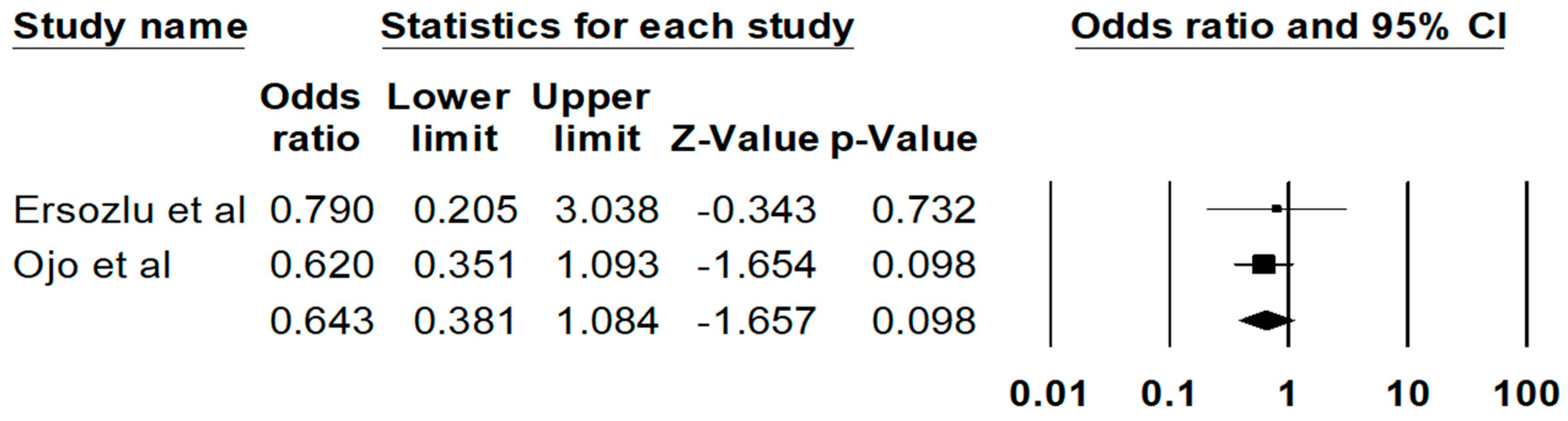
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germain, D.P.; Oliveira, J.P.; Bichet, D.G.; Yoo, H.W.; Hopkin, R.J.; Lemay, R.; Politei, J.; Wanner, C.; Wilcox, W.R.; Warnock, D.G. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: A consensus classification system by a multispecialty Fabry disease genotype-phenotype workgroup. J. Med. Genet. 2020, 57, 542–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashley, G.A.; Shabbeer, J.; Yasuda, M.; Eng, C.M.; Desnick, R.J. Fabry disease: Twenty novel alpha-galactosidase A mutations causing the classical phenotype. J. Hum. Genet. 2001, 46, 192–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton-Prolla, P.; Tong, B.; Shabbeer, J.; Astrin, K.H.; Eng, C.M.; Desnick, R.J. Fabry disease: Twenty-two novel mutations in the alpha-galactosidase A gene and genotype/phenotype correlations in severely and mildly affected hemizygotes and heterozygotes. J. Investig. Med. 2000, 48, 227–235. [Google Scholar] [PubMed]
- Davies, J.P.; Eng, C.M.; Hill, J.A.; Malcolm, S.; MacDermot, K.; Winchester, B.; Desnick, R.J. Fabry disease: Fourteen alpha-galactosidase A mutations in unrelated families from the United Kingdom and other European countries. Eur. J. Hum. Genet. 1996, 4, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Ashley, G.A.; Burgert, T.S.; Enriquez, A.L.; D’Souza, M.; Desnick, R.J. Fabry disease: Thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol. Med. 1997, 3, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.M.; Niehaus, D.J.; Enriquez, A.L.; Burgert, T.S.; Ludman, M.D.; Desnick, R.J. Fabry disease: Twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase A gene. Hum. Mol. Genet. 1994, 3, 1795–1799. [Google Scholar] [CrossRef]
- Germain, D.; Biasotto, M.; Tosi, M.; Meo, T.; Kahn, A.; Poenaru, L. Fluorescence-assisted mismatch analysis (FAMA) for exhaustive screening of the alpha-galactosidase A gene and detection of carriers in Fabry disease. Hum. Genet. 1996, 98, 719–726. [Google Scholar] [CrossRef]
- Germain, D.P. A new phenotype of Fabry disease with intermediate severity between the classical form and the cardiac variant. Contrib. Nephrol. 2001, 234–240. [Google Scholar] [CrossRef]
- Germain, D.P.; Poenaru, L. Fabry disease: Identification of novel alpha-galactosidase A mutations and molecular carrier detection by use of fluorescent chemical cleavage of mismatches. Biochem. Biophys. Res. Commun. 1999, 257, 708–713. [Google Scholar] [CrossRef]
- Germain, D.P.; Shabbeer, J.; Cotigny, S.; Desnick, R.J. Fabry disease: Twenty novel alpha-galactosidase A mutations and genotype-phenotype correlations in classical and variant phenotypes. Mol. Med. 2002, 8, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Kase, R.; Sakuraba, H.; Suzuki, Y. Characterization of a mutant alpha-galactosidase gene product for the late-onset cardiac form of Fabry disease. Biochem. Biophys. Res. Commun. 1993, 197, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Nakao, S.; Minamikawa-Tachino, R.; Desnick, R.J.; Fan, J.Q. Alternative splicing in the alpha-galactosidase A gene: Increased exon inclusion results in the Fabry cardiac phenotype. Am. J. Hum. Genet. 2002, 70, 994–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, S.; Sakuraba, H.; Suzuki, Y. Point mutations in the upstream region of the alpha-galactosidase A gene exon 6 in an atypical variant of Fabry disease. Hum. Genet. 1992, 89, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Ploos van Amstel, J.K.; Jansen, R.P.; de Jong, J.G.; Hamel, B.C.; Wevers, R.A. Six novel mutations in the alpha-galactosidase A gene in families with Fabry disease. Hum. Mol. Genet. 1994, 3, 503–505. [Google Scholar] [CrossRef]
- Sakuraba, H.; Oshima, A.; Fukuhara, Y.; Shimmoto, M.; Nagao, Y.; Bishop, D.F.; Desnick, R.J.; Suzuki, Y. Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease. Am. J. Hum. Genet. 1990, 47, 784–789. [Google Scholar]
- Shabbeer, J.; Yasuda, M.; Luca, E.; Desnick, R.J. Fabry disease: 45 novel mutations in the alpha-galactosidase A gene causing the classical phenotype. Mol. Genet. Metab. 2002, 76, 23–30. [Google Scholar] [CrossRef]
- Shabbeer, J.; Yasuda, M.; Benson, S.D.; Desnick, R.J. Fabry disease: Identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum. Genom. 2006, 2, 297–309. [Google Scholar] [CrossRef]
- Topaloglu, A.K.; Ashley, G.A.; Tong, B.; Shabbeer, J.; Astrin, K.H.; Eng, C.M.; Desnick, R.J. Twenty novel mutations in the alpha-galactosidase A gene causing Fabry disease. Mol. Med. 1999, 5, 806–811. [Google Scholar] [CrossRef] [Green Version]
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.H.; Park, M.H.; Byeon, J.J.; Lee, B.I.; Park, Y.; Ko, A.R.; Seong, M.R.; Lee, S.; Kim, M.R.; Seo, J.; et al. A Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometric Assay for the Quantification of Fabry Disease Biomarker Globotriaosylceramide (GB3) in Fabry Model Mouse. Pharmaceutics 2018, 10, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, R.; Russo, C.; Santoro, C.; Cocozza, S.; Riccio, E.; Sorrentino, R.; Pontillo, G.; Luciano, F.; Imbriaco, M.; Brunetti, A.; et al. Association between Left Atrial Deformation and Brain Involvement in Patients with Anderson-Fabry Disease at Diagnosis. J. Clin. Med. 2020, 9, 2741. [Google Scholar] [CrossRef] [PubMed]
- Levstek, T.; Vujkovac, B.; Trebusak Podkrajsek, K. Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes 2020, 11, 1091. [Google Scholar] [CrossRef] [PubMed]
- Barba-Romero, M.; Pintos-Morell, G. Gender Differences in the Application of Spanish Criteria for Initiation of Enzyme Replacement Therapy for Fabry Disease in the Fabry Outcome Survey. Int. J. Mol. Sci. 2016, 17, 1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veroux, M.; Monte, I.P.; Rodolico, M.S.; Corona, D.; Bella, R.; Basile, A.; Palmucci, S.; Pistorio, M.L.; Lanza, G.; De Pasquale, C.; et al. Screening for Fabry Disease in Kidney Transplant Recipients: Experience of a Multidisciplinary Team. Biomedicines 2020, 8, 396. [Google Scholar] [CrossRef] [PubMed]
- Capelli, I.; Aiello, V.; Gasperoni, L.; Comai, G.; Corradetti, V.; Ravaioli, M.; Biagini, E.; Graziano, C.; La Manna, G. Kidney Transplant in Fabry Disease: A Revision of the Literature. Medicina 2020, 56, 284. [Google Scholar] [CrossRef]
- Ravarotto, V.; Simioni, F.; Carraro, G.; Bertoldi, G.; Pagnin, E.; Calò, L.A. Oxidative Stress and Cardiovascular-Renal Damage in Fabry Disease: Is There Room for a Pathophysiological Involvement? J. Clin. Med. 2018, 7, 409. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.A.; Aguiar, P.; Deegan, P.B.; Ezgu, F.; Frustaci, A.; Lidove, O.; Linhart, A.; Lubanda, J.C.; Moon, J.C.; Nicholls, K.; et al. Early indicators of disease progression in Fabry disease that may indicate the need for disease-specific treatment initiation: Findings from the opinion-based PREDICT-FD modified Delphi consensus initiative. BMJ Open 2020, 10, e035182. [Google Scholar] [CrossRef]
- Pisani, A.; Visciano, B.; Roux, G.D.; Sabbatini, M.; Porto, C.; Parenti, G.; Imbriaco, M. Enzyme replacement therapy in patients with Fabry disease: State of the art and review of the literature. Mol. Genet. Metab. 2012, 107, 267–275. [Google Scholar] [CrossRef]
- Lambert, J.R.A.; Howe, S.J.; Rahim, A.A.; Burke, D.G.; Heales, S.J.R. Inhibition of Mitochondrial Complex I Impairs Release of α-Galactosidase by Jurkat Cells. Int. J. Mol. Sci. 2019, 20, 4349. [Google Scholar] [CrossRef] [Green Version]
- Duro, G.; Zizzo, C.; Cammarata, G.; Burlina, A.; Burlina, A.; Polo, G.; Scalia, S.; Oliveri, R.; Sciarrino, S.; Francofonte, D.; et al. Mutations in the GLA Gene and LysoGb3: Is It Really Anderson-Fabry Disease? Int. J. Mol. Sci. 2018, 19, 3726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardini, A.; Camporeale, A.; Pieroni, M.; Pieruzzi, F.; Figliozzi, S.; Lusardi, P.; Spada, M.; Mignani, R.; Burlina, A.; Carubbi, F.; et al. Atrial Dysfunction Assessed by Cardiac Magnetic Resonance as an Early Marker of Fabry Cardiomyopathy. JACC Cardiovasc. Imaging 2020, 13, 2262–2264. [Google Scholar] [CrossRef] [PubMed]
- Arends, M.; Körver, S.; Hughes, D.A.; Mehta, A.; Hollak, C.E.M.; Biegstraaten, M. Phenotype, disease severity and pain are major determinants of quality of life in Fabry disease: Results from a large multicenter cohort study. J. Inherit. Metab. Dis. 2018, 41, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branton, M.H.; Schiffmann, R.; Sabnis, S.G.; Murray, G.J.; Quirk, J.M.; Altarescu, G.; Goldfarb, L.; Brady, R.O.; Balow, J.E.; Austin Iii, H.A.; et al. Natural history of Fabry renal disease: Influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine 2002, 81, 122–138. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Visciano, B.; Imbriaco, M.; Di Nuzzi, A.; Mancini, A.; Marchetiello, C.; Riccio, E. The kidney in Fabry’s disease. Clin. Genet. 2014, 86, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desnick, R.J.; Simmons, R.L.; Allen, K.Y.; Woods, J.E.; Anderson, C.F.; Najarian, J.S.; Krivit, W. Correction of enzymatic deficiencies by renal transplantation: Fabry’s disease. Surgery 1972, 72, 203–211. [Google Scholar] [PubMed]
- Philippart, M.; Franklin, S.S.; Gordon, A. Reversal of an inborn sphingolipidosis (Fabry’s disease) by kidney transplantation. Ann. Intern. Med. 1972, 77, 195–200. [Google Scholar] [CrossRef]
- Philippart, M.; Franklin, S.S.; Leeber, D.A.; Hull, A.R.; Peters, P.C. Kidney transplantation in Fabry’s disease. N. Engl. J. Med. 1973, 289, 270–271. [Google Scholar] [CrossRef] [Green Version]
- Krivit, W.; Bernlohr, R.W.; Desnick, R.J. Enzyme replacement in genetic disease. Prospectus. Birth Defects Orig. Artic. Ser. 1973, 9, 232–233. [Google Scholar]
- Peces, R. Is there true recurrence of Fabry’s disease in the transplanted kidney? Nephrol. Dial. Transpl. 1996, 11, 561. [Google Scholar] [CrossRef]
- Mosnier, J.F.; Degott, C.; Bedrossian, J.; Molas, G.; Degos, F.; Pruna, A.; Potet, F. Recurrence of Fabry’s disease in a renal allograft eleven years after successful renal transplantation. Transplantation 1991, 51, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DerSimonian, R.; Laird, N. Meta-analysis in clinical trials. Control. Clin. Trials 1986, 7, 177–188. [Google Scholar] [CrossRef]
- Higgins, J.P.; Thompson, S.G.; Deeks, J.J.; Altman, D.G. Measuring inconsistency in meta-analyses. BMJ 2003, 327, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Easterbrook, P.J.; Berlin, J.A.; Gopalan, R.; Matthews, D.R. Publication bias in clinical research. Lancet 1991, 337, 867–872. [Google Scholar] [CrossRef]
- Barnes, B.A.; Bergan, J.J.; Braun, W.E. Renal transplantation in congenital and metabolic diseases. A report from the ASC/NIH renal transplant registry. J. Am. Med Assoc. 1975, 232, 148–153. [Google Scholar] [CrossRef]
- Donati, D.; Novario, R.; Gastaldi, L. Natural history and treatment of uremia secondary to Fabry’s disease: An European experience. Nephron 1987, 46, 353–359. [Google Scholar] [CrossRef]
- Tsakiris, D.; Simpson, H.K.L.; Jones, E.H.P.; Briggs, J.D.; Elinder, C.G.; Mendel, S.; Piccoli, G.; Dos Santos, J.P.; Tognoni, C.; Vanrenterghem, Y.; et al. Rare diseases in renal replacement therapy in the ERA-EDTA Registry. Nephrol. Dial. Transpl. 1996, 11, 4–20. [Google Scholar] [CrossRef]
- Mazzarella, V.; Splendiani, G.; Tozzo, C.; Tisone, G.; Pisani, F.; Iaria, G.; Casciani, C.U. Renal transplantation in patients with hereditary kidney disease: Our experience. Contrib. Nephrol. 1997, 122, 203–206. [Google Scholar]
- Inderbitzin, D.; Avital, I.; Largiadèr, F.; Vogt, B.; Candinas, D. Kidney transplantation improves survival and is indicated in Fabry’s disease. Transpl. Proc. 2005, 37, 4211–4214. [Google Scholar] [CrossRef]
- Mignani, R.; Feriozzi, S.; Pisani, A.; Cioni, A.; Comotti, C.; Cossu, M.; Foschi, A.; Giudicissi, A.; Gotti, E.; Lozupone, V.A.; et al. Agalsidase therapy in patients with Fabry disease on renal replacement therapy: A nationwide study in Italy. Nephrol. Dial. Transpl. 2008, 23, 1628–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulla, M.; Walter, K.N.; Schwarting, A.; Divito, R.; Feriozzi, S.; Sunder-Plassmann, G. Kidney transplantation in patients with Fabry disease. Transpl. Int. 2009, 22, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Gill, J.; Malhotra, N.; Takemoto, S.K.; Bunnapradist, S. Kidney transplant outcomes in patients with fabry disease. Transplantation 2009, 87, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Ojo, A.; Meier-Kriesche, H.U.; Friedman, G.; Hanson, J.; Cibrik, D.; Leichtman, A.; Kaplan, B. Excellent outcome of renal transplantation in patients with Fabry’s disease. Transplantation 2000, 69, 2337–2339. [Google Scholar] [CrossRef]
- Pineda-Galindo, L.F.; Moranchel-García, L. Beneficial effect of agalsidase beta on long term evolution of patients with Fabry disease and kidney transplant. Mol. Genet. Metab. 2016, 117, S95. [Google Scholar] [CrossRef]
- Ersözlü, S.; Huynh-Do, U.; Cippa, P.; Müller, T.; Nowak, A. Long-term outcome of kidney transplantation in Fabry disease. Swiss Med Wkly. 2016, 146, 8S–9S. [Google Scholar] [CrossRef]
- Baek, C.H.; Kim, H.; Baek, S.D.; Jang, M.; Kim, W.; Yang, W.S.; Han, D.J.; Park, S.K. Outcomes of living donor kidney transplantation in diabetic patients: Age and sex matched comparison with non-diabetic patients. Korean J. Intern. Med. 2018, 33, 356–366. [Google Scholar] [CrossRef] [Green Version]
- Maizel, S.E.; Simmons, R.L.; Kjellstrand, C.; Fryd, D.S. Ten-year experience in renal transplantation for Fabry’s disease. Transpl. Proc. 1981, 13, 57–59. [Google Scholar]
- Clarke, J.T.R.; Guttmann, R.D.; Wolfe, L.S.; Beaudoin, J.G.; Morehouse, D.D. Enzyme Replacement Therapy by Renal Allotransplantation in Fabry’s Disease. N. Engl. J. Med. 1972, 287, 1215–1218. [Google Scholar] [CrossRef]
- Wang, A.M.; Desnick, R.J. Structural organization and complete sequence of the human alpha-N-acetylgalactosaminidase gene: Homology with the alpha-galactosidase A gene provides evidence for evolution from a common ancestral gene. Genomics 1991, 10, 133–142. [Google Scholar] [CrossRef]
- Wang, A.; Bishop, D.; Desnick, R. Human α-N-acetylgalactosaminidase-molecular cloning, nucleotide sequence, and expression of a full-length cDNA: Homology with human α-galactosidase a suggests evolution from a common ancestral gene. J. Biol. Chem. 1991, 265, 21859–21866. [Google Scholar]
- Touraine, J.L.; Malik, M.C.; Perrot, H.; Maire, I.; Revillard, J.P.; Grosshans, E.; Traeger, J. Fabry’s disease: Two patients improved by fetal liver cells (author’s transl). Nouv. Presse Med. 1979, 8, 1499–1503. [Google Scholar] [PubMed]
- Likhitsup, A.; Helzberg, J.H.; Alba, L.M.; Larkin, M.K.; Cummings, L.S.; Island, E.R.; Lustig, R.M.; Forster, J. Persistent Alpha-galactosidase A Deficiency After Simultaneous Liver-kidney Transplantation in a Patient With Fabry Disease. Transplantation 2018, 102, e361. [Google Scholar] [CrossRef]
- Mignani, R.; Panichi, V.; Giudicissi, A.; Taccola, D.; Boscaro, F.; Feletti, C.; Prof, G.; Cagnoli, L. Enzyme replacement therapy with agalsidase beta in kidney transplant patients with Fabry disease: A pilot study. Kidney Int. 2004, 65, 1381–1385. [Google Scholar] [CrossRef] [PubMed]
- Bénichou, B.; Goyal, S.; Sung, C.; Norfleet, A.M.; O’Brien, F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol. Genet. Metab. 2009, 96, 4–12. [Google Scholar] [CrossRef]
- Lenders, M.; Stypmann, J.; Duning, T.; Schmitz, B.; Brand, S.M.; Brand, E. Serum-Mediated Inhibition of Enzyme Replacement Therapy in Fabry Disease. J. Am. Soc. Nephrol. 2016, 27, 256–264. [Google Scholar] [CrossRef]
- Lenders, M.; Oder, D.; Nowak, A.; Canaan-Kühl, S.; Arash-Kaps, L.; Drechsler, C.; Schmitz, B.; Nordbeck, P.; Hennermann, J.B.; Kampmann, C.; et al. Impact of immunosuppressive therapy on therapy-neutralizing antibodies in transplanted patients with Fabry disease. J. Intern. Med. 2017, 282, 241–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Study | Year | N | Male Sex | Mean Age at Diagnosis | Mean Age at Transplant | Enzyme Replacement Therapy | Age at Enzyme Replacement Therapy | Follow-Up Time | Patient Death | Graft Failure Before Death | All-Cause Graft Failure | Graft Rejection | Recurrence of FD in Kidney Allograft |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Barnes et al. [46] | 1975 | 9 | 8/9 (89%) | N/A | 41 years | 0/0 (0%) | N/A | N/A | 6 (67%) | 0 (0%) | 6 (67%) | N/A | N/A |
| Donati et al. [47] | 1987 | 8 | 8/8 (100%) | 29.9 years | 36.8 years | 0/8 (0%) | N/A | 3.6 years | 0 | 5 (63%) | 5 (63%) | N/A | N/A |
| Tsakiris et al. [48] | 1996 | 33 | 73/83 (88%) | N/A | - | 0/0 (0%) | N/A | 3 years | 5 (15%) | N/A | 9 (27%) | N/A | N/A |
| Mazzarella et al. [49] | 1997 | 2 | 2/2 (100%) | N/A | 32 years | 0/0 (0%) | N/A | 3.8 years | 0 (0%) | 0 (0%) | 0 (0) | 0 (0%) | N/A |
| Inderbitzin et al. [50] | 2005 | 10 | 10/10 (100%) | 26 years | 36 years | 1/10 (10%) | N/A | 10.2 years | 4 (40%) | 1 (10%) | 5 (50%) | 1 (10%) | 0—clinically 1—in autopsy biopsy |
| Mignani et al. [51] | 2008 | 17 | 16/17 (94%) | 37.1 years | 39.8 years | 17/17 (100%) | 44.6 years | 6 years | 0 (0%) | 2 (12%) | 2 (12%) | N/A | N/A |
| Cybulla et al. [52] | 2009 | 36 | 34/36 (94%) | 31.1 years | 37.6 years | 24/36 (67%) | N/A | 7.7 years | 4 (11%) | 3 (8%) | N/A | N/A | N/A |
| Shah et al. [53] | 2009 | FD 197 Control 1970 | 177/197 (90%) | N/A | N/A | N/A | N/A | 5 years | FD 37 (19%) | FD 24 (12%) | FD 51 (26%) | FD 41 (21%) Control 528 (27%) | N/A |
| Ojo et al. [54] | 2000 | FD 93 Control 186 | 83/93 (89) | N/A | 40 years | N/A | N/A | 5 years | FD 16 (17%) Control 34 (18%) | N/A | FD 22 (24%) Control 62 (33%) | N/A | N/A |
| Pineda-Galindo et al. [55] | 2016 | 2 | 2/2 (100%) | N/A | N/A | 2/2 (100%) | N/A | 6 years | 0 (0%) | 0 (0%) | 0 (0%) | N/A | N/A |
| Ersozlu et al. [56] | 2018 | FD 17 Control 17 | 15/17 (88%) | 34 years | 39.5 years | 14/17 (82%) | 40.3 years | 11.5 years | FD 7 (41%) Control 2 (12%) | FD 2 (12%) Control 9 (53%) | FD 8 (47%) Control 9 (53%) | FD 3 (18%) | FD 2 (12%)—kidney biopsy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suarez, M.L.G.; Thongprayoon, C.; Hansrivijit, P.; Medaura, J.; Vaitla, P.; Mao, M.A.; Bathini, T.; Boonpheng, B.; Kanduri, S.R.; Kovvuru, K.; et al. Outcomes of Kidney Transplantation in Fabry Disease: A Meta-Analysis. Diseases 2021, 9, 2. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases9010002
Suarez MLG, Thongprayoon C, Hansrivijit P, Medaura J, Vaitla P, Mao MA, Bathini T, Boonpheng B, Kanduri SR, Kovvuru K, et al. Outcomes of Kidney Transplantation in Fabry Disease: A Meta-Analysis. Diseases. 2021; 9(1):2. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases9010002
Chicago/Turabian StyleSuarez, Maria L. Gonzalez, Charat Thongprayoon, Panupong Hansrivijit, Juan Medaura, Pradeep Vaitla, Michael A. Mao, Tarun Bathini, Boonphiphop Boonpheng, Swetha R. Kanduri, Karthik Kovvuru, and et al. 2021. "Outcomes of Kidney Transplantation in Fabry Disease: A Meta-Analysis" Diseases 9, no. 1: 2. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases9010002