
Potential Effects of Melatonin and Micronutrients on Mitochondrial Dysfunction during a Cytokine Storm Typical of Oxidative/Inflammatory Diseases

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Meaning, Etiology, Features, and Consequences of “Cytokine Storm”
3. Interrelation between Cytokine Storm and Mitochondrial Dysfunction
3.1. Mitochondrion: Function, Dynamics, and Dysfunction
3.2. Inflammation and Mitochondrial Dysfunction
3.2.1. Inflammation Alters Mitochondrial Energetics
3.2.2. Inflammation Alters Mitochondrial Dynamics and Cell Death Pathways
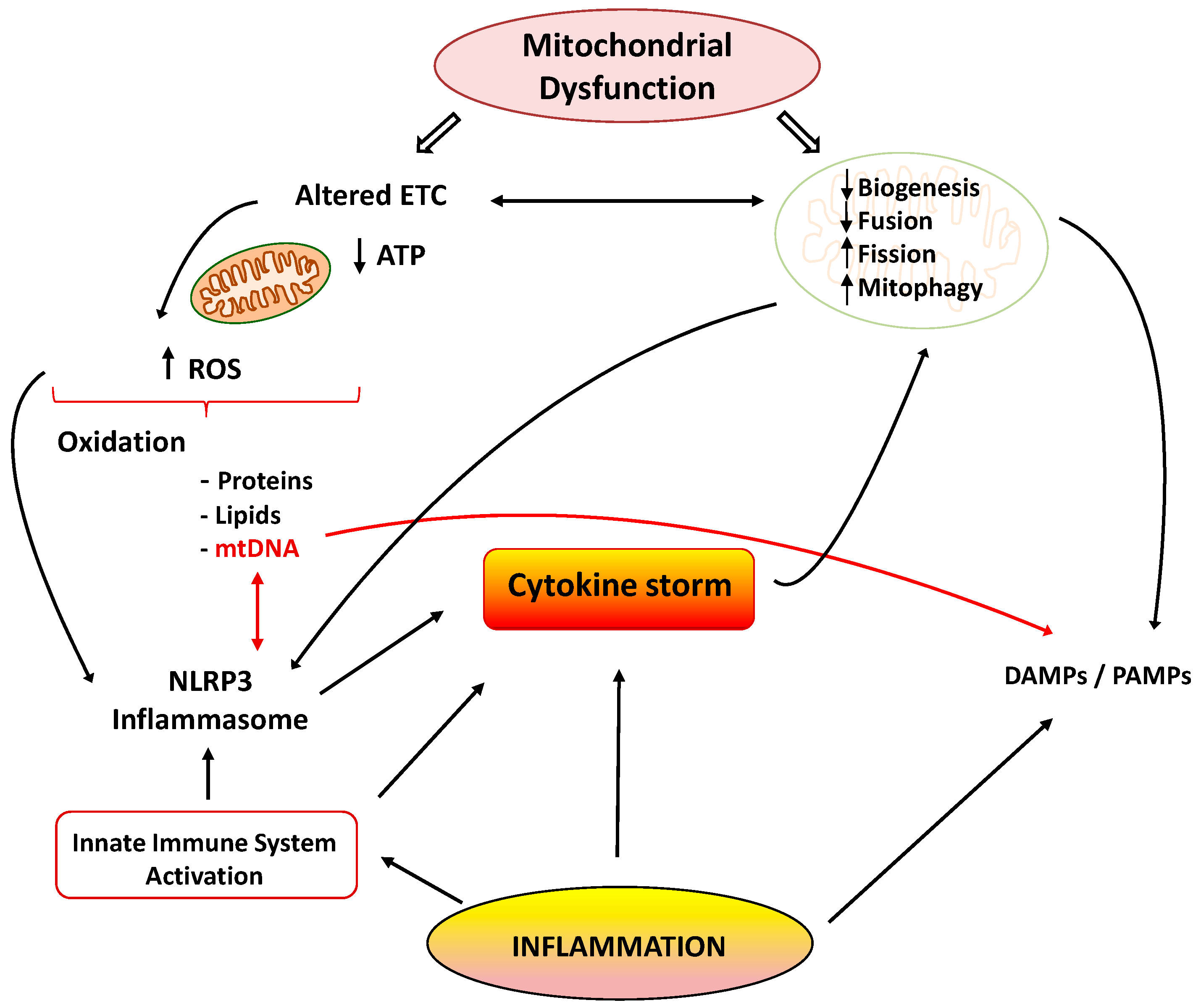
3.2.3. Mitochondrial Dysfunction Promotes Inflammation
3.3. Relevance of Mitochondrial Dysfunction in the Pathogenesis of Inflammation in Sepsis and COVID-19
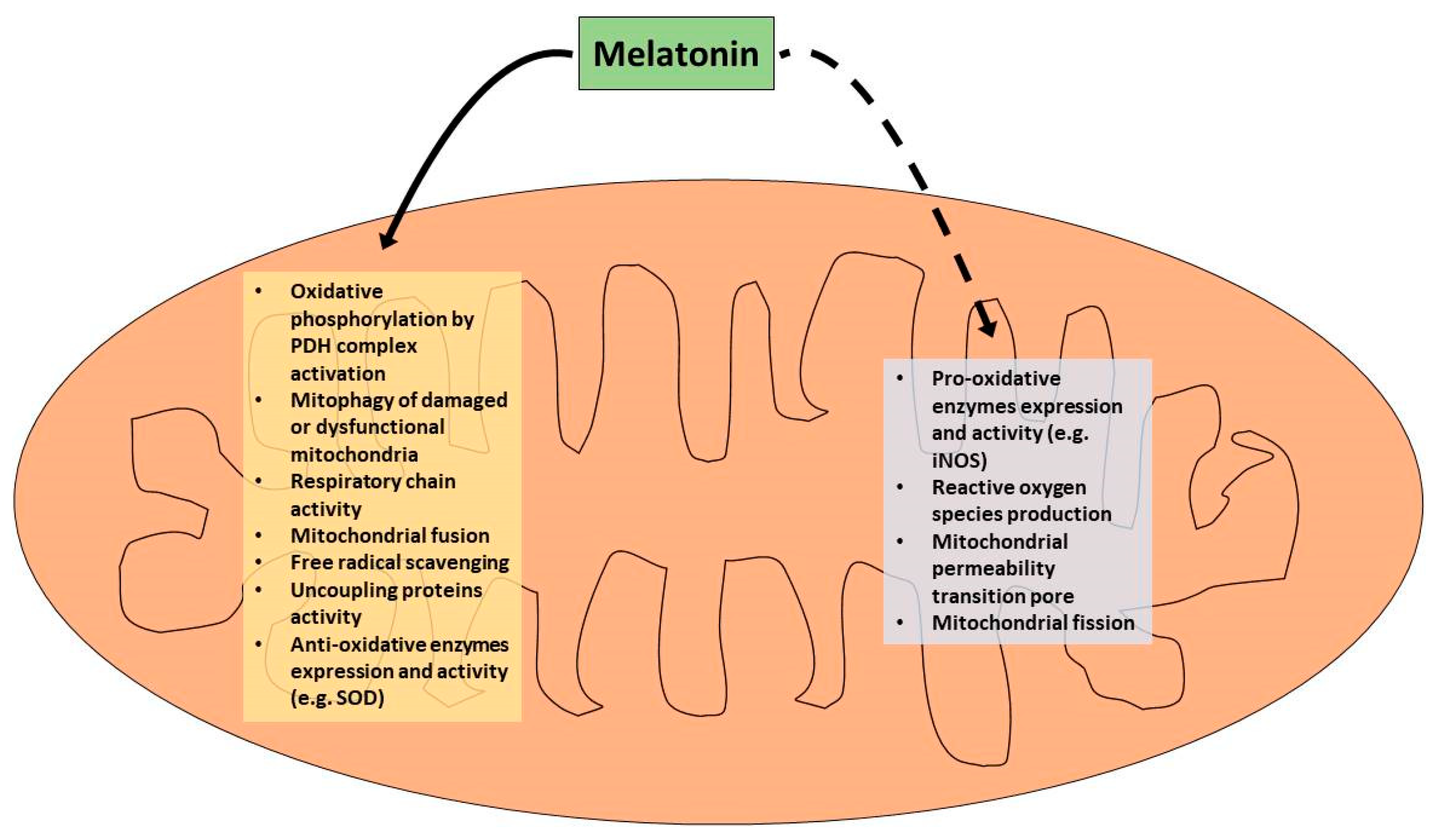
4. Role of Melatonin in the Attenuation of Mitochondrial Dysfunction Associated with the Cytokine Storm
5. Contribution of Micronutrients to the Therapy of Oxidative/Inflammatory Diseases through their Mitochondrial Actions
5.1. Vitamins
5.2. Minerals
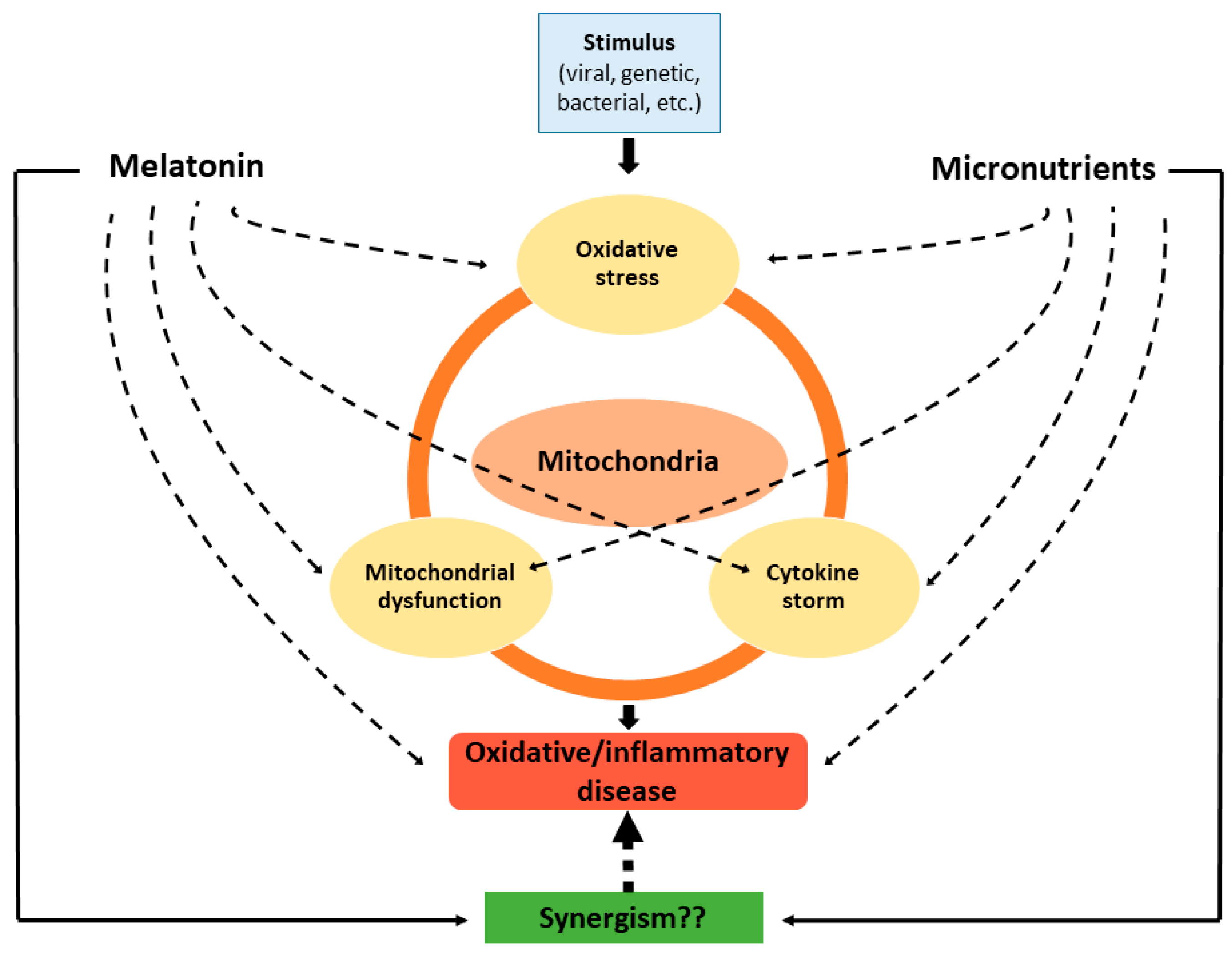
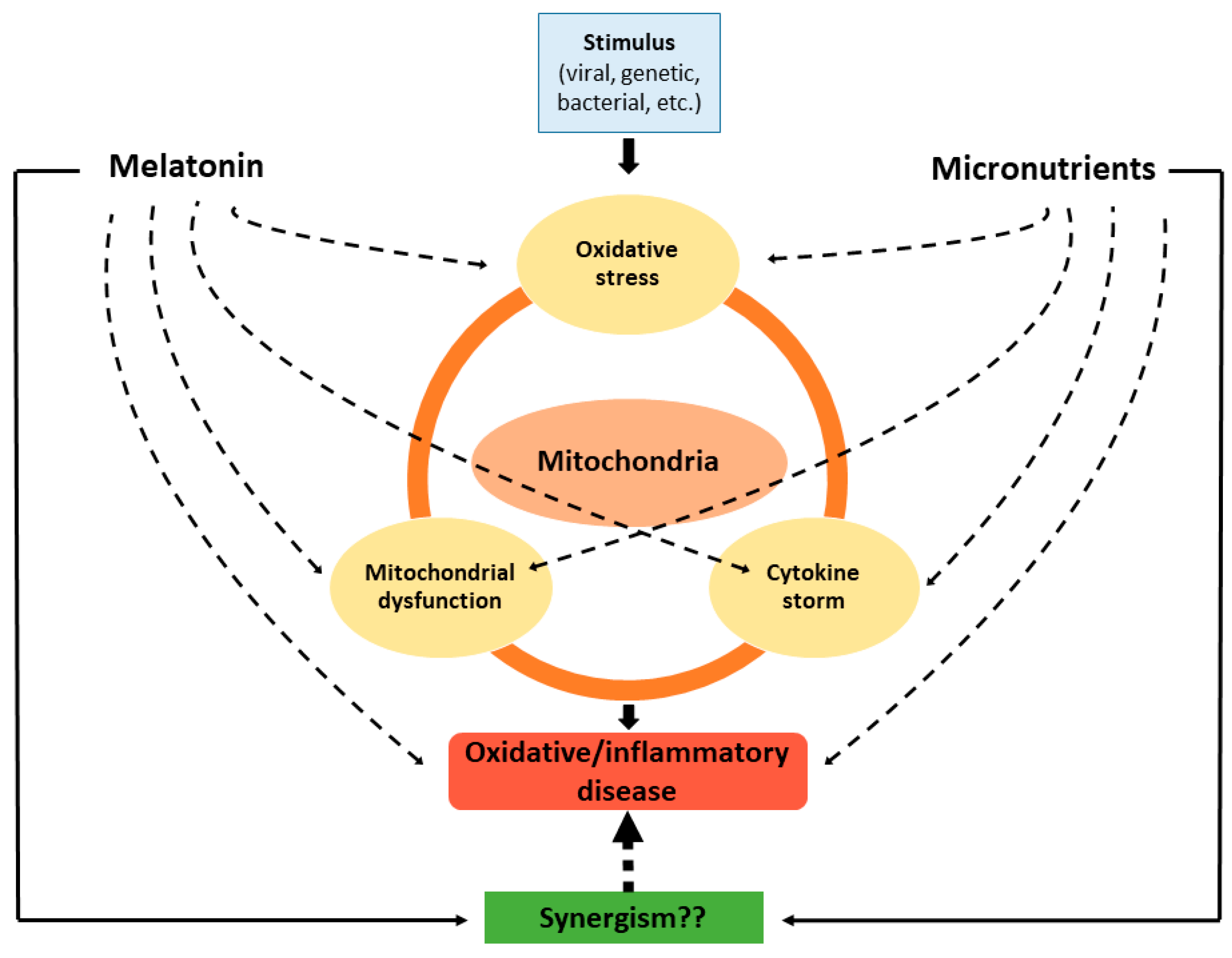
6. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Fernández-Ayala, D.J.M.; Navas, P.; López-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.H.; Franceschi, C.; Ferrucci, L. A public health perspective of aging: Do hyper-inflammatory syndromes such as COVID-19, SARS, ARDS, cytokine storm syndrome, and post-ICU syndrome accelerate short- and long-term inflammaging? Immun. Ageing 2020, 17, 1–10. [Google Scholar] [CrossRef]
- Maggini, S.; Pierre, A.; Calder, P.C. Immune Function and Micronutrient Requirements Change over the Life Course. Nutrients 2018, 10, 1531. [Google Scholar] [CrossRef] [Green Version]
- Shenkin, A. Micronutrients in health and disease. Postgrad. Med. J. 2006, 82, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Wesselink, E.; Koekkoek, W.; Grefte, S.; Witkamp, R.; van Zanten, A. Feeding mitochondria: Potential role of nutritional components to improve critical illness convalescence. Clin. Nutr. 2019, 38, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, J.L.; Abhyankar, S.; Gilliland, D.G. Cytokine storm of graft-versus-host disease: A critical effector role for interleukin-1. Transplant. Proc. 1993, 25, 1216–1217. [Google Scholar]
- Jazayeri, J.A.; Carroll, G.J.; Vernallis, A.B. Interleukin-6 subfamily cytokines and rheumatoid arthritis: Role of antagonists. Int. Immunopharmacol. 2010, 10, 1–8. [Google Scholar] [CrossRef]
- Pechous, R.D.; Sivaraman, V.; Price, P.A.; Stasulli, N.M.; Goldman, W.E. Early Host Cell Targets of Yersinia pestis during Primary Pneumonic Plague. PLoS Pathog. 2013, 9, e1003679. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.-H.; Yang, Z.-Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, T.S.; Christman, J.W. Sepsis and cytokines: Current status. Br. J. Anaesth. 1996, 77, 110–117. [Google Scholar] [CrossRef]
- Oppenheim, J.J. Cytokines: Past, Present, and Future. Int. J. Hematol. 2001, 74, 3–8. [Google Scholar] [CrossRef]
- Matsuda, N.; Hattori, Y. Systemic Inflammatory Response Syndrome (SIRS): Molecular Pathophysiology and Gene Therapy. J. Pharmacol. Sci. 2006, 101, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Scheller, J.; Rose-John, S. Interleukin-6 and its receptor: From bench to bedside. Med. Microbiol. Immunol. 2006, 195, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tanaka, T.; Narazaki, M.; Kishimoto, T. Targeting Interleukin-6 Signaling in Clinic. Immunity 2019, 50, 1007–1023. [Google Scholar] [CrossRef] [PubMed]
- Brocker, C.; Thompson, D.; Matsumoto, A.; Nebert, D.W.; Vasiliou, V. Evolutionary divergence and functions of the human interleukin (IL) gene family. Hum. Genom. 2010, 5, 30–55. [Google Scholar] [CrossRef] [PubMed]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell 2020, 183, 982–995.e14. [Google Scholar] [CrossRef]
- Hussell, T.; Pennycook, A.; Openshaw, P.J.M. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur. J. Immunol. 2001, 31, 2566–2573. [Google Scholar] [CrossRef]
- Shi, X.; Zhou, W.; Huang, H.; Zhu, H.; Zhou, P.; Zhu, H.; Ju, D. Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice. Crit. Care 2013, 17, R301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Karin, M. Is NF-kappaB the sensor of oxidative stress? FASEB J. 1999, 13, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.N.; Amstad, P.; Cerutti, P.; Baeuerle, P.A. The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κB. Chem. Biol. 1995, 2, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Toll-like receptors; their physiological role and signal transduction system. Int. Immunopharmacol. 2001, 1, 625–635. [Google Scholar] [CrossRef]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Moore, B.J.B.; June, C.H. Cytokine release syndrome in severe COVID-19. Science 2020, 368, 473–474. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rokni, M.; Ghasemi, V.; Tavakoli, Z. Immune responses and pathogenesis ofSARS-CoV-2 during an outbreak in Iran: Comparison with SARS and MERS. Rev. Med. Virol. 2020, 30, 2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Ruan, Q.; Yang, K.; Wang, W.; Jiang, L.; Song, J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensiv. Care Med. 2020, 46, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Prompetchara, E.; Ketloy, C.; Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pac. J. Allergy Immunol. 2020, 38, 1–9. [Google Scholar] [CrossRef]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahera, V.; de Las Heras, N.; López-Farré, A.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, Á.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, Oxidative Stress, and Obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef] [Green Version]
- Crimi, E.; Sica, V.; Slutsky, A.S.; Zhang, H.; Williams-Ignarro, S.; Ignarro, L.J.; Napoli, C. Role of oxidative stress in experimental sepsis and multisystem organ dysfunction. Free. Radic. Res. 2006, 40, 665–672. [Google Scholar] [CrossRef]
- Bartz, R.R.; Suliman, H.B.; Fu, P.; Welty-Wolf, K.; Carraway, M.S.; MacGarvey, N.C.; Withers, C.M.; Sweeney, T.E.; Piantadosi, C.A. Staphylococcus aureusSepsis and Mitochondrial Accrual of the 8-Oxoguanine DNA Glycosylase DNA Repair Enzyme in Mice. Am. J. Respir. Crit. Care Med. 2011, 183, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Escames, G.; López, L.C.; Ortiz, F.; López, A.; García, J.A.; Ros, E.; Acuña-Castroviejo, D. Attenuation of cardiac mitochondrial dysfunction by melatonin in septic mice. FEBS J. 2007, 274, 2135–2147. [Google Scholar] [CrossRef]
- Zell, R.; Geck, P.; Werdan, K.; Boekstegers, P. Tnf-α and IL-1α inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: Evidence for primary impairment of mitochondrial function. Mol. Cell. Biochem. 1997, 177, 61–67. [Google Scholar] [CrossRef]
- Vary, T.C.; Hazen, S. Sepsis alters pyruvate dehydrogenase kinase activity in skeletal muscle. Mol. Cell. Biochem. 1999, 198, 113–118. [Google Scholar] [CrossRef]
- Reiter, R.J.; Ma, Q.; Sharma, R. Melatonin in Mitochondria: Mitigating Clear and Present Dangers. Physiology 2020, 35, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Sharma, R.; de Campos Zuccari, D.A.P.; de Almeida Chuffa, L.G.; Manucha, W.; Rodriguez, C. Melatonin synthesis in and uptake by mitochondria: Implications for diseased cells with dysfunctional mitochondria. Futur. Med. Chem. 2021, 13, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Sheu, K.-F.R.; Blass, J.P. The alpha-Ketoglutarate Dehydrogenase Complex. Ann. N. Y. Acad. Sci. 1999, 893, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Mastrogiacomo, F.; Bergeron, C.; Kish, S.J. Brain α-Ketoglutarate Dehydrotenase Complex Activity in Alzheimer’s Disease. J. Neurochem. 1993, 61, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Hüttemann, M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Suliman, H.B.; Welty-Wolf, K.E.; Carraway, M.; Tatro, L.; Piantadosi, C.A. Lipopolysaccharide induces oxidative cardiac mitochondrial damage and biogenesis. Cardiovasc. Res. 2004, 64, 279–288. [Google Scholar] [CrossRef]
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-α regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomer, X.; Álvarez-Guardia, D.; Rodríguez-Calvo, R.; Coll, T.; Laguna, J.C.; Davidson, M.M.; Chan, T.O.; Feldman, A.M.; Vázquez-Carrera, M. TNF-α reduces PGC-1α expression through NF-κB and p38 MAPK leading to increased glucose oxidation in a human cardiac cell model. Cardiovasc. Res. 2008, 81, 703–712. [Google Scholar] [CrossRef]
- Chen, X.-H.; Zhao, Y.-P.; Xue, M.; Ji, C.-B.; Gao, C.-L.; Zhu, J.-G.; Qin, D.-N.; Kou, C.-Z.; Qin, X.-H.; Tong, M.-L.; et al. TNF-α induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 2010, 328, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Hahn, W.S.; Kuzmicic, J.; Burrill, J.S.; Donoghue, M.A.; Foncea, R.; Jensen, M.D.; Lavandero, S.; Arriaga, E.A.; Bernlohr, D.A. Proinflammatory cytokines differentially regulate adipocyte mitochondrial metabolism, oxidative stress, and dynamics. Am. J. Physiol. Metab. 2014, 306, E1033–E1045. [Google Scholar] [CrossRef] [Green Version]
- White, J.P.; Puppa, M.J.; Sato, S.; Gao, S.; Price, R.L.; Baynes, J.W.; Kostek, M.C.; Matesic, L.E.; Carson, J.A. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet. Muscle 2012, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Moro, L. Mitochondria at the Crossroads of Physiology and Pathology. J. Clin. Med. 2020, 9, 1971. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial Disorders in the Nervous System. Annu. Rev. Neurosci. 2008, 31, 91–123. [Google Scholar] [CrossRef] [PubMed]
- Cardaci, S.; Ciriolo, M.R. TCA Cycle Defects and Cancer: When Metabolism Tunes Redox State. Int. J. Cell Biol. 2012, 2012, 161837. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J.-I. ROS-Generating Mitochondrial DNA Mutations Can Regulate Tumor Cell Metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbini, A.A.; Guerra, F.; Greco, M.; Marra, E.; Gandee, L.; Xiao, G.; Lotan, Y.; Gasparre, G.; Hsieh, J.-T.; Moro, L. Mitochondrial DNA depletion sensitizes cancer cells to PARP inhibitors by translational and post-translational repression of BRCA2. Oncogenesis 2013, 2, e82. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, S. Coronavirus (Covid-19) sepsis: Revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020, 69, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shalova, I.N.; Lim, J.Y.; Chittezhath, M.; Zinkernagel, A.S.; Beasley, F.; Hernández-Jiménez, E.; Toledano, V.; Cubillos-Zapata, C.; Rapisarda, A.; Chen, J.; et al. Human Monocytes Undergo Functional Re-programming during Sepsis Mediated by Hypoxia-Inducible Factor-1α. Immunity 2015, 42, 484–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Buechler, N.L.; Woodruff, A.G.; Long, D.L.; Zabalawi, M.; Yoza, B.K.; McCall, C.E.; Vachharajani, V. Sirtuins and Immuno-Metabolism of Sepsis. Int. J. Mol. Sci. 2018, 19, 2738. [Google Scholar] [CrossRef] [Green Version]
- Vachharajani, V.; McCall, C.E. Sirtuins: Potential therapeutic targets for regulating acute inflammatory response? Expert Opin. Ther. Targets 2020, 24, 489–497. [Google Scholar] [CrossRef]
- Mueller, A.L.; McNamara, M.S.; Sinclair, D.A. Why does COVID-19 disproportionately affect older people? Aging 2020, 12, 9959–9981. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Dominquez-Rodriguez, A.; Marik, P.E.; Abreu-Gonzalez, P. Melatonin Inhibits COVID-19-induced Cytokine Storm by Reversing Aerobic Glycolysis in Immune Cells: A Mechanistic Analysis. Med. Drug Discov. 2020, 6, 100044. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. Melatonin: Roles in influenza, Covid-19, and other viral infections. Rev. Med. Virol. 2020, 30, e2109. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.; Acuna-Castroviejo, D.; Escames, G. Inhibition of mitochondrial pyruvate dehydrogenase kinase: A proposed mechanism by which melatonin causes cancer cells to overcome cytosolic glycolysis, reduce tumor biomass and reverse insensitivity to chemotherapy. Melatonin Res. 2019, 2, 105–119. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Prado, N.; Diez, E.; Manucha, W.; Reiter, R.J. New proposal involving nanoformulated melatonin targeted to the mitochondria as a potential COVID-19 treatment. Nanomedicine 2020, 15, 2819–2821. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Liu, C.; Manucha, W.; Abreu-Gonzalez, P.; Dominguez-Rodriguez, A. Plasticity of glucose metabolism in activated immune cells: Advantages for melatonin inhibition of COVID-19 disease. Melatonin Res. 2020, 3, 362–379. [Google Scholar] [CrossRef]
- Acuña-Castroviejo, D.; Noguiera-Navarro, M.T.; Reiter, R.J.; Escames, G. Melatonin actions in the heart; more than a hormone. Melatonin Res. 2018, 1, 21–26. [Google Scholar] [CrossRef]
- Acuna-Castroviejo, D. Melatonin role in the mitochondrial function. Front. Biosci. 2007, 12, 947–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, J.; Acuna-Castroviejo, D.; Escames, G.; Tan, D.-X.; Reiter, R.J. Melatonin mitigates mitochondrial malfunction. J. Pineal Res. 2004, 38, 1–9. [Google Scholar] [CrossRef]
- Leon, J.; Acuña-Castroviejo, D.; Sainz, R.M.; Mayo, J.C.; Tan, D.-X.; Reiter, R.J. Melatonin and mitochondrial function. Life Sci. 2004, 75, 765–790. [Google Scholar] [CrossRef]
- Ramis, M.R.; Esteban, S.; Miralles, A.; Tan, D.-X.; Reiter, R.J. Protective Effects of Melatonin and Mitochondria-targeted Antioxidants Against Oxidative Stress: A Review. Curr. Med. Chem. 2015, 22, 2690–2711. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.-X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.; Tan, D.X.; Jou, M.J.; Galano, A.; Xu, B. Melatonin as a mitochondria-targeted antioxidant: One of evolution’s best ideas. Cell. Mol. Life Sci. 2017, 74, 3863–3881. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Zhou, X.J.; Xu, B. Mitochondria: Central Organelles for Melatonin′s Antioxidant and Anti-Aging Actions. Molecules 2018, 23, 509. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.-X.; Manchester, L.C.; Qin, L.; Reiter, R.J. Melatonin: A Mitochondrial Targeting Molecule Involving Mitochondrial Protection and Dynamics. Int. J. Mol. Sci. 2016, 17, 2124. [Google Scholar] [CrossRef]
- Basile, M.; Romeo, C.; Gitto, E.; Spitz, L.; Pierro, A.; Eaton, S. Melatonin protects from, but does not reverse, the effects of mediators of sepsis on liver bioenergetics. Pediatr. Surg. Int. 2004, 20, 263–266. [Google Scholar] [CrossRef]
- Gitto, E.; Karbownik, M.; Reiter, R.J.; Tan, D.X.; Cuzzocrea, S.; Chiurazzi, P.; Cordaro, S.; Corona, G.; Trimarchi, A.G.; Barberi, I. Effects of Melatonin Treatment in Septic Newborns. Pediatr. Res. 2001, 50, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Doerrier, C.; García, J.A.; Volt, H.; Díaz-Casado, M.E.; Lima-Cabello, E.; Ortiz, F.; Luna-Sánchez, M.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Identification of mitochondrial deficits and melatonin targets in liver of septic mice by high-resolution respirometry. Life Sci. 2015, 121, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; Acuña-Castroviejo, D.; López, L.C.; Tan, D.-X.; Maldonado, M.D.; Sánchez-Hidalgo, M.; León, J.; Reiter, R.J. Pharmacological utility of melatonin in the treatment of septic shock: Experimental and clinical evidence. J. Pharm. Pharmacol. 2010, 58, 1153–1165. [Google Scholar] [CrossRef]
- Escames, G.; López, L.C.; Tapias, V.; Utrilla, P.; Reiter, R.J.; Hitos, A.B.; León, J.; Rodriguez, M.I.; Acuña-Castroviejo, D. Melatonin counteracts inducible mitochondrial nitric oxide synthase-dependent mitochondrial dysfunction in skeletal muscle of septic mice. J. Pineal Res. 2005, 40, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Lowes, D.A.; Allen, L.; Cameron, G.; Aucott, L.S.; Webster, N.R. Melatonin as a potential therapy for sepsis: A phase I dose escalation study and an ex vivo whole blood model under conditions of sepsis. J. Pineal Res. 2014, 56, 427–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, J.A.; Ortiz, F.; Miana, J.; Doerrier, C.; Fernández-Ortiz, M.; Rusanova, I.; Escames, G.; Acuña-Castroviejo, D. Contribution of inducible and neuronal nitric oxide synthases to mitochondrial damage and melatonin rescue in LPS-treated mice. J. Physiol. Biochem. 2017, 73, 235–244. [Google Scholar] [CrossRef]
- López, L.C.; Escames, G.; Tapias, V.; Utrilla, P.; León, J.; Acuña-Castroviejo, D. Identification of an inducible nitric oxide synthase in diaphragm mitochondria from septic miceIts relation with mitochondrial dysfunction and prevention by melatonin. Int. J. Biochem. Cell Biol. 2006, 38, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Lowes, D.A.; Almawash, A.M.; Webster, N.R.; Reid, V.; Galley, H.F. Melatonin and structurally similar compounds have differing effects on inflammation and mitochondrial function in endothelial cells under conditions mimicking sepsis. Br. J. Anaesth. 2011, 107, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Doerrier, C.; García, J.A.; Volt, H.; Díaz-Casado, M.E.; Luna-Sánchez, M.; Fernández-Gil, B.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Permeabilized myocardial fibers as model to detect mitochondrial dysfunction during sepsis and melatonin effects without disruption of mitochondrial network. Mitochondrion 2016, 27, 56–63. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Wang, X.; Chen, X.; Long, Y.; Chai, W.; Zhou, X.; Rui, X.; Zhang, Q.; Wang, H.; et al. Melatonin improved rat cardiac mitochondria and survival rate in septic heart injury. J. Pineal Res. 2013, 55, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lowes, D.; Webster, N.; Murphy, M.; Galley, H. Antioxidants that protect mitochondria reduce interleukin-6 and oxidative stress, improve mitochondrial function, and reduce biochemical markers of organ dysfunction in a rat model of acute sepsis. Br. J. Anaesth. 2013, 110, 472–480. [Google Scholar] [CrossRef] [Green Version]
- Galano, A.; Reiter, R.J. Melatonin and its metabolites vs oxidative stress: From individual actions to collective protection. J. Pineal Res. 2018, 65, e12514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Wang, X.; Ni, L.; Di, X.; Ma, B.; Niu, S.; Liu, C.; Reiter, R.J. COVID-19: Melatonin as a potential adjuvant treatment. Life Sci. 2020, 250, 117583. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; Díaz-Casado, M.E.; Doerrier, C.; Luna-Sánchez, M.; López, L.C.; Acuña-Castroviejo, D. Early gender differences in the redox status of the brain mitochondria with age: Effects of melatonin therapy. Horm. Mol. Biol. Clin. Investig. 2013, 16, 91–100. [Google Scholar] [CrossRef]
- Escames, G.; López, A.; García, J.A.; García, L.; Acuña-Castroviejo, D.; García, J.J.; López, L.C. The Role of Mitochondria in Brain Aging and the Effects of Melatonin. Curr. Neuropharmacol. 2010, 8, 182–193. [Google Scholar] [CrossRef] [Green Version]
- García, S.; Giménez, V.M.M.; Marón, F.J.M.; Reiter, R.J.; Manucha, W. Melatonin and cannabinoids: Mitochondrial-targeted molecules that may reduce inflammaging in neurodegenerative diseases. Histol. Histopathol. 2020, 35, 18212. [Google Scholar]
- Reiter, R.J.; Tan, D.X.; Manchester, L.C.; El-Sawi, M.R. Melatonin Reduces Oxidant Damage and Promotes Mitochondrial Respiration. Ann. N. Y. Acad. Sci. 2002, 959, 238–250. [Google Scholar] [CrossRef]
- Favero, G.; Rodella, L.F.; Reiter, R.J.; Rezzani, R. Melatonin and its atheroprotective effects: A review. Mol. Cell. Endocrinol. 2014, 382, 926–937. [Google Scholar] [CrossRef]
- Lu, K.; Liu, X.; Guo, W. Melatonin attenuates inflammation-related venous endothelial cells apoptosis through modulating the MST1–MIEF1 pathway. J. Cell. Physiol. 2019, 234, 23675–23684. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Paulis, L.; Simko, F. Peripheral and Central Effects of Melatonin on Blood Pressure Regulation. Int. J. Mol. Sci. 2014, 15, 17920–17937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardinali, D.P.; Vigo, D.E. Melatonin, mitochondria, and the metabolic syndrome. Cell. Mol. Life Sci. 2017, 74, 3941–3954. [Google Scholar] [CrossRef] [PubMed]
- Prado, N.J.; Ferder, L.; Manucha, W.; Diez, E.R. Anti-Inflammatory Effects of Melatonin in Obesity and Hypertension. Curr. Hypertens. Rep. 2018, 20, 45. [Google Scholar] [CrossRef] [PubMed]
- Promsan, S.; Lungkaphin, A. The roles of melatonin on kidney injury in obese and diabetic conditions. BioFactors 2020, 46, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Jiao, Y.; Wang, J.; Zhang, Y.; Shen, M.; Reiter, R.J.; Xi, Q.; Chen, Y. Cardioprotective Role of Melatonin in Acute Myocardial Infarction. Front. Physiol. 2020, 11, 366. [Google Scholar] [CrossRef]
- Halladin, N.L. Oxidative and inflammatory biomarkers of ischemia and reperfusion injuries. Dan. Med. J. 2015, 62, 5054. [Google Scholar]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y.; Fu, Z. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef]
- Lin, C.; Chao, H.; Li, Z.; Xu, X.; Liu, Y.; Hou, L.; Liu, N.; Ji, J. Melatonin attenuates traumatic brain injury-induced inflammation: A possible role for mitophagy. J. Pineal Res. 2016, 61, 177–186. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Kamrava, S.K.; Joghataei, M.T.; Darabi, R.; Shakeri-Zadeh, A.; Shahriari, M.; Reiter, R.J.; Ghaznavi, H.; Mehrzadi, S. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J. Pineal Res. 2016, 61, 411–425. [Google Scholar] [CrossRef]
- Das, N.; Mandala, A.; Naaz, S.; Giri, S.; Jain, M.; Bandyopadhyay, D.; Reiter, R.J.; Roy, S.S. Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. J. Pineal Res. 2017, 62, e12404. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lu, C.; Zhao, W.; Shao, X. Melatonin attenuates TNF-α-mediated hepatocytes damage via inhibiting mitochondrial stress and activating the Akt-Sirt3 signaling pathway. J. Cell. Physiol. 2019, 234, 20969–20979. [Google Scholar] [CrossRef] [PubMed]
- Crooke, A.; Huete-Toral, F.; Colligris, B.; Pintor, J. The role and therapeutic potential of melatonin in age-related ocular diseases. J. Pineal Res. 2017, 63, e12430. [Google Scholar] [CrossRef] [Green Version]
- Dehdashtian, E.; Mehrzadi, S.; Yousefi, B.; Hosseinzadeh, A.; Reiter, R.J.; Safa, M.; Ghaznavi, H.; Naseripour, M. Diabetic retinopathy pathogenesis and the ameliorating effects of melatonin; involvement of autophagy, inflammation and oxidative stress. Life Sci. 2018, 193, 20–33. [Google Scholar] [CrossRef]
- Juybari, K.B.; Hosseinzadeh, A.; Ghaznavi, H.; Kamali, M.; Sedaghat, A.; Mehrzadi, S.; Naseripour, M. Melatonin As a Modulator of Degenerative and Regenerative Signaling Pathways in Injured Retinal Ganglion Cells. Curr. Pharm. Des. 2019, 25, 3057–3073. [Google Scholar] [CrossRef]
- Mehrzadi, S.; Hemati, K.; Reiter, R.J.; Hosseinzadeh, A. Mitochondrial dysfunction in age-related macular degeneration: Melatonin as a potential treatment. Expert Opin. Ther. Targets 2020, 24, 359–378. [Google Scholar] [CrossRef]
- Mahalanobish, S.; Dutta, S.; Saha, S.; Sil, P.C. Melatonin induced suppression of ER stress and mitochondrial dysfunction inhibited NLRP3 inflammasome activation in COPD mice. Food Chem. Toxicol. 2020, 144, 111588. [Google Scholar] [CrossRef] [PubMed]
- Feitosa, E.L.; Júnior, F.; Neto, J.A.O.N.; Matos, L.F.L.; Moura, M.H.S.; Rosales, T.O.; De Freitas, G.B.L. COVID-19: Rational discovery of the therapeutic potential of Melatonin as a SARS-CoV-2 main Protease Inhibitor. Int. J. Med. Sci. 2020, 17, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Kleszczyński, K.; Slominski, A.T.; Steinbrink, K.; Reiter, R.J. Clinical Trials for Use of Melatonin to Fight against COVID-19 Are Urgently Needed. Nutrients 2020, 12, 2561. [Google Scholar] [CrossRef] [PubMed]
- Sehirli, A.O.; Sayiner, S.; Serakinci, N. Role of melatonin in the treatment of COVID-19; as an adjuvant through cluster differentiation 147 (CD147). Mol. Biol. Rep. 2020, 47, 8229–8233. [Google Scholar] [CrossRef]
- Zweers, H.; Janssen, M.C.H.; Leij, S.; Wanten, G. Patients With Mitochondrial Disease Have an Inadequate Nutritional Intake. J. Parenter. Enter. Nutr. 2017, 42, 581–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendivil, C.O. Dietary Fish, Fish Nutrients, and Immune Function: A Review. Front. Nutr. 2021, 7, 617652. [Google Scholar] [CrossRef] [PubMed]
- Pecora, F.; Persico, F.; Argentiero, A.; Neglia, C.; Esposito, S. The Role of Micronutrients in Support of the Immune Response against Viral Infections. Nutrients 2020, 12, 3198. [Google Scholar] [CrossRef]
- Quiles, J.L.; Rivas-García, L.; Varela-López, A.; Llopis, J.; Battino, M.; Sánchez-González, C. Do nutrients and other bioactive molecules from foods have anything to say in the treatment against COVID-19? Environ. Res. 2020, 191, 110053. [Google Scholar] [CrossRef]
- Batllori, M.; Molero-Luis, M.; Ormazabal, A.; Montero, R.; Sierra, C.; Ribes, A.; Montoya, J.; Ruiz-Pesini, E.; O’Callaghan, M.; Pias, L.; et al. Cerebrospinal fluid monoamines, pterins, and folate in patients with mitochondrial diseases: Systematic review and hospital experience. J. Inherit. Metab. Dis. 2018, 41, 1147–1158. [Google Scholar] [CrossRef]
- Ormazabal, A.; Casado, M.; Molero-Luis, M.; Montoya, J.; Rahman, S.; Aylett, S.-B.; Hargreaves, I.; Heales, S.J.R.; Artuch, R. Can folic acid have a role in mitochondrial disorders? Drug Discov. Today 2015, 20, 1349–1354. [Google Scholar] [CrossRef]
- Piquereau, J.; Moulin, M.; Zurlo, G.; Mateo, P.; Gressette, M.; Paul, J.-L.; Lemaire, C.; Ventura-Clapier, R.; Veksler, V.; Garnier, A. Cobalamin and folate protect mitochondrial and contractile functions in a murine model of cardiac pressure overload. J. Mol. Cell. Cardiol. 2017, 102, 34–44. [Google Scholar] [CrossRef]
- Rodríguez-Varela, C.; Labarta, E. Clinical Application of Antioxidants to Improve Human Oocyte Mitochondrial Function: A Review. Antioxidants 2020, 9, 1197. [Google Scholar] [CrossRef]
- Bielli, A.; Scioli, M.G.; Mazzaglia, D.; Doldo, E.; Orlandi, A. Antioxidants and vascular health. Life Sci. 2015, 143, 209–216. [Google Scholar] [CrossRef]
- Belsky, J.B.; Wira, C.R.; Jacob, V.; Sather, J.E.; Lee, P.J. A review of micronutrients in sepsis: The role of thiamine, l-carnitine, vitamin C, selenium and vitamin D. Nutr. Res. Rev. 2018, 31, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Guidarelli, A.; Cantoni, O. Mitochondrial reactive oxygen species: The effects of mitochondrial ascorbic acid vs. untargeted and mitochondria-targeted antioxidants. Int. J. Radiat. Biol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Leite, H.P.; De Lima, L.F.P. Metabolic resuscitation in sepsis: A necessary step beyond the hemodynamic? J. Thorac. Dis. 2016, 8, E552–E557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantzarlis, K.; Tsolaki, V.; Zakynthinos, E. Role of Oxidative Stress and Mitochondrial Dysfunction in Sepsis and Potential Therapies. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Marik, P.E. Vitamin C for the treatment of sepsis: The scientific rationale. Pharmacol. Ther. 2018, 189, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.M. Do micronutrient deficiencies contribute to mitochondrial failure in critical illness? Curr. Opin. Clin. Nutr. Metab. Care 2020, 23, 102–110. [Google Scholar] [CrossRef]
- Ramanathan, N.; Tan, E.; Loh, L.J.; Soh, B.S.; Yap, W.N. Tocotrienol is a cardioprotective agent against ageing-associated cardiovascular disease and its associated morbidities. Nutr. Metab. 2018, 15, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrogini, P.; Torquato, P.; Bartolini, D.; Albertini, M.C.; Lattanzi, D.; Di Palma, M.; Marinelli, R.; Betti, M.; Minelli, A.; Cuppini, R.; et al. Excitotoxicity, neuroinflammation and oxidant stress as molecular bases of epileptogenesis and epilepsy-derived neurodegeneration: The role of vitamin E. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1098–1112. [Google Scholar] [CrossRef]
- Minter, B.E.; Lowes, D.A.; Webster, N.R.; Galley, H.F. Differential Effects of MitoVitE, α-Tocopherol and Trolox on Oxidative Stress, Mitochondrial Function and Inflammatory Signalling Pathways in Endothelial Cells Cultured under Conditions Mimicking Sepsis. Antioxidants 2020, 9, 195. [Google Scholar] [CrossRef] [Green Version]
- Manucha, W.; Ritchie, B.; Ferder, L. Hypertension and Insulin Resistance: Implications of Mitochondrial Dysfunction. Curr. Hypertens. Rep. 2014, 17, 504. [Google Scholar] [CrossRef]
- Longoni, A.; Kolling, J.; Dos Santos, T.M.; Dos Santos, J.P.; Da Silva, J.S.; Pettenuzzo, L.; Gonçalves, C.; De Assis, A.M.; Quincozes-Santos, A.; Wyse, A.T. 1,25-Dihydroxyvitamin D3 exerts neuroprotective effects in an ex vivo model of mild hyperhomocysteinemia. Int. J. Dev. Neurosci. 2015, 48, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hussien, N.I.; El-Wakeel, H.S.; Souror, S.M.; Ahmed, I.A. Alleviation of cardiac mitochondrial dysfunction and oxidative stress underlies the protective effect of vitamin D in chronic stress-induced cardiac dysfunction in rats. Gen. Physiol. Biophys. 2019, 38, 51–61. [Google Scholar] [CrossRef]
- Longoni, A.; Kolling, J.; Siebert, C.; Dos Santos, J.P.; Da Silva, J.S.; Pettenuzzo, L.F.; Meira-Martins, L.A.; Gonçalves, C.-A.; De Assis, A.M.; Wyse, A.T. 1,25-Dihydroxyvitamin D 3 prevents deleterious effects of homocysteine on mitochondrial function and redox status in heart slices. Nutr. Res. 2017, 38, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Vitamin D deficiency accelerates ageing and age-related diseases: A novel hypothesis. J. Physiol. 2017, 595, 6825–6836. [Google Scholar] [CrossRef] [PubMed]
- Marón, F.J.M.; Ferder, L.; Reiter, R.J.; Manucha, W. Daily and seasonal mitochondrial protection: Unraveling common possible mechanisms involving vitamin D and melatonin. J. Steroid Biochem. Mol. Biol. 2020, 199, 105595. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Bi, X.; Ling, L.; Ding, W. 1,25-Dihydroxyvitamin D3 Attenuates Angiotensin II-Induced Renal Injury by Inhibiting Mitochondrial Dysfunction and Autophagy. Cell. Physiol. Biochem. 2018, 51, 1751–1762. [Google Scholar] [CrossRef]
- de Las Heras, N.; Giménez, V.M.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef] [PubMed]
- Ferder, L.; Giménez, V.M.M.; Inserra, F.; Tajer, C.; Antonietti, L.; Mariani, J.; Manucha, W. Vitamin D supplementation as a rational pharmacological approach in the COVID-19 pandemic. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L941–L948. [Google Scholar] [CrossRef]
- Mariani, J.; Giménez, V.M.M.; Bergam, I.; Tajer, C.; Antonietti, L.; Inserra, F.; Ferder, L.; Manucha, W. Association Between Vitamin D Deficiency and COVID-19 Incidence, Complications, and Mortality in 46 Countries: An Ecological Study. Health Secur. 2020. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Ferder, L.; Inserra, F.; García, J.; Manucha, W. Differences in RAAS/vitamin D linked to genetics and socioeconomic factors could explain the higher mortality rate in African Americans with COVID-19. Ther. Adv. Cardiovasc. Dis. 2020, 14, 1753944720977715. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Inserra, F.; Ferder, L.; García, J.; Manucha, W. Vitamin D deficiency in African Americans is associated with a high risk of severe disease and mortality by SARS-CoV-2. J. Hum. Hypertens. 2020, 1–3. [Google Scholar] [CrossRef]
- Giménez, V.M.M.; Inserra, F.; Tajer, C.D.; Mariani, J.; Ferder, L.; Reiter, R.J.; Manucha, W. Lungs as target of COVID-19 infection: Protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci. 2020, 254, 117808. [Google Scholar] [CrossRef] [PubMed]
- Farbood, Y.; Sarkaki, A.; Mahdavinia, M.; Ghadiri, A.; Teimoori, A.; Seif, F.; Dehghani, M.A.; Navabi, S.P. Protective Effects of Co-administration of Zinc and Selenium Against Streptozotocin-Induced Alzheimer’s Disease: Behavioral, Mitochondrial Oxidative Stress, and GPR39 Expression Alterations in Rats. Neurotox. Res. 2020, 38, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Avery, J.C.; Hoffmann, P.R. Selenium, Selenoproteins, and Immunity. Nutrients 2018, 10, 1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, P.R.; Berry, M.J. The influence of selenium on immune responses. Mol. Nutr. Food Res. 2008, 52, 1273–1280. [Google Scholar] [CrossRef]
- Ma, Y.-M.; Guo, Y.-Z.; Ibeanu, G.; Wang, L.-Y.; Dong, J.-D.; Wang, J.; Jing, L.; Zhang, J.-Z.; Li, P.A. Overexpression of selenoprotein H prevents mitochondrial dynamic imbalance induced by glutamate exposure. Int. J. Biol. Sci. 2017, 13, 1458–1469. [Google Scholar] [CrossRef] [Green Version]
- Colle, D.; Santos, D.B.; De Souza, V.; Lopes, M.W.; Leal, R.B.; de Souza Brocardo, P.; Farina, M. Sodium selenite protects from 3-nitropropionic acid-induced oxidative stress in cultured primary cortical neurons. Mol. Biol. Rep. 2018, 46, 751–762. [Google Scholar] [CrossRef]
- Mccarty, M.F.; Assanga, S.B.I.; Luján, L.L.; O’Keefe, J.H.; Di Nicolantonio, J.J. Nutraceutical Strategies for Suppressing NLRP3 Inflammasome Activation: Pertinence to the Management of COVID-19 and Beyond. Nutrients 2020, 13, 47. [Google Scholar] [CrossRef]
- Yan, J.; Pang, Y.; Zhuang, J.; Lin, H.; Zhang, Q.; Han, L.; Ke, P.; Zhuang, J.; Huang, X. Selenepezil, a Selenium-Containing Compound, Exerts Neuroprotective Effect via Modulation of the Keap1–Nrf2–ARE Pathway and Attenuates Aβ-Induced Cognitive Impairment in Vivo. ACS Chem. Neurosci. 2019, 10, 2903–2914. [Google Scholar] [CrossRef]
- Mertens, K.; Lowes, D.; Webster, N.; Talib, J.; Hall, L.; Davies, M.; Beattie, J.; Galley, H. Low zinc and selenium concentrations in sepsis are associated with oxidative damage and inflammation. Br. J. Anaesth. 2015, 114, 990–999. [Google Scholar] [CrossRef] [Green Version]
- Prauchner, C.A. Oxidative stress in sepsis: Pathophysiological implications justifying antioxidant co-therapy. Burns 2017, 43, 471–485. [Google Scholar] [CrossRef]
- Bajčetić, M.; Spasić, S.; Spasojević, I. Redox Therapy in Neonatal Sepsis. Shock 2014, 42, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, O.; Levy, E.; Reifen, R. High selenium diet protects against TNBS-induced acute inflammation, mitochondrial dysfunction, and secondary necrosis in rat colon. Nutrition 2007, 23, 878–886. [Google Scholar] [CrossRef]
- Hiffler, L.; Rakotoambinina, B. Selenium and RNA Virus Interactions: Potential Implications for SARS-CoV-2 Infection (COVID-19). Front. Nutr. 2020, 7, 164. [Google Scholar] [CrossRef]
- Zhang, J.; Saad, R.; Taylor, E.W.; Rayman, M.P. Selenium and selenoproteins in viral infection with potential relevance to COVID-19. Redox Biol. 2020, 37, 101715. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Kim, H. Mini-Review on the Roles of Vitamin C, Vitamin D, and Selenium in the Immune System against COVID-19. Molecules 2020, 25, 5346. [Google Scholar] [CrossRef]
- Costagliola, G.; Spada, E.; Comberiati, P.; Peroni, D.G. Could nutritional supplements act as therapeutic adjuvants in COVID-19? Ital. J. Pediatr. 2021, 47, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Detopoulou, P.; Demopoulos, C.; Antonopoulou, S. Micronutrients, Phytochemicals and Mediterranean Diet: A Potential Protective Role against COVID-19 through Modulation of PAF Actions and Metabolism. Nutrition 2021, 13, 462. [Google Scholar] [CrossRef]
- Mrityunjaya, M.; Pavithra, V.; Neelam, R.; Janhavi, P.; Halami, P.M.; Ravindra, P.V. Immune-Boosting, Antioxidant and Anti-inflammatory Food Supplements Targeting Pathogenesis of COVID-19. Front. Immunol. 2020, 11, 570122. [Google Scholar] [CrossRef]
- Shakoor, H.; Feehan, J.; Al Dhaheri, A.S.; Ali, H.I.; Platat, C.; Ismail, L.C.; Apostolopoulos, V.; Stojanovska, L. Immune-boosting role of vitamins D, C, E, zinc, selenium and omega-3 fatty acids: Could they help against COVID-19? Maturitas 2021, 143, 1–9. [Google Scholar] [CrossRef]
- Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxid. Med. Cell. Longev. 2018, 2018, 9156285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaghari-Tabari, M.; Jafari-Gharabaghlou, D.; Sadeghsoltani, F.; Hassanpour, P.; Qujeq, D.; Rashtchizadeh, N.; Ghorbanihaghjo, A. Zinc and Selenium in Inflammatory Bowel Disease: Trace Elements with Key Roles? Biol. Trace Element Res. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-W.; Huang, T.-C.; Hu, Y.-C.; Hsieh, B.-S.; Chiu, P.-R.; Cheng, H.-L.; Chang, K.-L. Zinc protects chondrocytes from monosodium iodoacetate-induced damage by enhancing ATP and mitophagy. Biochem. Biophys. Res. Commun. 2020, 521, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Nutrition, immunity and COVID-19. BMJ Nutr. Prev. Heal. 2020, 3, 74–92. [Google Scholar] [CrossRef]
- Celik, C.; Gencay, A.; Ocsoy, I. Can food and food supplements be deployed in the fight against the COVID 19 pandemic? Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129801. [Google Scholar] [CrossRef] [PubMed]
- Gröber, U.; Holick, M.F. The coronavirus disease (COVID-19)—A supportive approach with selected micronutrients. Int. J. Vitam. Nutr. Res. 2021, 1–22. [Google Scholar] [CrossRef]
- Pallath, M.M.; Ahirwar, A.K.; Tripathi, S.C.; Asia, P.; Sakarde, A.; Gopal, N. COVID-19 and nutritional deficiency: A review of existing knowledge. Horm. Mol. Biol. Clin. Investig. 2021, 42, 77–85. [Google Scholar] [CrossRef]
- Skalny, A.V.; Rink, L.; Ajsuvakova, O.P.; Aschner, M.; Gritsenko, V.A.; Alekseenko, S.I.; Svistunov, A.A.; Petrakis, D.; Spandidos, D.A.; Aaseth, J.; et al. Zinc and respiratory tract infections: Perspectives for COVID‑19 (Review). Int. J. Mol. Med. 2020, 46, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Zabetakis, I.; Lordan, R.; Norton, C.; Tsoupras, A. COVID-19: The Inflammation Link and the Role of Nutrition in Potential Mitigation. Nutrients 2020, 12, 1466. [Google Scholar] [CrossRef]
- Abdallah, N.H.; Baulies, A.; Bouhlel, A.; Bejaoui, M.; Zaouali, M.A.; Ben Mimouna, S.; Messaoudi, I.; Fernandez-Checa, J.C.; Ruiz, C.G.; Ben Abdennebi, H. The effect of zinc acexamate on oxidative stress, inflammation and mitochondria induced apoptosis in rat model of renal warm ischemia. Biomed. Pharmacother. 2018, 105, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Filipović, D.; Mandić, L.M.; Kanazir, D.; Pajovic, S.B. Acute and/or chronic stress models modulate CuZnSOD and MnSOD protein expression in rat liver. Mol. Cell. Biochem. 2009, 338, 167–174. [Google Scholar] [CrossRef]
- Xu, W.; Barrientos, T.; Andrews, N.C. Iron and Copper in Mitochondrial Diseases. Cell Metab. 2013, 17, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cobine, P.A.; Pierrel, F.; Winge, D.R. Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim. Biophys. Acta Bioenerg. 2006, 1763, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Trivedi, P.P.; Timbalia, S.A.; Griffin, A.T.; Rahn, J.J.; Chan, S.S.L.; Gohil, V.M. Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency. Hum. Mol. Genet. 2014, 23, 3596–3606. [Google Scholar] [CrossRef]
- Zeng, H.; Saari, J.T.; Johnson, W.T. Copper Deficiency Decreases Complex IV but Not Complex I, II, III, or V in the Mitochondrial Respiratory Chain in Rat Heart. J. Nutr. 2007, 137, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Baker, Z.N.; Cobine, P.A.; Leary, S.C. The mitochondrion: A central architect of copper homeostasis. Metallomics 2017, 9, 1501–1512. [Google Scholar] [CrossRef]
- Zischka, H.; Einer, C. Mitochondrial copper homeostasis and its derailment in Wilson disease. Int. J. Biochem. Cell Biol. 2018, 102, 71–75. [Google Scholar] [CrossRef]
- Villa-Bellosta, R. Dietary magnesium supplementation improves lifespan in a mouse model of progeria. EMBO Mol. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, M.M.; Zhu, M.; Maitra, S.R. Alterations in cellular calcium and magnesium during circulatory/septic shock. Magnesium 1989, 8, 179–189. [Google Scholar]
- Zheltova, A.A.; Kharitonova, M.V.; Iezhitsa, I.N.; Spasov, A.A. Magnesium deficiency and oxidative stress: An update. Biomedicine 2016, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M.; Dominguez, L. Magnesium and Aging. Curr. Pharm. Des. 2010, 16, 832–839. [Google Scholar] [CrossRef]
- Liu, M.; Dudley, S.C., Jr. Magnesium, Oxidative Stress, Inflammation, and Cardiovascular Disease. Antioxidants 2020, 9, 907. [Google Scholar] [CrossRef] [PubMed]
- Spasov, A.A.; Zheltova, A.A.; Kharitonov, M.V. Magnesium and the oxidative stress. Ross. Fiziol. Zhurnal Im. I.M. Sechenova 2012, 98, 915–923. (In Russian) [Google Scholar] [PubMed]
- Shindo, Y.; Yamanaka, R.; Suzuki, K.; Hotta, K.; Oka, K. Intracellular magnesium level determines cell viability in the MPP+ model of Parkinson’s disease. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 3182–3191. [Google Scholar] [CrossRef] [Green Version]
- Shindo, Y.; Yamanaka, R.; Suzuki, K.; Hotta, K.; Oka, K. Altered expression of Mg2+ transport proteins during Parkinson’s disease-like dopaminergic cell degeneration in PC12 cells. Biochim. Biophys. Acta Bioenerg. 2016, 1863, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jeong, E.-M.; Liu, H.; Xie, A.; So, E.Y.; Shi, G.; Jeong, G.E.; Zhou, A.; Dudley, S.C. Magnesium supplementation improves diabetic mitochondrial and cardiac diastolic function. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef]
- Di Nicolantonio, J.J.; O’Keefe, J.H. Magnesium and Vitamin D Deficiency as a Potential Cause of Immune Dysfunction, Cytokine Storm and Disseminated Intravascular Coagulation in Covid-19 patients. Mo. Med. 2021, 118, 68–73. [Google Scholar]
- van Kempen, T.; Deixler, E. SARS-CoV-2: Influence of phosphate and magnesium, moderated by vitamin D, on energy (ATP) metabolism and on severity of COVID-19. Am. J. Physiol. Metab. 2021, 320, E2–E6. [Google Scholar] [CrossRef]
- Duarte, T.; Da Cruz, I.B.M.; Barbisan, F.; Capelleto, D.; Moresco, R.N.; Duarte, M.M. The effects of rosuvastatin on lipid-lowering, inflammatory, antioxidant and fibrinolytics blood biomarkers are influenced by Val16Ala superoxide dismutase manganese-dependent gene polymorphism. Pharm. J. 2016, 16, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhou, H.-M. The Role of Manganese Superoxide Dismutase in Inflammation Defense. Enzym. Res. 2011, 2011, 387176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Benedictis, C.A.; Vilella, A.; Grabrucker, A.M. The Role of Trace Metals in Alzheimer’s Disease. In Alzheimer’s Disease; Wisniewski, T., Ed.; Codon Publications: Brisbane, QLD, Australia, 2019. [Google Scholar]
- Li, L.; Yang, X. The Essential Element Manganese, Oxidative Stress, and Metabolic Diseases: Links and Interactions. Oxidative Med. Cell. Longev. 2018, 2018, 7580707. [Google Scholar] [CrossRef] [Green Version]
- Mezzaroba, L.; Alfieri, D.F.; Simão, A.N.C.; Reiche, E.M.V. The role of zinc, copper, manganese and iron in neurodegenerative diseases. NeuroToxicology 2019, 74, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Nam, E.; Han, J.; Suh, J.-M.; Yi, Y.; Lim, M.H. Link of impaired metal ion homeostasis to mitochondrial dysfunction in neurons. Curr. Opin. Chem. Biol. 2018, 43, 8–14. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín Giménez, V.M.; de las Heras, N.; Ferder, L.; Lahera, V.; Reiter, R.J.; Manucha, W. Potential Effects of Melatonin and Micronutrients on Mitochondrial Dysfunction during a Cytokine Storm Typical of Oxidative/Inflammatory Diseases. Diseases 2021, 9, 30. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases9020030
Martín Giménez VM, de las Heras N, Ferder L, Lahera V, Reiter RJ, Manucha W. Potential Effects of Melatonin and Micronutrients on Mitochondrial Dysfunction during a Cytokine Storm Typical of Oxidative/Inflammatory Diseases. Diseases. 2021; 9(2):30. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases9020030
Chicago/Turabian StyleMartín Giménez, Virna Margarita, Natalia de las Heras, León Ferder, Vicente Lahera, Russel J. Reiter, and Walter Manucha. 2021. "Potential Effects of Melatonin and Micronutrients on Mitochondrial Dysfunction during a Cytokine Storm Typical of Oxidative/Inflammatory Diseases" Diseases 9, no. 2: 30. https://0-doi-org.brum.beds.ac.uk/10.3390/diseases9020030