Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond


Department of Pharmacy-Center for Drug Research, Ludwig-Maximilians University, Butenandtstr. 5-13, 81377 Munich, Germany
*
Author to whom correspondence should be addressed.
Sci. Pharm. 2018, 86(4), 43; https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm86040043
Submission received: 22 August 2018
/
Revised: 19 September 2018
/
Accepted: 20 September 2018
/
Published: 27 September 2018
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Pharmacokinetic enhancers (boosters) are compounds used in combination with a primary therapeutic agent (drug) and are not used for their direct effects on the disease but because they enhance or restore the activity of the primary agent. Hence, in certain cases, they represent an indispensable escort for enzyme-labile drugs. Pharmacokinetic enhancers can exert their activity on different ways. In the most common case, they inhibit enzymes such as human cytochrome P450 enzymes in the liver or other organs and, thereby, block or reduce undesired metabolism and inactivation of the primary drug. In this review, an overview will be given on the therapeutically most important classes of pharmacokinetic enhancers like β-lactamase inhibitors, inhibitors of CYP (cytochrome P450) enzymes in HIV therapy and hepatitis C, boosters for fluoropyrimidine-type anticancer agents, compounds utilized for enabling therapy of Parkinson’s disease with levodopa, and others. Inhibitors of efflux pumps in both pathogenic bacteria and tumor cells will be addresses shortly.
1. Introduction
Pharmacokinetic enhancers, which are also named boosters or boosting agents, are compounds used in combination with a primary therapeutic agent (drug) not for their direct effects on the disease itself but because they enhance or restore the activity, increase the levels, and/or prolong the half-life of the primary agent. Hence, in certain cases, they represent an indispensable escort for drugs that preserve or restore their therapeutic potential. Pharmacokinetic enhancers can exert their activity in different ways. In the most common case, they inhibit enzymes such as human cytochrome P450 enzymes in the liver or other organs and, thereby, block or at least reduce undesired metabolism and inactivation of the primary drug. As a result, the required therapeutic dosage of the primary therapeutic agent can typically be lowered significantly, which ends up with reduced side effects and better tolerability of the therapy. The pharmacokinetic enhancers themselves should be well tolerated. Nevertheless, new drug-drug interactions can arise especially with enhancers influencing the hepatic clearance of other drugs in multimorbid patients as well.
Pharmacokinetic enhancers in current use can be categorized mainly into three groups, which are outlined below.
- inhibitors of hepatic drug-metabolizing enzymes such as CYP3A4
- inhibitors of drug-specific metabolizing enzymes
- inhibitors of bacterial β-lactamases as escort for β-lactam antibiotics
Comparable effects can further be achieved by using compounds affecting transport processes, e.g., during absorption from the intestine or during renal elimination, as well as efflux pumps in both tumor cells or pathogenic bacteria. However, these phenomena are beyond the closer scope of this review article and will be only mentioned in this paper.
This review mainly deals with the targets and molecular modes of action of pharmacokinetic enhancers in current use. The details of clinical aspects of the compounds and their combinations with drugs are beyond the scope of this review.
2. Antimicrobial Chemotherapy
2.1. Enhancers Targeting Bacterial Enzymes: β-Lactamase Inhibitors
2.1.1. Background
The β-lactam antibiotics are among the most important classes of antibacterial agents. However, resistance to β-lactam antibiotics in pathogenic bacteria is an emerging problem. The number of resistant strains has increased dramatically over the last few decades. Consequently, the identification of new antibiotics from known classes and even preferably new targets for antibiotics is an urgent need. However, the development of antibiotics with innovative molecular modes of action for fighting resistant pathogens is time-consuming, expensive, and risky. An alternative approach is to prolong the lifespan of marketed antibiotics by combining them with pharmacokinetic enhancers. In the field of antibiotics, these enhancers are also called “antibiotic adjuvants”, “resistance breakers”, or “antibiotic potentiators”. Typically, these enhancers have little or no intrinsic antibiotic activity and their only mission when co-administered with the antibiotic is to protect the antibiotic from degradation by the resistant bacteria by blocking the specific inactivation mechanism of the bacterium [1].
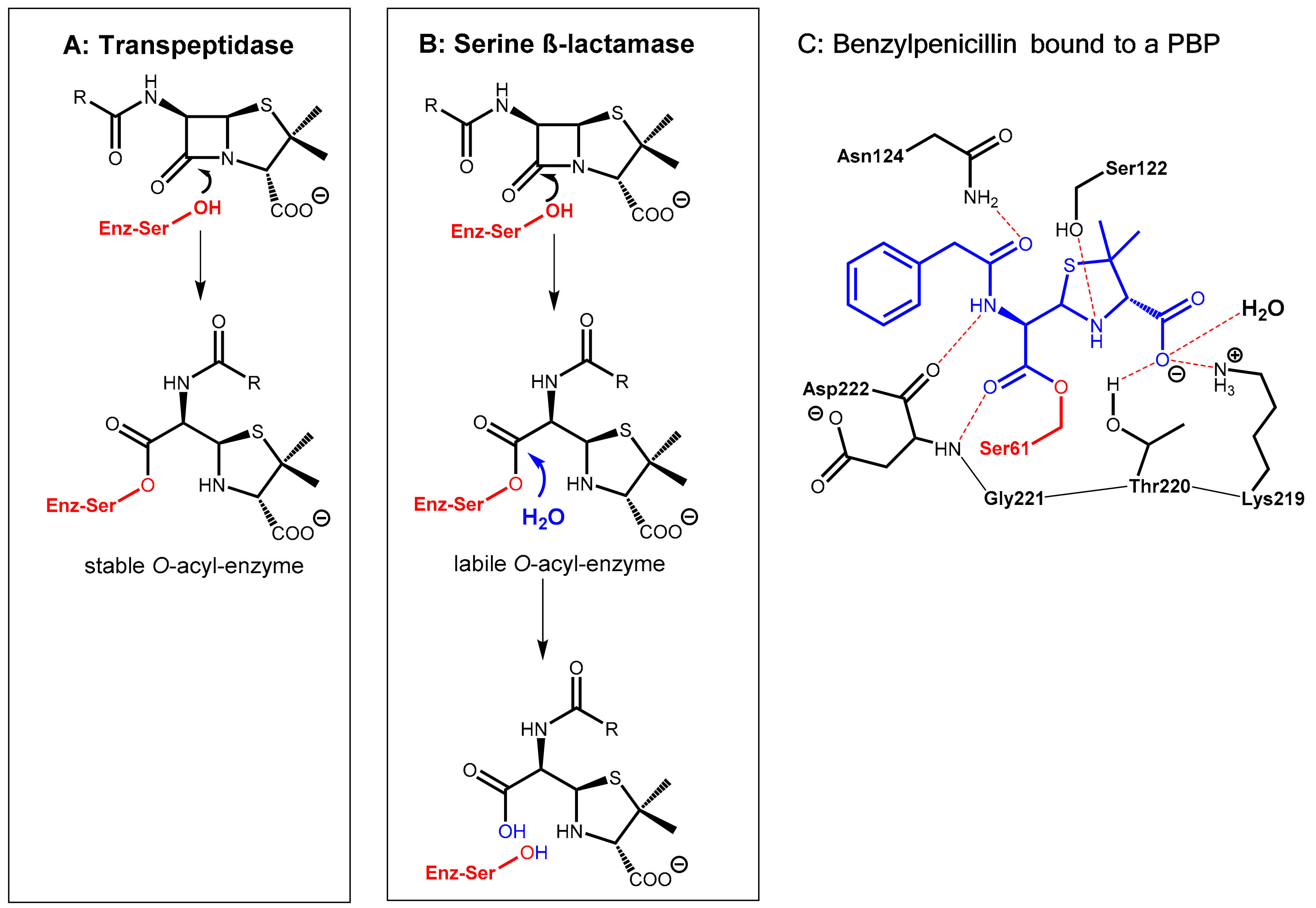
Inactivation of β-lactam antibiotics (penicillins, cephalosporins, and others) by bacterial enzymes named β-lactamases is the most prominent bacterial resistance mechanism that can be met with pharmacokinetic enhancers. Presently, the ability of certain bacteria to produce β-lactamases is the dominant cause of antibiotic resistance in Gram-negative bacteria. These enzymes hydrolyze the labile β-lactam ring of the β-lactam antibiotics in an acylation-deacylation-based process (Figure 1). Four major classes of β-lactamases (A, B, C, and D) are known. The classes A, C, and D are so-called serine-β-lactamases. In contrast, class B enzymes are zinc-containing metallo-β-lactamases [1,2].
The β-lactam antibiotics principally interfere with bacterial target proteins (penicillin-binding proteins, PBP, e.g., transpeptidases involved the biosynthesis of the cell wall component murein) by forming a stable O-acyl–enzyme complex upon reaction with a serine residue in the active site (Figure 1A,C). In a similar manner, hydrolysis of the β-lactam ring by bacterial serine-β-lactamases occurs via nucleophilic attack of the hydroxyl group of a serine hydroxyl group in the active site of the enzyme at the β-lactam carbonyl group of the antibiotic giving, under ring opening, an O-acyl derivative of the serine side chain. In a consecutive step, this ester is rapidly hydrolyzed to give an inactive hydrolyzed antibiotic under regeneration of the active site of the β-lactamase enzyme (Figure 1B).
Pharmacokinetic enhancers (β-lactamase inhibitors) suppress hydrolytic inactivation of β-lactam antibiotics. They are able to inactivate the bacterial β-lactamase enzyme, which is the most convenient approach for prolonging the lifespan of marketed β-lactam antibiotics. Consequently, β-lactamase inhibitors are the most widely clinically used adjuvants to overcome resistance caused by β-lactamases. The carbapenem-hydrolyzing class D β-lactamases represent the most rapidly growing and diverse group of β-lactamases with over 500 reported enzymes. These are found in the most clinically challenging resistant species, e.g., Acinetobacter baumannii, Pseudomonas aeruginosa, and Proteus mirabilis. They are able to hydrolyze penicillin but also other β-lactam-type antibiotics like cephalosporins and aztreonam [1].
2.1.2. Irreversibly Binding β-Lactam-Type β-Lactamase Inhibitors
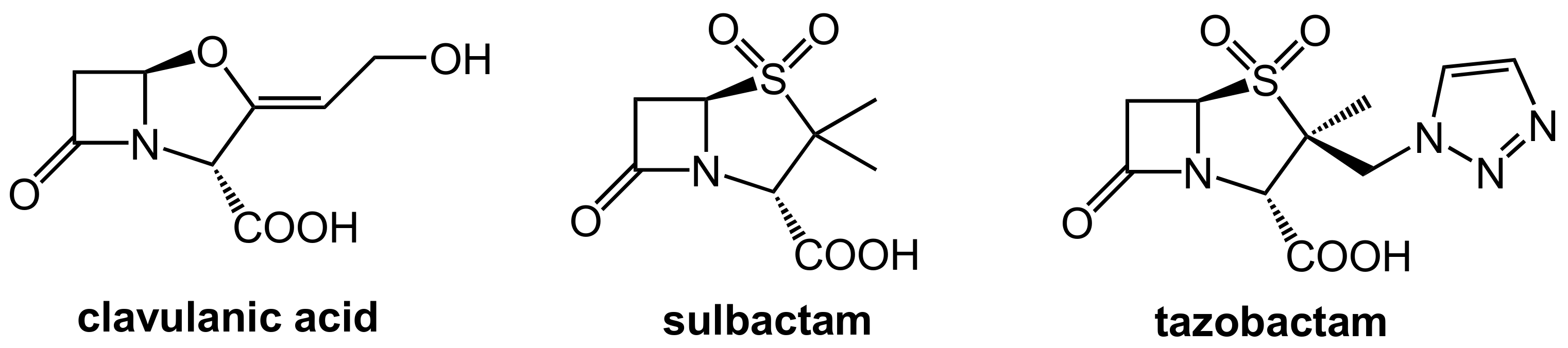
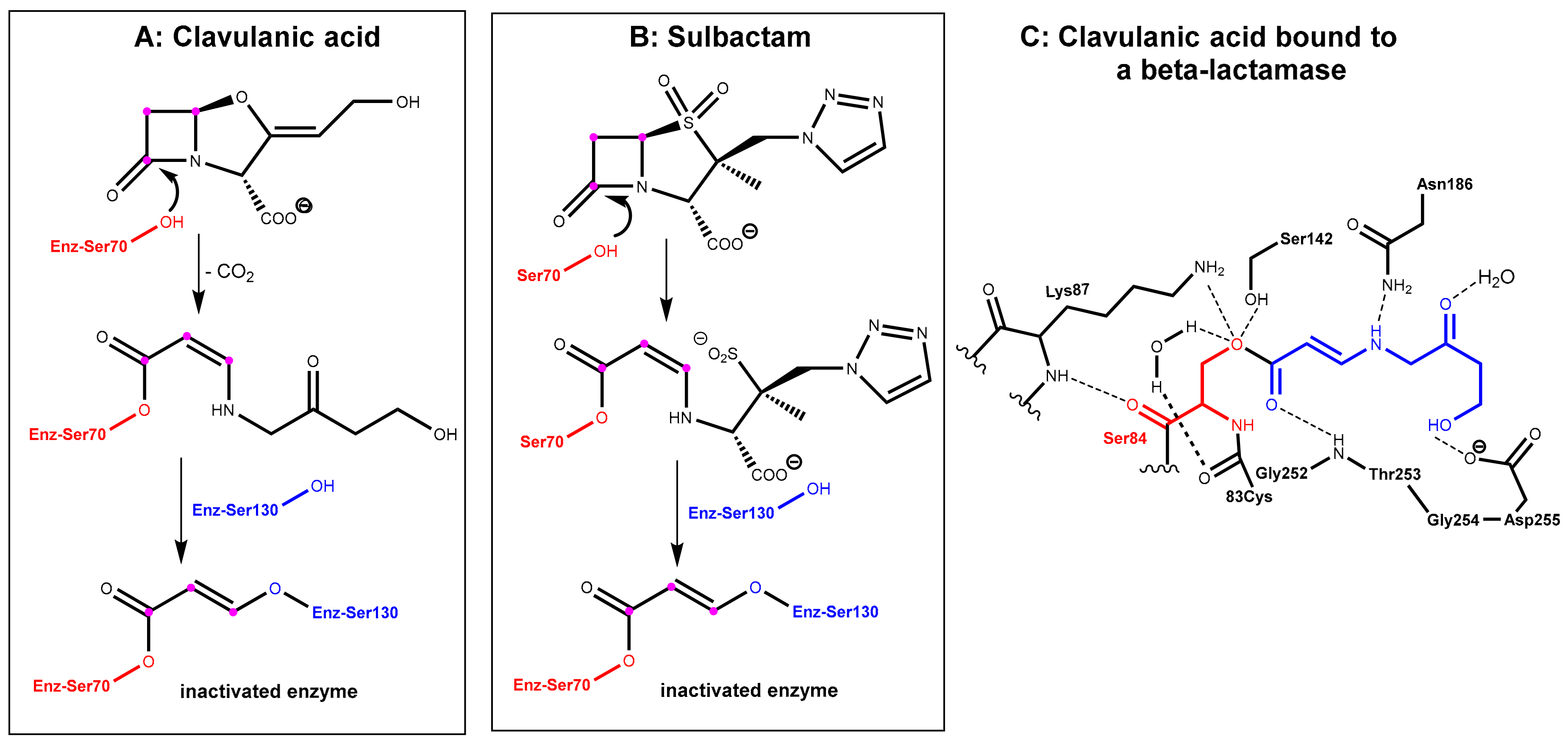
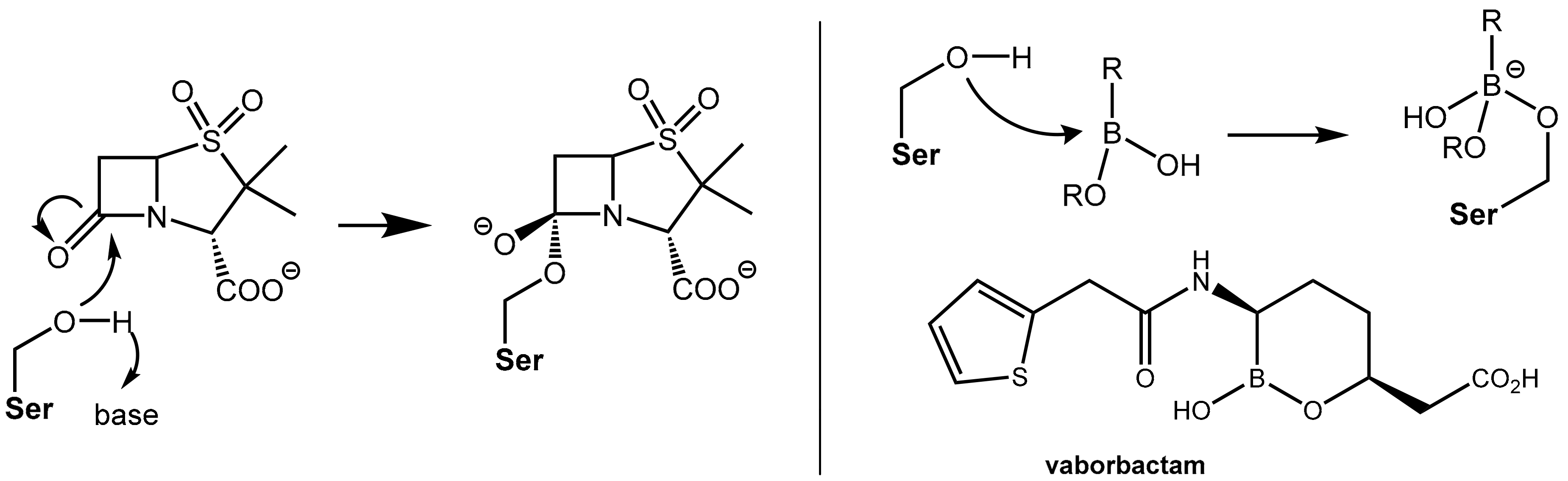
The first generation of β-lactamase inhibitors in clinical use are clavulanic acid, sulbactam, and tazobactam (Figure 2). These inhibitors contain a β-lactam moiety as well and they represent mechanism-based covalent inhibitors. They form a stable acyl–enzyme complex with crucial serine residues in the active sites of the β-lactamases.
In the first step, which is mechanistically closely related to the crucial step of the mode of action of the β-lactam antibiotics (Figure 1A), the β-lactamase inhibitors react under the opening of the four-membered ring with the active site serine (Ser70 here) of the β-lactamase to give an O-acyl derivative. The respective intermediates undergo further transformations (e.g., decarboxylation in the clavulanic acid adduct, see Figure 3A. Tazobactam reacts in a closely related manner, see Figure 3B). The common second central step is a reaction of an intermediate enamino ester with a second serine residue (Ser130 here). In the end, a total of three carbon atoms of the inhibitor is bound covalently to the enzyme and interconnects two serine residues in the active site under the destruction of the catalytic activity of the β-lactamase, as demonstrated in Figure 3C [3,4].
Typical combinations on the market are clavulanic acid/amoxicillin (co-amoxiclav) for oral application and tazobactam/piperacillin and sulbactam/ampicillin for parenteral therapy.
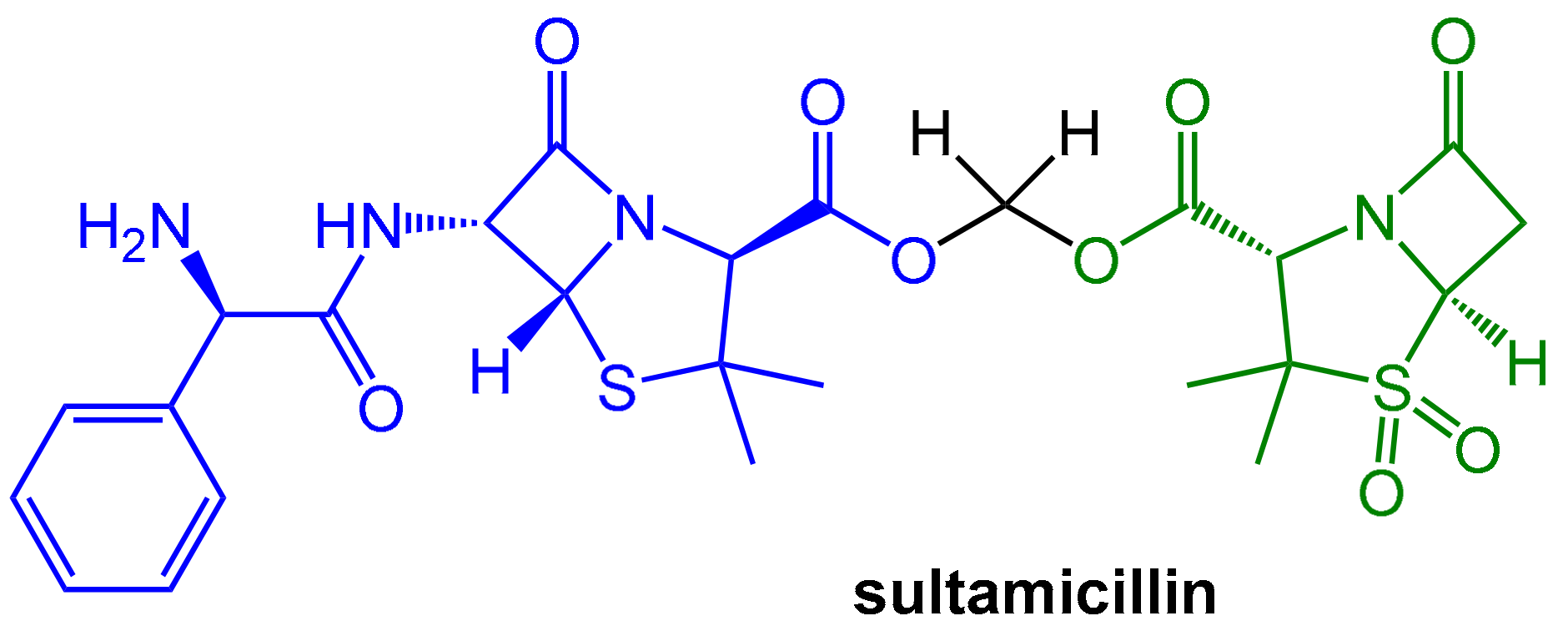
However, both components of the sulbactam/ampicillin couple are not well absorbed upon oral application. A noteworthy improvement was achieved with sultamicillin (Figure 4). In this drug, both ampicillin and sulbactam are connected covalently in the form of a mixed di-ester of formaldehyde hydrate. This compound is a “mutual pro-drug”, which is readily absorbed when administered orally. The pro-drug is rapidly hydrolyzed by esterases to provide high equimolar levels (bioavailability > 80%) of its two constituents (plus formaldehyde). The pharmacokinetic parameters of both resulting active components are similar and, fortunately, the inevitable equimolar concentrations of both components are therapeutically useful [5].
However, these first generation β-lactam-derived inhibitors are clinically ineffective against class C and D-type β-lactamases. Fortunately, several innovative types of inhibitors have been developed recently to overcome this limitation, as pointed out below.
2.1.3. Reversibly Binding β-Lactamase Inhibitors
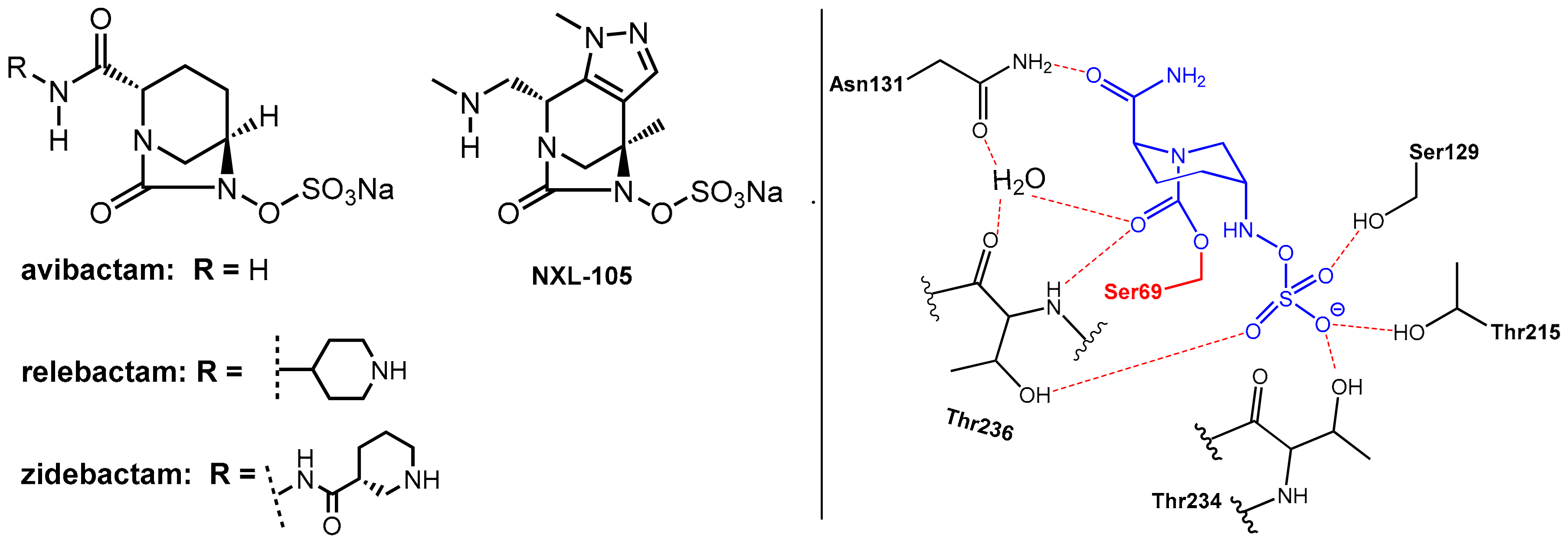
• Avibactam and Related Diazabicyclooctanes (DBOs)
The acyl–enzyme complexes from reactive β-lactams (antibiotics as well as β-lactam-type β-lactamase inhibitors, see Section 2.1.1 and Section 2.1.2) with the active site serines of PBPs and β-lactamases are generally considered to be irreversible. In contrast, appropriately functionalized γ-lactam analogues of the abovementioned β-lactam-type inhibitors can react reversibly with the nucleophilic serine residue [2]. Work from the 1980s, already revealed that bicyclic compounds containing five-membered lactam moieties can exhibit antibacterial activity comparable to β-lactam antibiotics especially if the electron density on the nitrogen of the γ-lactam ring was reduced by appropriate substituents.
Avibactam, which is a 1,6-diazabicyclo[3.2.1]octane (DBO)-based bicyclic γ-lactam marketed in combination with the third generation cephalosporin ceftazidime [2,6] in 2015 for the treatment of complicated urinary tract and intra-abdominal infections, represents the first member of non-β-lactam-type inhibitors of serine β-lactamases. It is a potent inhibitor of class A, class C, and some class D serine β-lactamases. Furthermore, it is closely related DBOs such as relebactam and zidebactam (Figure 5), which are presently used in clinical trials [1].
These DBOs are able to acylate nucleophilic serine residues in a manner analogous to β-lactams. As the abovementioned β-lactam-type inhibitors, avibactam does not exhibit clinically relevant antibacterial activity. Interestingly, some related DBO derivatives (e.g., NXL-105, Figure 5) also display antibacterial activity.
Avibactam reacts with the nucleophilic serine in the active site of the β-lactamase to form, once again, under ring opening, which is a relatively stable carbamoyl–enzyme complex. In contrast to the β-lactam-based serine-β-lactamase inhibitors (clavulanic acid, sulbactam, tazobactam), there is no evidence for subsequent fragmentation reactions of the O-acyl–enzyme products. Rather, this O-acylation reaction is generally reversible. However, as revealed by a crystal structure, the ring-opened adduct is stabilized by interactions with a series of amino acids, which are relevant in catalysis. The primary amide of avibactam is positioned to interact via a network of hydrogen bonds with a water molecule (which is engaged in subsequent hydrolysis reactions in the case of complexes built with β-lactam-derived β-lactamase inhibitors). The organosulfate residue undergoes interactions similar to those of the carboxylate group of β-lactam substrates. This network of interactions can explain the observed long-lived nature of the avibactam complex (t1/2 > 7 days, compared with 5 h for the tazobactam-derived complex) [2].
Combinations of avibactam with other β-lactam antibiotics (e.g., ceftaroline, aztreonam) are in development. Hereby, the aztreonam–avibactam combination is of special interest since aztreonam is the only β-lactam antibiotic non-susceptible to inactivation by the class B metallo-β-lactamases [1].
• Vaborbactam, a Novel Non-Acylating Serine β-Lactamase Inhibitor
Both the action of β-lactam antibiotics on PBP and the action of a serine β-lactamases on β-lactam antibiotics involve, from a mechanistic point of view, an identical first reaction step, which is the nucleophilic attack of an “active” (what means in situ deprotonated) serine residue in the active site of the enzyme at the β-lactam carbonyl group of the β-lactam antibiotic under formation of a tetrahedral known as a negatively charged transition state. Further fractionation leads to cleavage of the β-lactam ring (see Figure 1, Figure 3 and Figure 5). The β- lactam-type and γ-lactam-type serine β-lactamase inhibitors described above exert their protective activity by undergoing O-acylation of active site serine hydroxyl groups of the β-lactamases, which results in an irreversible inhibition of these enzymes. In a novel chemotype of non-acylating β-lactamase inhibitors (“transition state analogues”), attack of the active site serine is prevented not by acylation of the hydroxyl group but by reversible reaction with an organoboronate group (boronic acid or derivative thereof). The Lewis-acidic boronate group of the inhibitor undergoes reversible coupling with the serine alkoxide of the β-lactamase to give a tetrahederal, negatively charged boronate adduct (Figure 6). By improving the affinity of the lead structures with the incorporation of side chains similar to those found in launched penicillin (e.g., the (thiophen-2-yl)acetamido group), the first inhibitor vaborbactam (RPX7009), which is a potent inhibitor of clinically relevant class A and C β-lactamases like carbapenemases, was obtained. This compound was launched in the U.S. in 2017 in fixed combination with the carbapenem antibiotic meropenem for the treatment of complicated urinary tract infections [7]). Presently, some additional boronate-based β-lactamase inhibitors are undergoing clinical trials [1_BMCL].
2.2. Enhancers Targeting Host Enzymes: Cilastatin
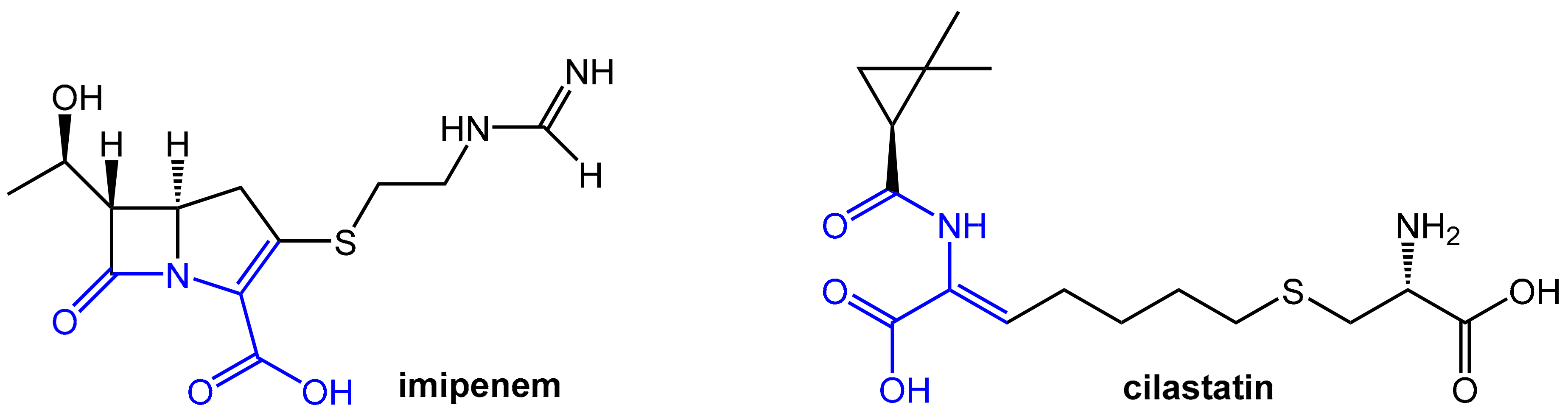
The carbapenem-type β-lactam antibiotic imipenem (Figure 7) has a broad spectrum of activity against aerobic and anaerobic Gram-positive and Gram-negative pathogenic bacteria. It is a derivative of the natural product thienamycin produced by Streptomyces cattleya, which was not qualified for further development due to limited chemical stability [8]. Imipenem is of very high clinical relevance due to its outstanding activity against Pseudomonas aeruginosa and Enterococci. Like other carbapenems, it is highly resistant to β-lactamases produced by many multiple drug-resistant Gram-negative bacteria and exhibits no cross-resistance with other β-lactam antibiotics like penicillin and cephalosporins.
Unfortunately, imipenem is rapidly degraded by the renal enzyme dehydropeptidase-I, which is a zinc metalloenzyme located in the microvilli of kidney proximal tubular cells in man and, therefore, this antibiotic is not suitable for monotherapy. Comparable sensitivity to dehydropeptidase-I was found for lead structure thienamycin, but, in contrast, the carbapenem antibiotic meropenem (see above; launched in fixed combination with the novel boronic acid-derived β-lactamase inhibitor vaborbactam) is stable against hydrolysis by this renal enzyme. Dehydropeptidase-I acts as an unprecedented mammalian (!) β-lactamase upon imipenem by performing a hydrolytic cleavage of the β-lactam moiety. This is very remarkable considering the stability of this antibiotic toward classical bacterial β-lactamases. Conversely, the typical penicillin and cephalosporins, part of which show significant sensitivity to bacterial β-lactamases, show little or no detectable susceptibility to β-lactam hydrolysis by human dehydropeptidase-I [8].
In general, the enzyme dehydropeptidase-I exhibits versatile substrate specificity by hydrolyzing dipeptides and dehydropeptides (what means N-acylated α, β-unsaturated α-amino acids) as well as β-lactam antibiotics having the unique double bond geometry such as imipenem [9].
Efficient therapy with imipenem is not possible without protection from degradation by dehydropeptidase-I. A systematic search for inhibitors of this degrading enzyme on the basis of the structural similarities between the scissile bonds in imipenem and dehydropeptides led to cilastatin. This compound also bears a dehydropeptide moiety (Figure 7) and represents a very potent reversible, competitive inhibitor of renal dehydropeptidase-I [10,11]. Cilastatin itself does not exhibit any antibacterial activity, but it matches in its pharmacokinetic properties well for co-administration with imipenem. It is, therefore, combined intravenously with imipenem in order to protect the antibiotic from dehydropeptidase-I and prolong its antibacterial effect. This combination provides clinically relevant concentrations of imipenem in the urine for 8 to 10 h [8].
2.3. Fruitful Drug-Drug Interactions in Helicobacter pylori Eradication
Infection of gastric mucosa by the bacterium Helicobacter pylori is a causative agent of chronic gastritis but also is an essential co-factor in the etiology of gastroduodenal ulcer and gastric carcinoma. Eradication of this bacterium using triple or quadruple therapies with a proton pump inhibitor in combination with two or three antibiotics is the treatment of choice and eradication rates of 80% to 90% can be achieved. A proton pump inhibitor is included in this regimen for two main reasons: first, at an elevated gastric pH, the portion of H. pylori cells, which are in the proliferative phase is enhanced. This results in improved susceptibility for an antibiotic attack. Second, the half-life of acid-sensitive antibiotics (penicillins, macrolides) is enhanced at a higher gastric pH [12].
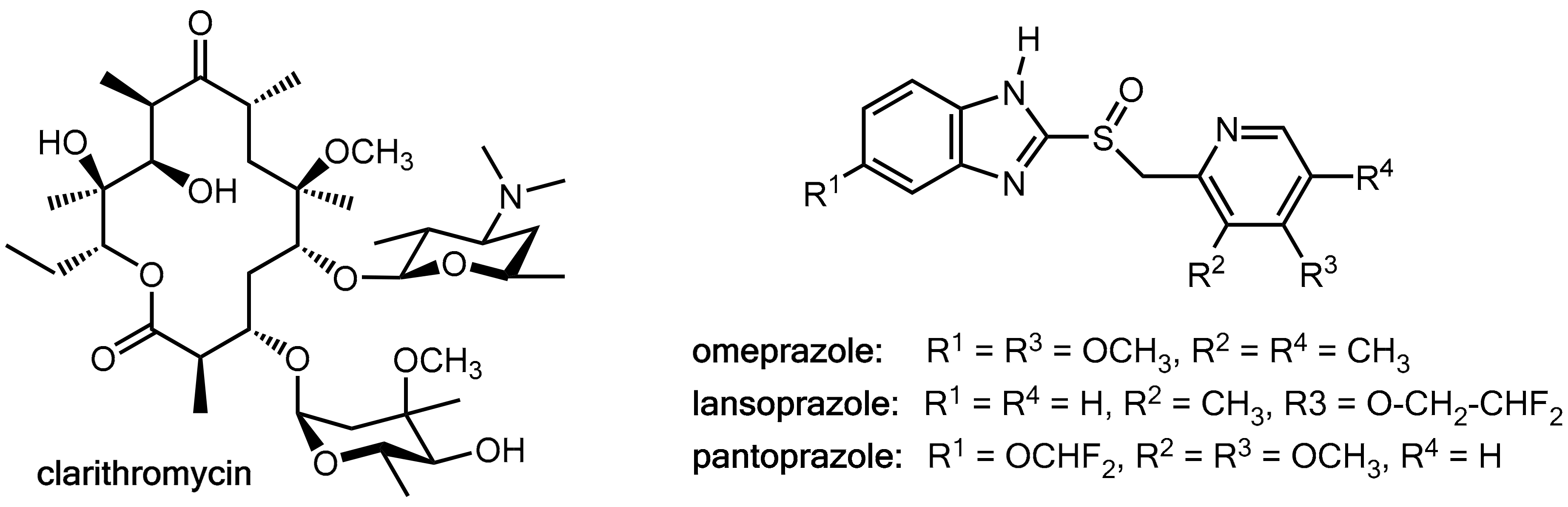
Clarithromycin (Figure 8), which is a macrolide antibiotic commonly used in these regimens, is a potent inhibitor of CYP3A4 and CYP2C19 but also a substrate of these liver enzymes [13,14,15]. On the other hand, the proton pump inhibitors omeprazole, lansoprazole, and pantoprazole (Figure 8) are substrates of both CYP3A4 and CYP2C19 [16] and omeprazole and its metabolites are also inhibitors of both of these CYP enzymes [17]. Therefore, at least in principle, clarithromycin and the prazoles have the potential for mutually influencing their pharmacokinetics. Combination of clarithromycin with these prazoles did not result in enhanced clarithromycin levels [12], but significant inhibition of the metabolism of both omeprazole [15,18] and lansoprazole [13] were recorded while pantoprazole levels remained almost unchanged [18]. Amoxicillin levels were not influenced in a lansoprazole-clarithromycin-amoxicillin triple therapy [12]. In conclusion, these data suggest that clarithromycin acts as a pharmacokinetic enhancer for omeprazole and lansoprazole. However, the clinical relevance of this effect appears to be of a limited extent.
2.4. Enhancers for Antibiotics Beyond Enzyme Inhibition: Probenecid and Efflux Pump Inhibitors
Albeit beyond the closer scope of this review article, some additional principles of enhancing the therapeutic effects of antibiotics will be mentioned below.
2.4.1. Probenecid
In the 1940s, the effect of simple benzamides on renal elimination of ionizable drugs like penicillin was observed [19] and due to the limited amounts of penicillin available during World War II, probenecid (Figure 9) was developed with the goal of reducing the renal excretion of penicillin. Co-administration of probenecid was intended in order to reduce the consumption of the very precious antibiotic. Unfortunately, the development was not finished before the end of the war [20]. Probenecid was used as adjunct therapy with penicillin. Probenecid is still used for its uricosuric properties in the treatment of gout, but its clinical relevance is poor. The compound was found to inhibit the active transport of both anionic and cationic drug molecules at various sites in the body [21].
Subsequent studies revealed that probenecid was effective in enhancing the retention of other antibiotics as well. It decreases the renal secretion of numerous quinolones such as ciprofloxacin, gatifloxacin, levofloxacin, norfloxacin, fleroxacin, and enoxacin while moxifloxacin and sparfloxacin are not affected by probenecid co-administration [22].
2.4.2. Inhibitors of Bacterial Efflux Pumps (“Antibiotic Escort Molecules”, “Antibiotic Adjuvants”)
Intrinsic resistance in Gram-negative bacteria is largely attributed to the activity of multidrug resistance (MDR) efflux pumps, but these pumps also play a significant role in acquired clinical resistance to antibiotics. This indicated a significant potential for developing small molecule inhibitors against bacterial efflux pumps [23]. Efflux pump inhibitors were expected to render resistant bacteria increasingly more sensitive to a given antibiotic or even reverse the multidrug resistant phenotype [24].
A large number of efflux pump inhibitors with in vitro activity for efflux pumps like NorA in Staphylococcus aureus, MexAB-OprM in Pseudomonas aeruginosa, and AcrAB-TolC in Enterobacteriaceae, Mycobacterium tuberculosis, and others have been discovered including natural products, antibiotics, and synthetic molecules. However, these efforts did not result in a clinically useful compound yet for various reasons including in vivo toxicity and side effects [24,25].
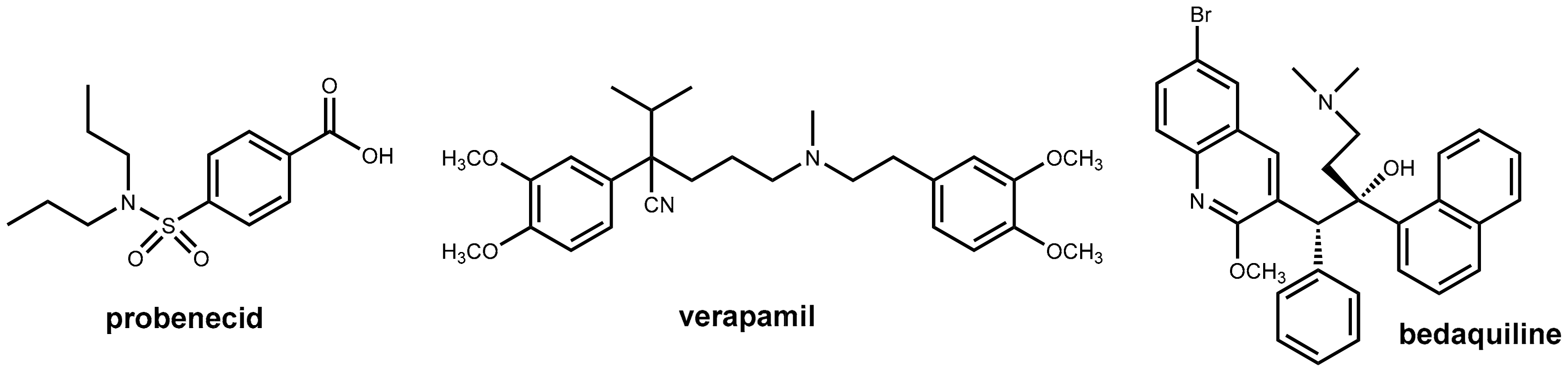
2.4.3. Pharmacokinetic Enhancement in Antitubercular Therapy (?): Bedaquiline Plus Verapamil
Bedaquiline (Figure 9) is the first new antitubercular drug approved by the FDA in 40 years and is an effective treatment option for multidrug-resistant Mycobacterium tuberculosis infection. However, this occurs at a very early stage after approval efflux-mediated bedaquiline resistance, which has been identified in treatment failures. This resistance is due to an efflux of the drug (and a variety of other anti-tubercular drugs) from the mycobacterial cell. This decreases the intracellular drug concentrations below the required levels. It was observed that the addition of efflux pump inhibitors partially restored drug susceptibility in vitro and in vivo [26,27]. The well-known efflux pump inhibitor verapamil (Figure 9) showed beneficial effects in vivo when co-administered with bedaquiline, but subsequent investigations demonstrated that this effect is likely due to enhanced systemic exposure to bedaquiline via the effects on mammalian transporters (resulting in increased oral bioavailability) rather than inhibition of bacterial efflux pumps. Therefore, it was claimed recently that there may be no advantage to administering verapamil versus simply increasing the dose of bedaquiline [28].
3. HIV Therapy: Inhibition of CYP3A4 for Boosting Antiretroviral Drugs
3.1. HIV Proteases
Maximal and durable suppression of the viral load is the predominant goal of HIV therapy. By this means, novel antiretroviral therapy (ART) regimens were developed, which have transformed HIV-1 infection from a fatal to a kind of chronic disease. The success of ART is directly related to adequate and permanent drug exposure and patient-friendly dosage schedules [29,30].
Numerous classes of antiretroviral drugs for ART like entry inhibitors, reverse transcriptase inhibitors, integrase inhibitors, and protease inhibitors (PIs) have been developed over the decades. Among them, protease inhibitors are of outstanding importance [31].
HIV proteases are aspartyl proteases, which catalyze the proteolytic cleavage of viral polypeptide precursors into mature enzymes and structural proteins. This process is essential for the conversion of HIV particles into their mature infectious form. Inhibitors of HIV proteases were developed by a structure-based design created utilizing the knowledge of these enzymes’ molecular modes of action, which ends up with peptidomimetic drugs suitable for oral application. However, PIs were historically associated with poor oral bioavailability and a high pill burden [31].
The poor oral bioavailability of PIs is mainly due to their susceptibility to oxidation by CYP3A4 enzymes in both human small intestinal enterocytes and hepatocytes while biotransformation in the gut wall substantially contributes to the overall first-pass metabolism. Consequently, pharmacokinetic enhancement of protease inhibitors can be achieved by intentional inhibition of CYP3A4 leading to higher drug exposure, lower pill burden, and, therefore, simplified dosing schedules. Since virtually all PIs are CYP3A4 substrates, co-administration of a boosting agent to increase drug concentrations is commonly employed. Furthermore, the high content of P-glycoprotein efflux transporters on the apical surface of enterocytes shuttles the protease inhibitors out of the gut wall back into the intestinal lumen and, hence, decreases oral bioavailability further. This supports the theory that this efflux transporter may act in concert with CYP3A4 to limit oral drug bioavailability [29,32].
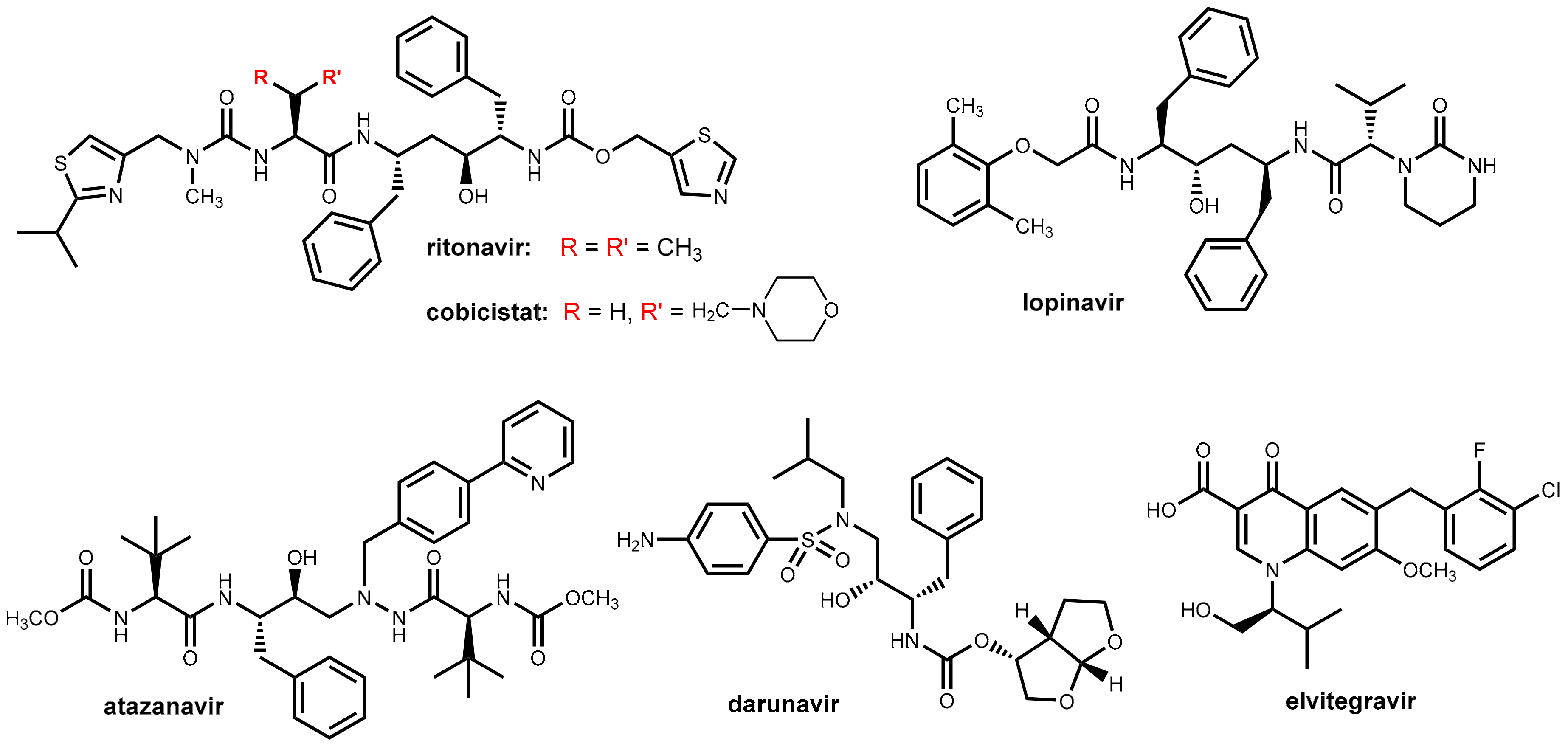
3.2. Ritonavir and Cobicistat
Ritonavir (Figure 10) was initially developed as an HIV inhibitor, but its use at the originally approved dose (600 mg twice daily) was limited by dose-related side effects like diarrhea and nausea. In the course of its initial application in combination with other antiretroviral drugs, it was observed that ritonavir inhibits CYP enzymes especially CYP3A4 as well as P-glycoprotein efflux transporters and, thus, acts as a pharmacokinetic enhancer (booster) for some of the co-administered drugs [33]. Nowadays, ritonavir is being used almost exclusively at well-tolerated doses of 100 to 200 mg/day as a booster for other HIV protease inhibitors (e.g., lopinavir, atazanavir, darunavir; Figure 10) for maintaining therapeutic serum levels of these drugs. Ritonavir is marketed both as a single entity (Norvir®) for combination with other antiretroviral drugs and as a fixed dose combination with lopinavir (Kaletra®). Boosting the protease inhibitors with ritonavir allows for less-frequent dosing (once or twice daily) and a lower pill burden and results in a reduced impact of food on bioavailability, reduced variability of systemic drug exposure, and improved treatment efficacy [34].
Later, the application of ritonavir has been extended to boosting the therapeutic effect of the integrase inhibitor elvitegravir (Figure 10). In contrast to other integrase inhibitors like raltegravir and dolutegravir, which possess minimal CYP involvement, elvitegravir is metabolized primarily by CYP3A4. Pharmacokinetic boosting of elvitegravir with ritonavir leads to systemic exposures that permit daily dosing. Alternatively, elvitegravir is co-formulated with cobicistat (see below) [35].
An additional application of ritonavir as a booster for a protease inhibitor in hepatitis C therapy is discussed below (Section 4).
In 2012, a second pharmacokinetic enhancer from this chemotype was launched. Cobicistat (Tybost®) (Figure 10), which is a compound structurally closely related to ritonavir, does, in contrast to ritonavir, no longer exhibit intrinsic inhibitory activity on HIV protease and acts exclusively as a booster due to the inhibition of CYP3A4 and blocking P-glycoprotein efflux transporters [33,34]. Cobicistat is further marketed as fixed dose combinations with protease inhibitors (atazanavir, darunavir) and the integrase inhibitor elvitegravir. Cobicistat (150 mg once daily) exhibits a boosting effect comparable with that of ritonavir (100 mg once daily).
Cobicistat was developed with the aim of reducing the incidence of undesired side effects believed to be caused by ritonavir like alterations in lipid metabolism, gastrointestinal disturbances, and pronounced drug-drug interactions [33]. Cobicistat has improved physiochemical properties (e.g., water solubility) compared with ritonavir and is a potent inhibitor of CYP3A, but it is only a weak inhibitor of CYP2D6 and was claimed to have almost no effect on other CYP enzymes. In contrast, ritonavir inhibits, beyond CYP3A4, a number of additional CYP enzymes (2C19, 2C8, 2C9, 2D6, and 2E1). In addition, in contrast to ritonavir, which is also an inducer of CYP1A2 and CYP2C9, cobicistat does not demonstrate any inducing effects on CYP450 enzymes [34,36]. On the basis of these data, it was claimed that switching patients from ritonavir to cobicistat regimens may be problematic particularly with co-medications that have a very narrow therapeutic index [34]. So drug-drug interactions may be different for both pharmacokinetic enhancers and need to be evaluated for each drug individually. However, a recent in vitro investigation of the inhibition of diverse CYP enzymes by both compounds revealed that both ritonavir and cobicistat are highly potent inhibitors of CYP3A, but both compounds were weaker inhibitors of other investigated human CYP enzymes (2B6, 2C9, 2C19, 2D6) with IC50 values at least two orders of magnitude higher. There was no evidence of a meaningful difference in selectivity between ritonavir and cobicistat [37].
Like ritonavir, cobicistat further enhances the intestinal absorption of HIV protease inhibitors by the inhibition of the intestinal efflux transporter P-glycoprotein [38].
In clinical studies, the pharmacokinetic effects of cobicistat and ritonavir on the properties of atazanavir, darunavir, and elvitegravir were almost identical, but, in contrast, cobicistat was found to be an inadequate booster for the protease inhibitor tipranavir [34].
3.3. Development of New Boosters—A Long and Rocky Road
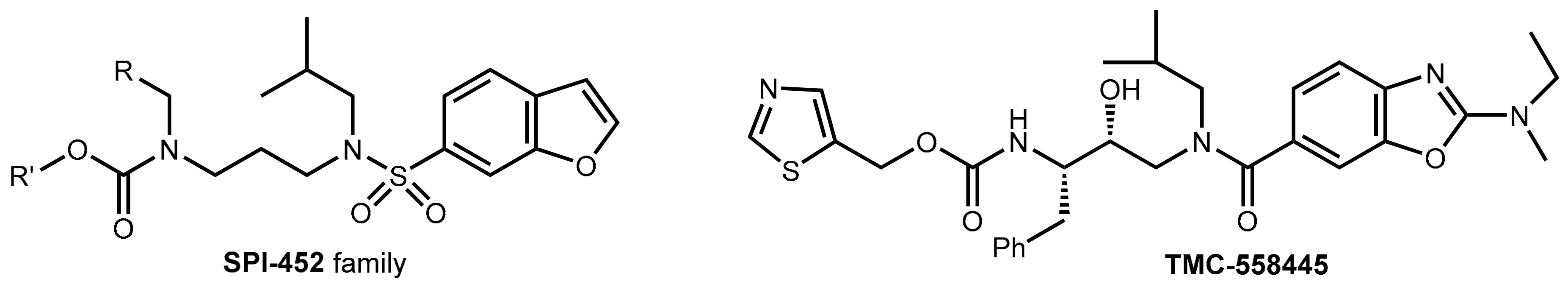
The uncontested therapeutic value and the economic success of pharmacokinetic enhancers for antiretroviral drugs led to immense efforts in the pharmaceutical industry and academia in order to develop new, improved boosters in addition to ritonavir and cobicistat. Several promising compounds like Sequoia’s SPI-452 and Tibotec’s TMC-558445 (Phase I study completed in 2010) were claimed, but none of them have been marketed yet. The reported developments appear to be cancelled or are on hold [39] (Figure 11).
Note: Concert Pharmaceuticals and GSK developed a novel HIV protease inhibitor named CTP-518 in which certain key hydrogen atoms of the established PI atazanavir are replaced with deuterium atoms [40]. Deuterium (2H) forms a stronger chemical bond to carbon atoms than hydrogen (1H) and deuterium modification at selected positions of the drug was expected to substantially enhance the metabolic stability. It was demonstrated in pre-clinical studies that sophisticated deuterium modification of atazanavir fully retains its antiviral potency, but can markedly reduce hepatic metabolism, which results in increased half-life and plasma levels. It has been claimed that CTP-518 could potentially avoid the need for co-administration of a booster such as ritonavir. However, the development appears to have been discontinued in 2013.
4. Ritonavir in Hepatitis C Therapy
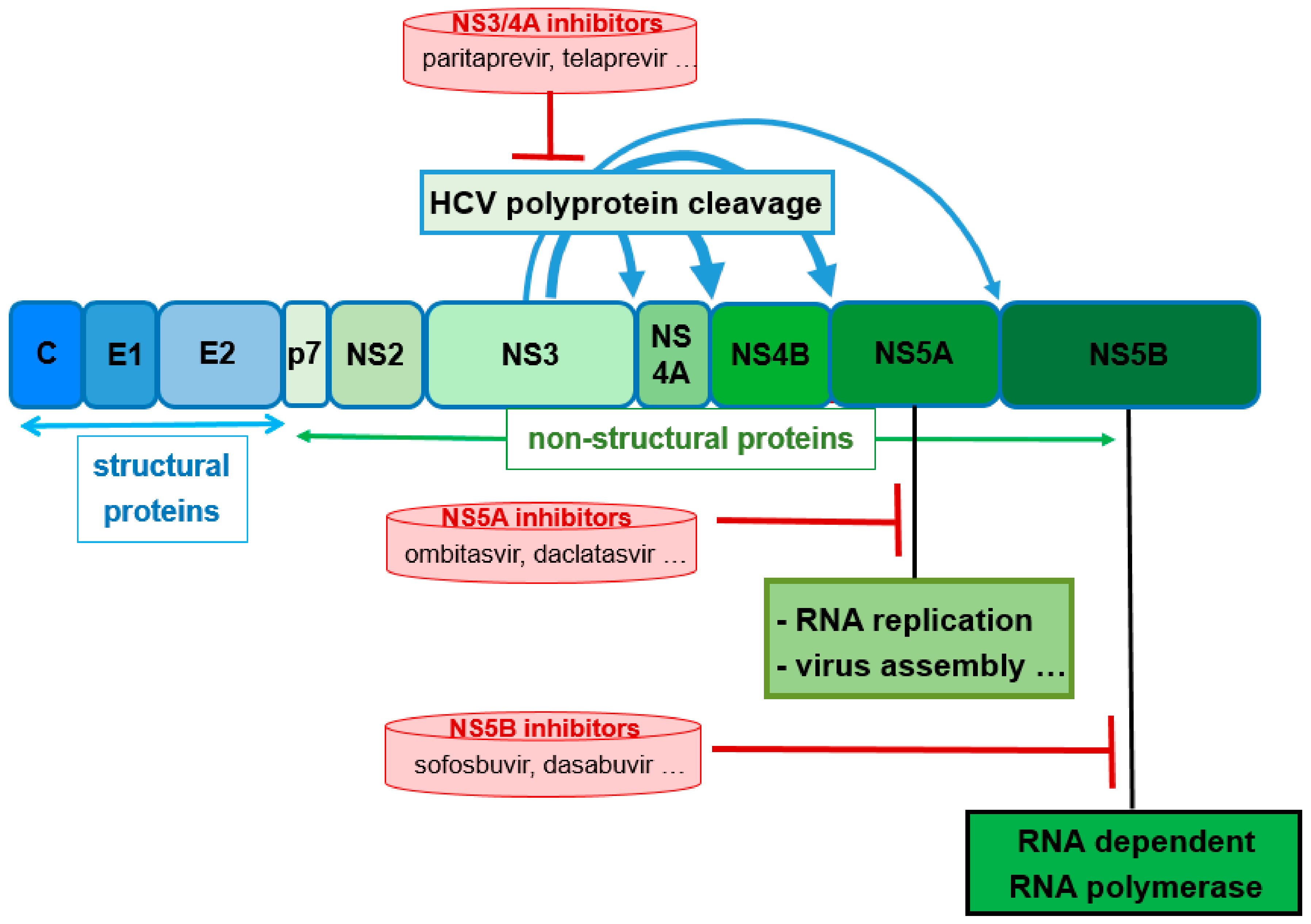
About 130 million to 150 million people worldwide are infected with the hepatitis C virus (HCV) with approximately 500,000 dying each year as a result of HCV complications. Previous standard treatments utilizing interferons and ribavirin had very poor response rates and poorly tolerable side effects. In the past decade, the development of direct-acting antivirals (DAAs) has revolutionized therapy of chronic hepatitis C. Novel DAA combinations with high efficacy, tolerable side effects, and short treatment courses are currently available. The DAAs target specific nonstructural (NS) proteins encoded by the single-stranded HCV RNA virus. The novel DAAs include NS3/4A protease inhibitors, NS5A inhibitors, and NS5B polymerase inhibitors (Figure 12) [36]. For the treatment of HCV genotypes 1 and four combinations of two or three DAAs targeting different proteins are applied. Typically, these combinations are supplemented by ribavirin and/or pegylated interferon with both exhibiting rather nonspecific antiviral activities. Due to better tolerability, interferon-free therapy regimens are strongly preferred [36,41,42,43].
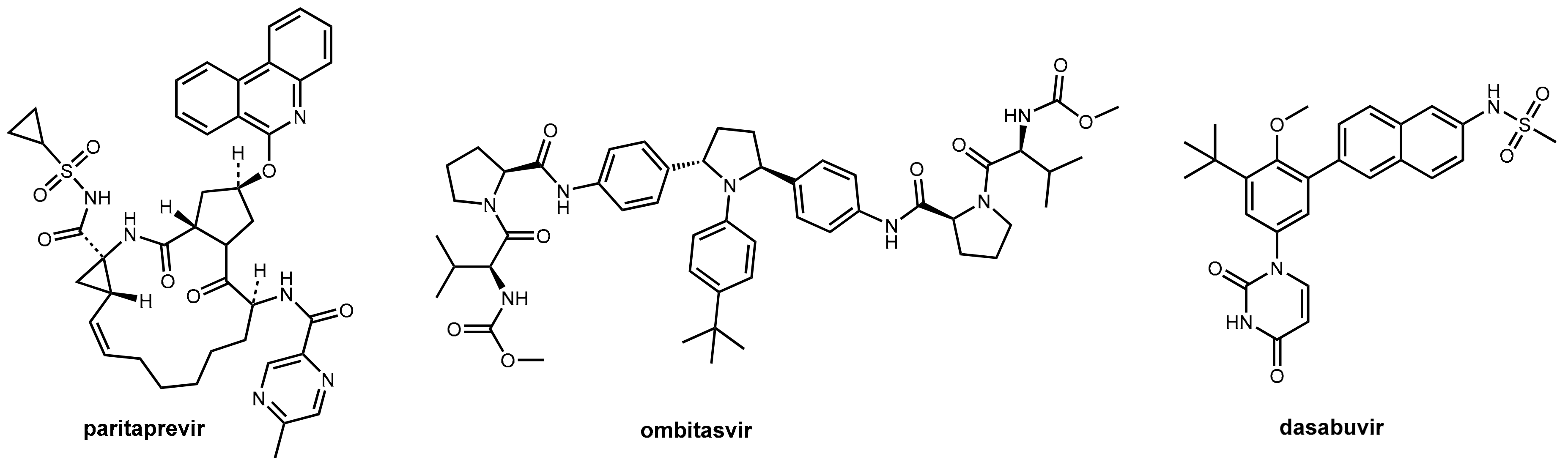
Inhibitors of the NS3/4A protease are indispensable components of the modern DAA therapy regimens. One recently launched fixed combination (Viekirax® Technivie®) contains the NS5A inhibitor ombitasvir and the NS3/4A inhibitor paritaprevir. For the treatment of HCV1 genotype-1 patients, this is combined with the NS5B polymerase inhibitor dasabuvir (Figure 13) and ribavirin [45] for genotype 4 with only ribavirin [42].
Paritaprevir (Figure 13) is a potent competitive macrocyclic inhibitor of the serine protease NS3 and its co-factor NS4A in HCV genotypes 1 and 4. Like other second-generation NS3/4A inhibitors (e.g., simeprevir [46]), it is a substrate of CYP3A4 and, thus, needs bolstering. The inclusion of ritonavir into the fixed combination guarantees sufficiently high and long-lasting plasma levels of paritaprevir that allows a therapy regimen of one tablet per day. However, and as discussed above (Section 3.2), ritonavir also increases the potential for drug–drug interactions for this combination regimen. Consequently, medications that are extensively metabolized by CYP3A4 are contraindicated during therapy, as are potent CYP3A4 inducers that would dramatically decrease plasma levels of paritaprevir. Ritonavir does not have own activity on HCV proteases (in contrast to HIV proteases, as described above). The other components of the combination regimen are not influenced by the ritonavir booster process: dasabuvir is metabolized by CYP2C8 and only to a lesser extent by CYP3A. Ombitasvir is predominantly metabolized by hydrolysis [36].
5. Probenecid Reloaded for Antiviral Therapy
5.1. Reduction of the Nephrotoxicity of Cidofovir
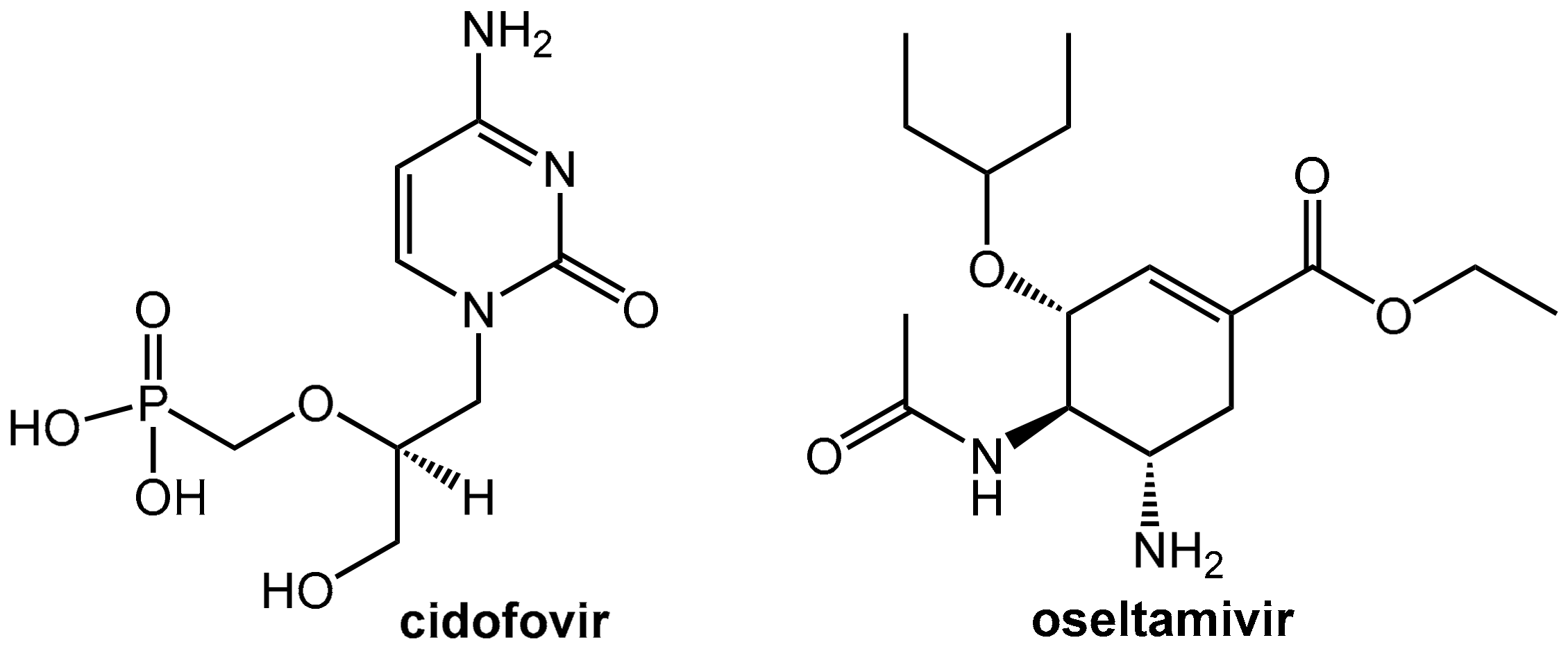
Cidofovir (Figure 14) is an injectable antiviral nucleotide analogon used exclusively as treatment for cytomegalovirus retinitis in AIDS-infected patients. This drug suppresses cytomegalovirus (CMV) replication by selective inhibition of viral DNA synthesis. It is an antimetabolite and, after phosphorylation by intracellular kinases to give cidofovir diphosphate, it inhibits the viral DNA polymerases. Incorporation of cidofovir into the growing viral DNA chain results in reduced viral DNA synthesis. After infusion, cidofovir is almost completely eliminated as the unchanged drug into the urine. The renal clearance of cidofovir is higher than creatinine clearance, which indicates that renal tubular secretion significantly contributes to its elimination. Therefore, the drug is taken up from the blood into the proximal tubular cells by means of “organic anion transporters” located on the basolateral side of the tubular cells while the following secretion into the tubular lumen is much slower. This rather slow efflux of cidofovir into the urine in comparison to fast uptake from the blood results in an accumulation of the drug in the proximal tubular cells and is most likely the reason for the observed dose-limiting nephrotoxicity of the drug [47].
This nephrotoxicity can be eliminated by co-administration of probenecid (see also Section 2.4.1, Figure 9), which is an inhibitor of the organic anion transporter. This reduces the rate of tubular uptake of cidofovir from blood into the tubular cells and so protects the kidneys by balancing uptake into the tubular cells and subsequent efflux into the urine through the brush boarder membrane. In the same course, probenecid also reduces the overall renal clearance of the drug and, thus, increases serum cidofovir concentrations significantly [48,49].
The recommended standard-dose probenecid regimen is 2 g at 3 h before cidofovir infusion and then 1 g at 2 and 8 h after cidofovir (total 4 g). As an additional prophylaxis, intravenous pre-hydration with 1 liter of 0.9% saline is performed [50].
Note: Probenecid is a banned drug for athletes for a related reason. Since it blocks entry of certain drugs into the urine, it can be used as a masking agent for other banned performance-enhancing drugs including steroids.
5.2. Probenecid Plus Oseltamivir
Oseltamivir (Tamiflu®) was approved in 2002 for the treatment of uncomplicated influenza within 48 h of the onset of symptoms and was regarded as a very promising drug for fighting long awaited influenza pandemics at that time. There were concerns whether the amounts of oseltamivir stockpiled by governments around the world would be sufficient in case of a worldwide H1N1 pandemic [51]. Since (like penicillin and other acidic antibiotics, see Section 2.4.1 and cidofovir, see above) oseltamivir is actively secreted by the kidneys. Probenecid was investigated as a co-medication for saving precious oseltamivir. In fact, this co-administration was able to enhance the patient’s total exposure to the drug by a factor of 2.5 [52]. Meanwhile, experts are significantly less enthusiastic about the therapeutic value of oseltamivir and, in 2017, the World Health Organization (WHO) downgraded oseltamivir in its list of essential medicines from a “core” drug to one that is “complementary.”
6. Tumor Therapy
6.1. Pharmacokinetic Enhancers For Fluoropyrimidine Antimetabolites
6.1.1. Metabolism of 5-Fluorouracil (5-FU) and Tegafur
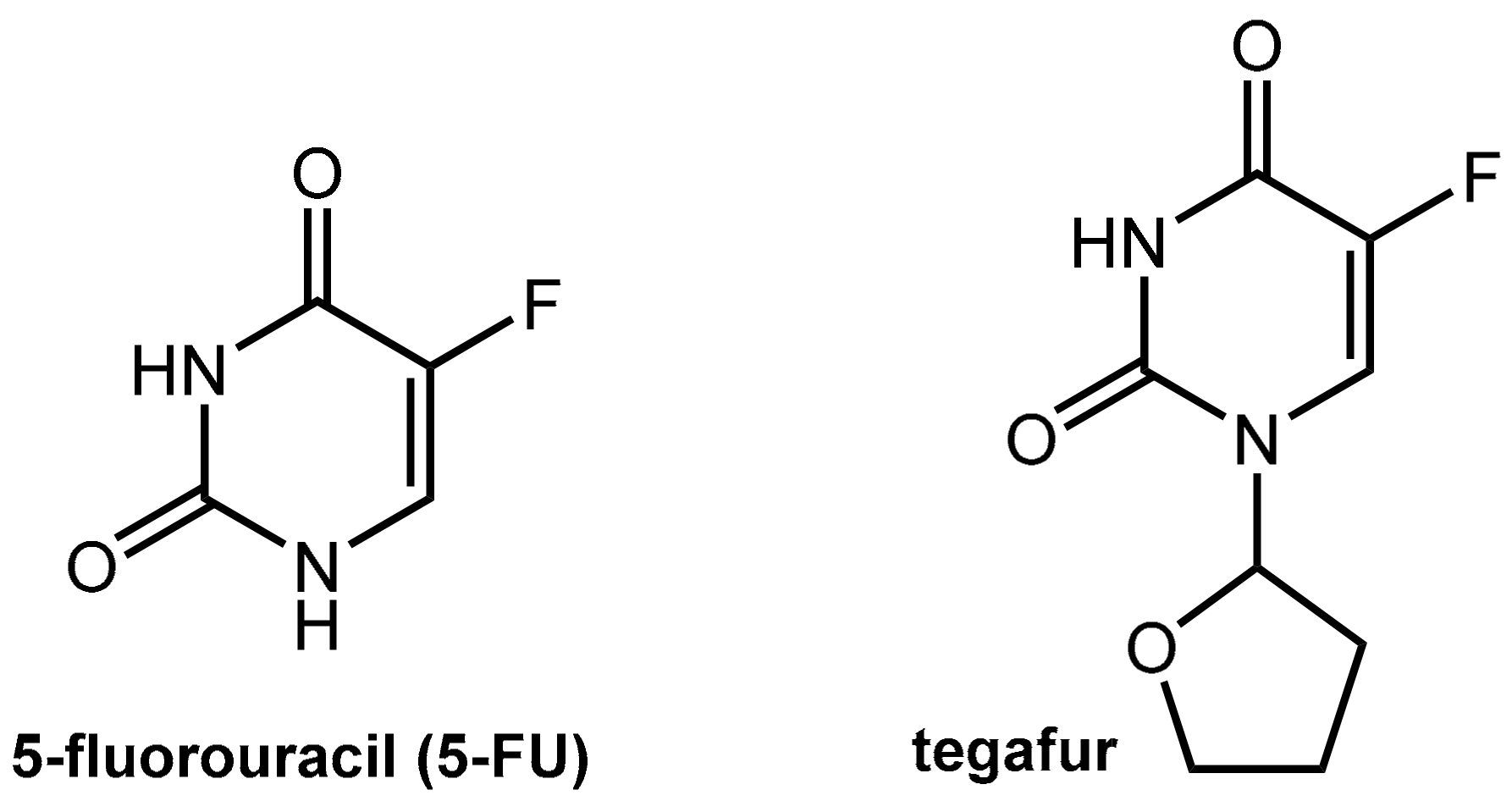
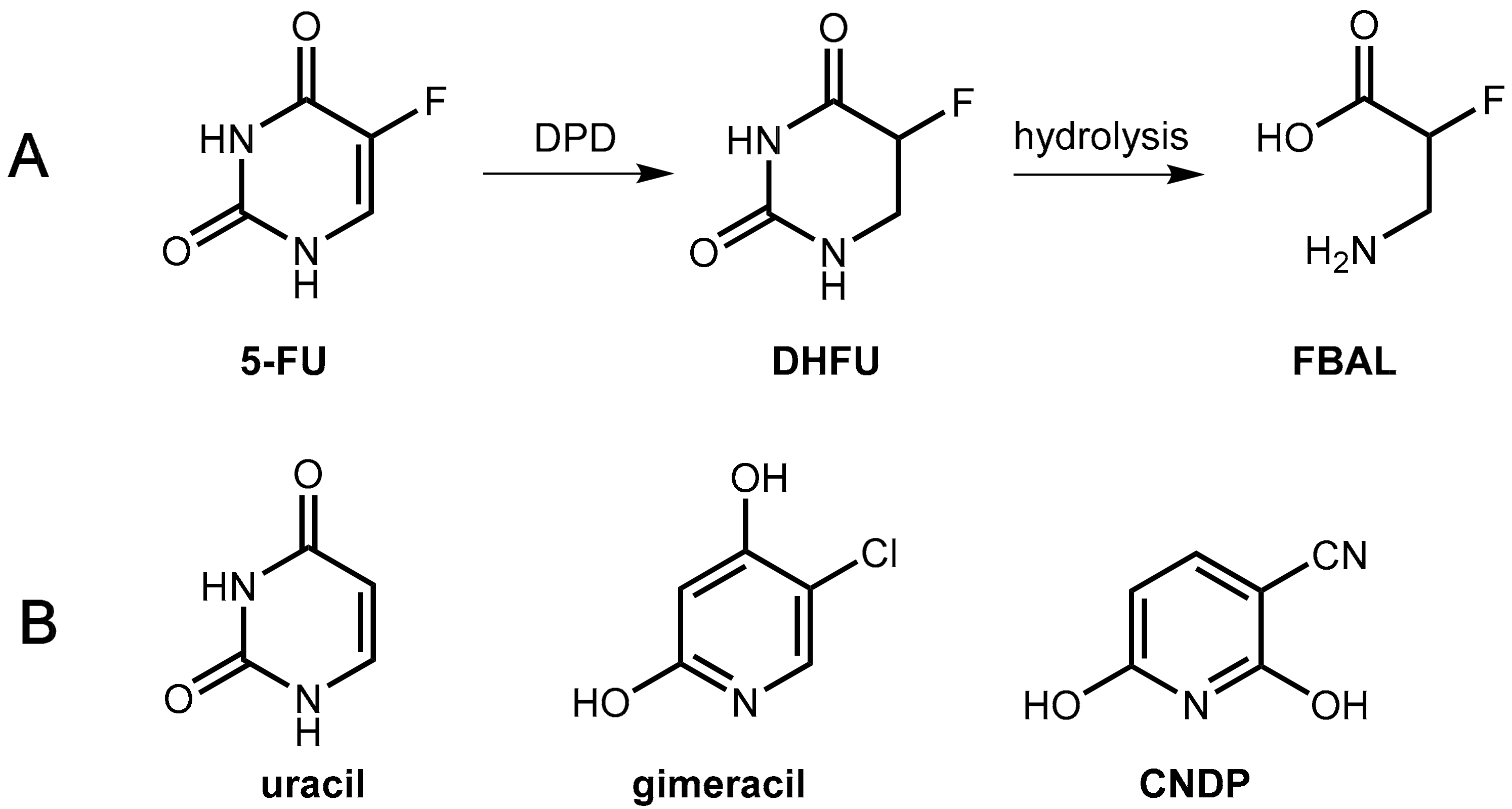
5-Fluorouracil (5-FU, Figure 15) is one of the world’s most widely used anticancer agents. It is widely used as the first or the second line therapy for a variety of cancers including colorectal and gastric cancer but also breast, head and neck, and ovarian cancers. 5-FU is administered via continuous i.v. infusion, which is both cumbersome and expensive. The less expensive and less stressful oral administration of 5-FU is not applicable due to incomplete and erratic bioavailability. The reason for this is due to the level of dihydropyrimidine dehydrogenase (DPD), which is the rate-limiting enzyme in 5-FU catabolism, varies among individuals and even within individuals during the course of a day. Furthermore, variable levels of intestinal DPD create erratic absorption of orally administered 5-FU and render efficacy and toxicity highly unpredictable [53,54].
Tegafur, which is a 5-FU pro-drug, is suitable for oral application but still shares the issues of 5-FU since its active metabolite is the parent 5-FU. The conversion of tegafur into 5-FU is performed mainly by hepatic oxidation at C-5’ catalyzed by CYP2A6 while the tetrahydrofuranyl residue is finally released as γ-hydroxybutyrolactone (which may account for the observed neurotoxicity of tegafur).
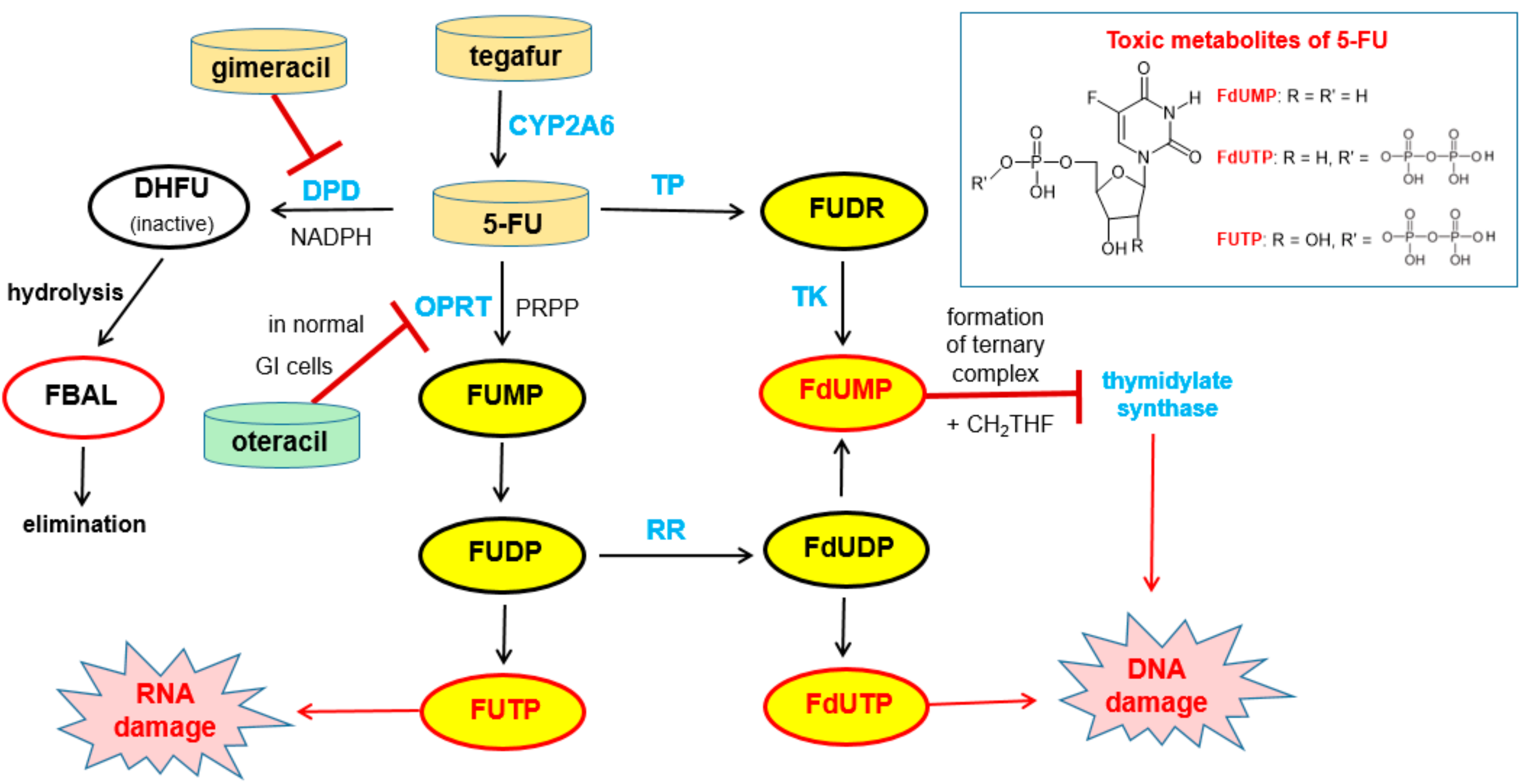
Free 5-FU is converted into three main active metabolites: the main activation is orotate phosphoribosyltransferase (OPRT)-mediated transformation into fluorouridine monophosphate (FUMP) utilizing phosphoribosyl pyrophosphate (PRPP) as the cofactor and two further phosphorylation steps give, via fluorouridine diphosphate (FUDP), fluorouridine triphosphate (FUTP), which is an antimetabolite that leads to RNA damage. The intermediate FUDP can also be converted into fluorodeoxyuridine diphosphate (FdUDP) by the enzyme ribonucleotide reductase (RR). Subsequent phosphorylation of FdUDP gives fluorodeoxyuridine triphosphate (FdUTP), which is an anti-metabolite that leads to DNA damages. Alternatively, FdUDP is dephosphorylated to the active compound fluorodeoxyuridine monophosphate (FdUMP). FdUMP can also be produced directly from 5-FU by the action of the enzymes thymidine phosphorylase (TP) and thymidine kinase (TK). FdUMP is a mechanism-based inhibitor (for details of the irreversible binding, see Reference [55]) of the enzyme thymidylate synthase (TS) since it forms a stable ternary complex with TS and its methyl donor methylene tetrahydrofolate (see note below). Since this enzymatic reaction is the sole de novo source of thymidylate for the cancer cells, DNA synthesis is effectively blocked [56] (Figure 16).
5-FU is degraded mainly in the liver and in the blood stream by the enzyme dihydropyrimidine dehydrogenase (DPD) to give dihydrofluorouracil (DHFU) as the first metabolite. Hydrolytic decomposition of the reduced pyrimidine ring gives fluoro-β-alanine (FBAL), which is excreted (Figure 16).
The very low and, moreover, strongly fluctuating bioavailability of 5-FU after oral application is due to the strong expression of dihydropyrimidine dehydrogenase (DPD) in gastrointestinal cells, which results in unpredictable absorption of an intact drug [57].
Note: Since methylene tetrahydrofolate is an indispensable component for the formation of the ternary complex of the 5-FU metabolite FdUMP with thymidylate synthase (Figure 16), the cytotoxic effect of 5-FU can be enhanced by co-administration of the folate precursor leucovorin (=formyltetrahydrofolate). However, this does not represent pharmacokinetic enhancement in the strict sense [58].
6.1.2. Inhibitors of Dihydropyrimidine Dehydrogenase (DPD)
As mentioned above, oral application of 5-FU as a monotherapy is precluded due to unpredictable bioavailability. However, even after IV application of 5-FU or oral application of its pro-drug tegafur, which circumvents the problems of undesired metabolic inactivation of 5-FU by DPD in gastric cells during the absorption step, fluctuating levels of 5-FU are observed due to inter-individual and intra-individual differences in DPD expression. This variability makes effective dosing of both drugs (5-FU, tegafur) difficult. Furthermore, a number of side effects of 5-FU/tegafur therapy are associated with metabolites like FBAL (Figure 16) arising from metabolism by DPD. For all of these reasons, inhibitors of dihydropyrimidine dehydrogenase (DPD) are valuable pharmacokinetic enhancers for both parenteral 5-FU and oral tegafur therapy [59].
First, success was achieved with a combination of tegafur and uracil (Figure 17) in a 1:4 molar ratio (UFT®). The physiologically and pharmacologically inert substance uracil is a natural substrate of DPD and, at sufficiently high levels, it competes in the saturable hepatic catabolism with the active tegafur metabolite 5-FU for reduction by DPD, which ends up with enhanced levels of 5-FU [60].
In the novel combination, Teysuno® tegafur is combined with the DPD inhibitor gimeracil (5-chloro-2,4-dihydroxypyridine, CDHP, Figure 17) as well as the orotate phosphoribosyltransferase inhibitor oteracil (see Section 6.1.3). Gimeracil is a significantly stronger inhibitor of DPD as uracil (factor about 180) [60]. An even stronger competitive inhibitor (2000 fold compared to uracil) is 3-cyano-2,6-dihydroxypyridine (CNDP) [61].
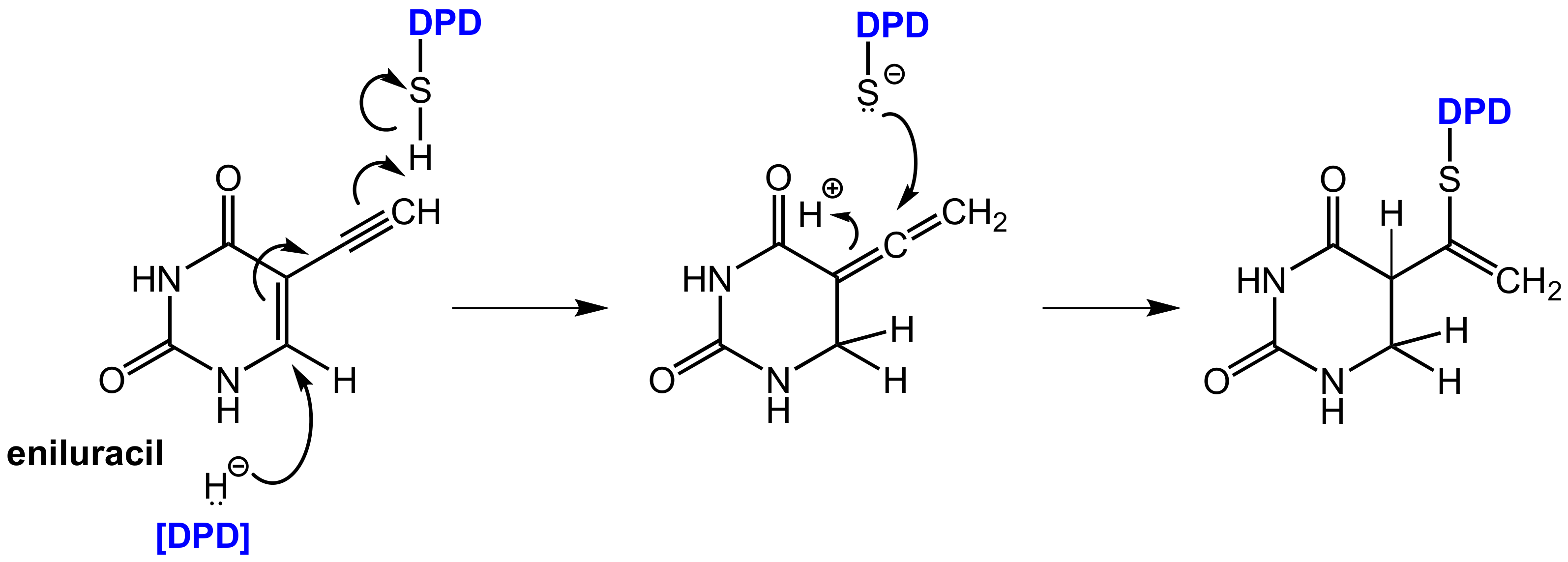
In contrast, eniluracil (5-ethynyluracil, Figure 18) is a covalent inhibitor of DPD. This compound irreversibly inhibits DPD and the enzyme is completely inactivated within 1 h of eniluracil administration. The compound leads to almost 100% oral bioavailability of 5-FU, which facilitates uniform absorption and predictable toxicity. Eniluracil itself is neither toxic nor active as a single agent and it improves the antitumor efficacy and therapeutic index of 5-FU significantly [53]. Eniluracil is a so-called mechanism-based inactivator of dihydropyrimidine dehydrogenase (DPD) since it requires catalytic activation by its target enzyme in an initial step. This makes the compound extremely specific for the target enzyme and brings advantages over reversible inhibitors, which must be present permanently and in relatively high concentrations to exert sustained DPD inhibition. After irreversible inhibition by eniluracil, further 5-FU degradation will only take place with newly synthesized DPD enzyme [54].
Bio-activation of eniluracil is initialized by reduction by DPD (in analogy to the reduction of 5-FU by this enzyme) whereby the initially formed reduction product most likely undergoes isomerization to an allenic intermediate. This compound contains an α,β-unsaturated lactam moiety, which can undergo a nucleophilic addition of an appropriate functional group in the enzyme in the sense of a Michael addition resulting in covalent binding. The reactive functional group in DPD was identified as a thiol group of a cysteine in the active site (Figure 18) [62].
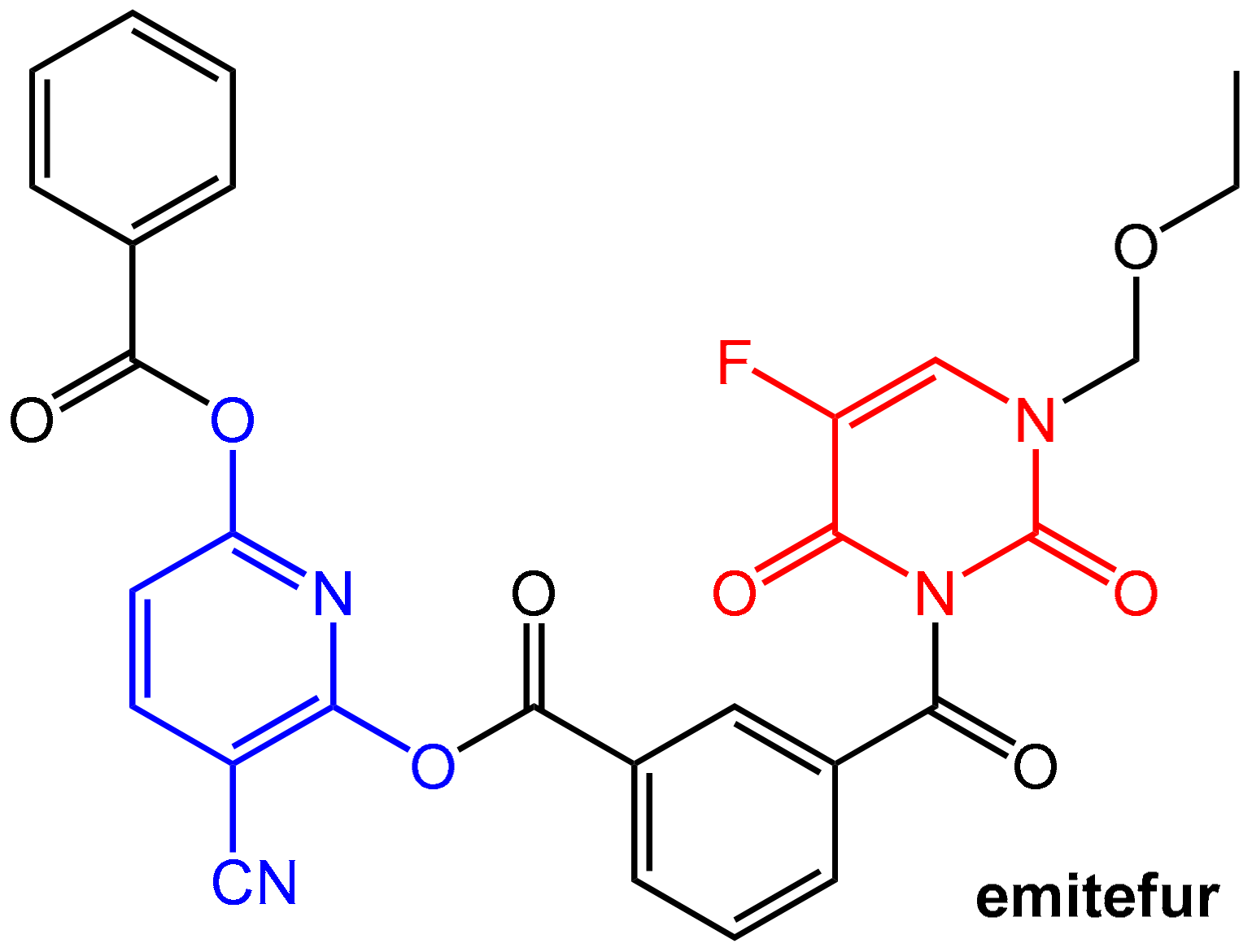
Emitefur (=BOF-A2) (Figure 19) is an investigational orally available antimetabolite composed of the 1-ethoxymethyl derivative of 5-FU and the reversible DPD inhibitor CNDP (see Figure 17). Both components are coupled via a di-ester linkage using an isophthalic acid unit (compare the mutual prodrug sultamicillin, Section 2.1.2, Figure 4). After oral application and absorption, the molecule is cleaved by esterases to give equimolar amounts of 5-FU and CNDP. Emitefur was considered to be a promising drug for treating advanced gastric cancer [61].
6.1.3. Inhibition of Orotate Phosphoribosyltransferase
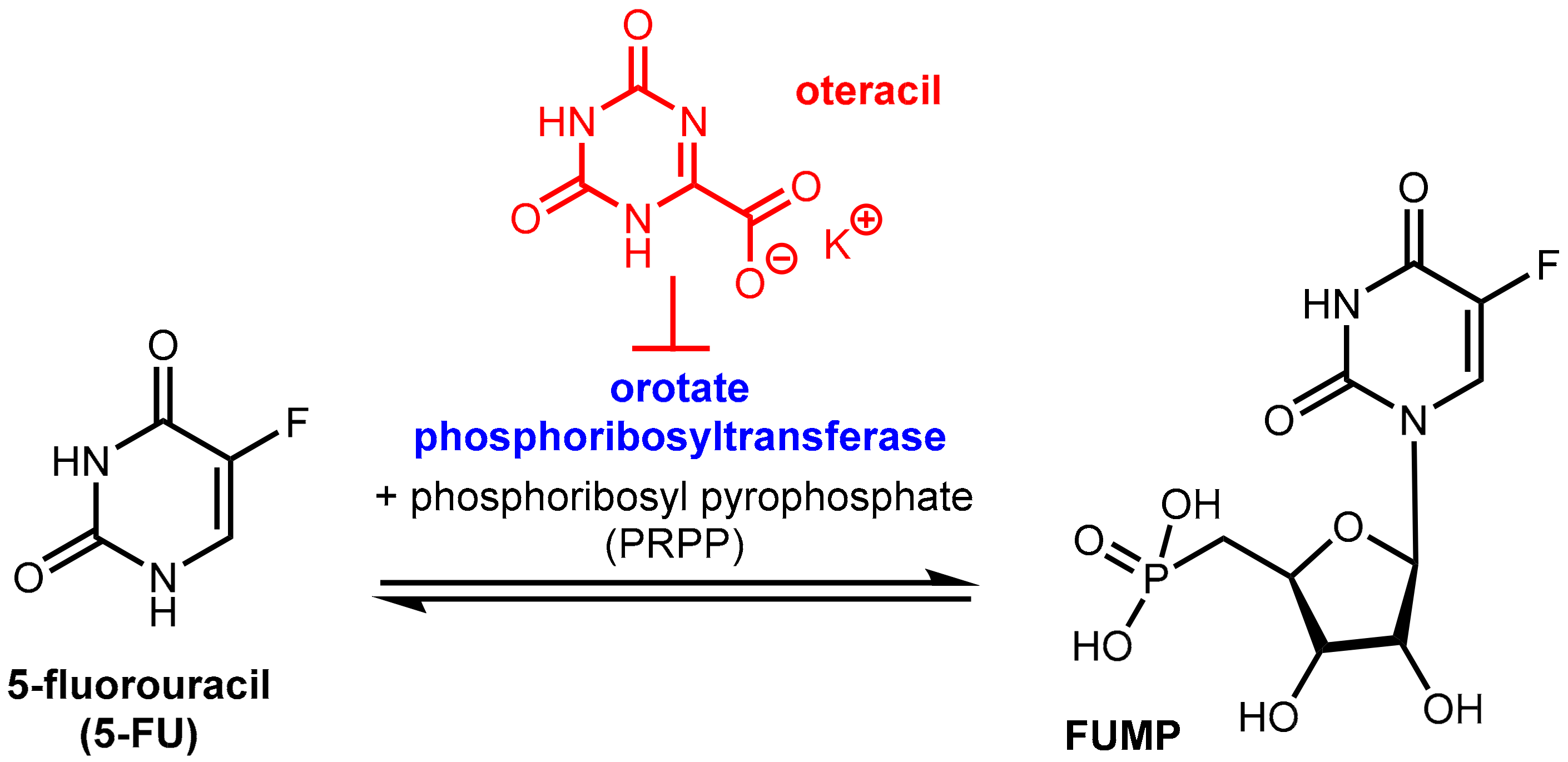
Tegafur when administered orally alone or in combination with the DPD inhibitor gimeracil (see above) exhibits gastrointestinal toxicity, which leads to diarrhea and other side effects. These side effects are mainly assigned to the high expression of the enzyme orotate phosphoribosyltransferase (OPRT, Figure 20) even in normal gastrointestinal mucosal cells. This enzyme is involved in pyrimidine biosynthesis and physiologically catalyzes the formation of orotidine-5’-monophosphate from orotate and phosphoribosyl diphosphate (FRPP). Oteracil (oxonic acid, 5-azaorotic acid) is an inhibitor of orotate phosphoribosyl transferase. It accumulates specifically in normal gut cells where it inhibits the (here undesired) conversion of 5-FU to 5-fluorouridine monophosphate (FUMP), which is a precursor of cytotoxic metabolites of 5-FU like fluorodeoxyuridine monophosphate (FdUMP) [63] with the result of suppressing the toxic effects of 5-FU in the mucosal cells. Since oteracil acid does not accumulate in gastric cells and other tumor cells, it has a selective protective effect against gastrointestinal toxicity, but it does not affect anti-tumor activity. Oteracil acid does not affect the pharmacokinetics of 5-FU [60]. The anticancer activity of 5-FU is maintained since the inhibitory effect of FdUMP on thymidylate synthase and DNA biosynthesis in tumor cells is unaffected and because inhibition of OPRT by oteracil in gastric cancer cells is incomplete since OPRT is overexpressed in the tumor cells (Figure 16) [57].
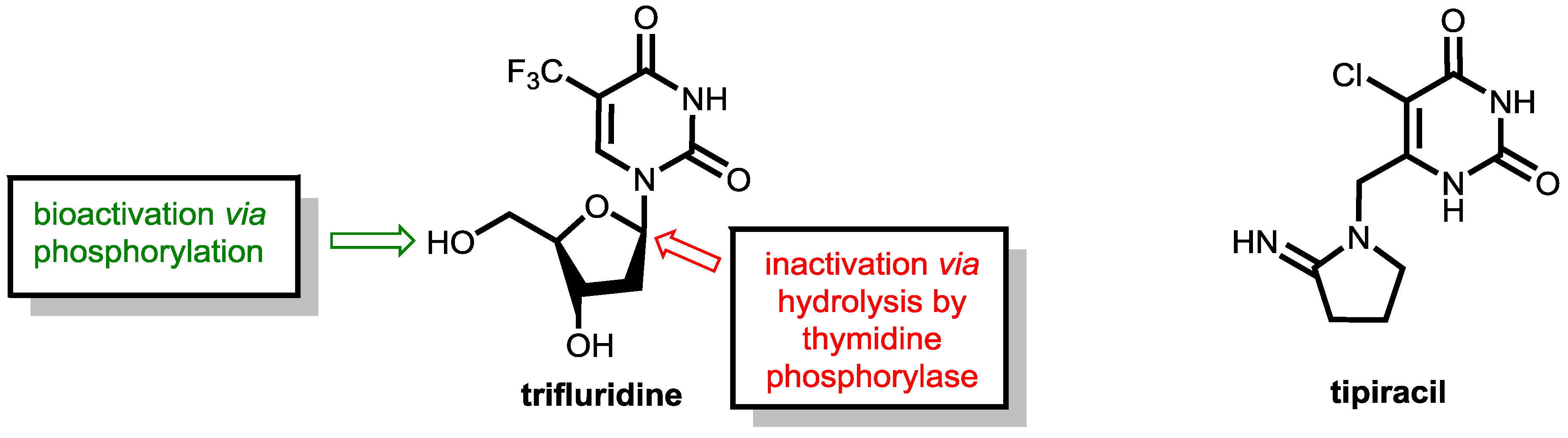
6.2. Inhibition of Thymidine Phosphorylase: Tipiracil As a Pharmacokinetic Enhancer for Trifluridine
As mentioned above, fluoropyrimidine-based therapies have outstanding relevance in the therapy of colorectal cancers. However, emerging resistance of tumor cells towards fluoropyrimidine treatment is a severe problem. The combination of trifluridine and tipiracil (as a 1:0.5 molar ratio in TAS-102, which is marketed as Lonsurf®, Figure 21) has shown clinical activity in patients resistant to 5-flurorouracil (5-FU) and offers a therapeutic alternative for pre-treated patients with metastatic colorectal cancer [64,65].
Trifluridine, which is a trifluoromethylated analogue of thymidine, is an anticancer compound that was developed as an alternative to 5-FU already in 1964 and inhibits tumor cell growth by affecting DNA synthesis. Upon administration, the molecule undergoes bioactivation in tumor cells by phosphorylation of the enzyme thymidine kinase. The resulting trifluridine monophosphate then inhibits the enzyme thymidylate synthase (TS, compare the action of 5-FU and tegafur shown in Figure 16) and prevents the methylation of deoxyuridine monophosphate (dUMP) into deoxythymidine monophosphate, which is a key step in DNA biosynthesis. Trifluridine like 5-FU is a mechanism-based inhibitor that causes irreversible inhibition of thymidylate synthase (TS). The inhibition is initiated by a nucleophilic attack of an active site nucleophilic group (such as thiol) at the C-6 position. Subsequent fluoride elimination converts the trifluoromethyl group into a highly reactive intermediate, which further reacts with another nucleophilic amino residue of the enzyme to lead to a stable adduct linking the inhibitor irreversibly to the enzyme (for details, see ref. [55]). Additionally, trifluridine monophosphate is further phosphorylated to the triphosphate, which acts as an antimetabolite and causes cell death via direct incorporation into DNA [65].
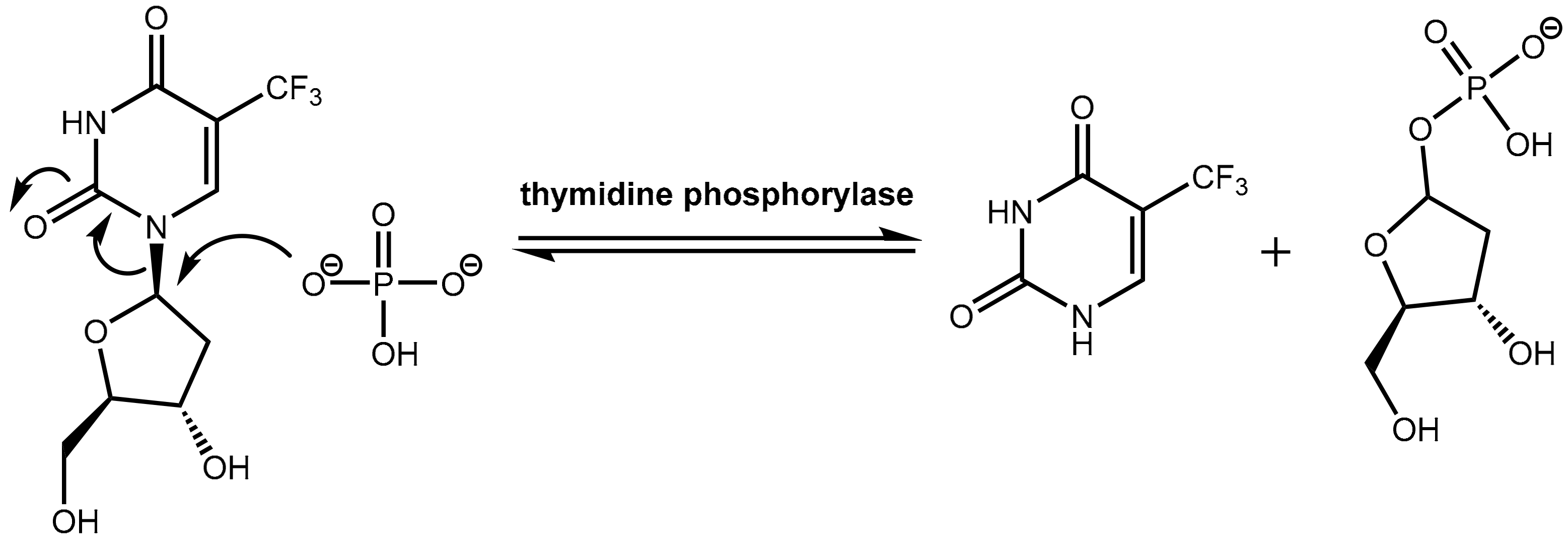
However, trifluridine has very poor oral bioavailability due to fast breakdown by action of the enzyme thymidine phosphorylase in a first-pass metabolism while trifluridine is converted under removal of the ribose unit into inactive 5-trifluoromethyl-pyrimidinedione. The enzyme thymidine phosphorylase catalyzes the reversible phosphorolysis of a number of natural and synthetic pyrimidine nucleosides (Figure 22), according to the following equation [66]:
Pyrimidine(deoxy)ribose + phosphate → pyrimidine base + (deoxy)ribose-1-phosphate.
Therapeutic levels of orally administered trifluridine are achieved upon co-administration with the pharmacokinetic enhancer tipiracil (Figure 21). Tipiracil is an effective inhibitor of the enzyme thymidine phosphorylase and boosts trifluridine so that trifluridine levels are achieved that are high enough for therapy of colorectal cancers [64].
6.3. Efflux Pump Inhibitors In Cancer Therapy
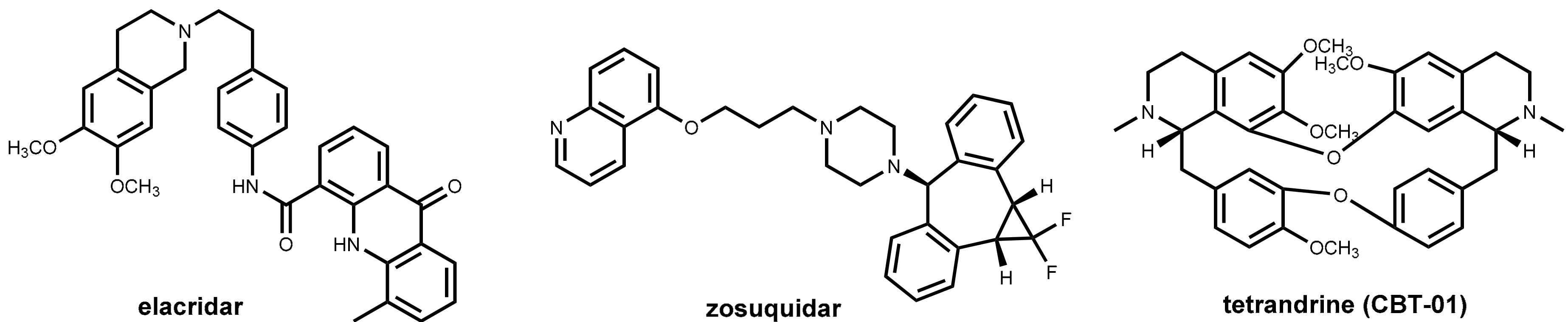
One of the major challenges in cancer chemotherapy is the development of multi-drug resistance to the anticancer drugs. One major factor for emerging resistance is the efflux of the anticancer drugs by P-glycoprotein (P-gp) pumps. This phenomenon has affected not only long-established chemotherapeutics like anthracyclines, Vinca alkaloids, and cisplatin but also newer drugs such as docetaxel and protein kinase inhibitors [67]. Like in the field of “antibiotic adjuvants” (see Section 2.4.2), considerable effort has been undertaken to develop inhibitors of efflux pumps of tumor cells aimed at restoring the original antitumor activity of drugs by co-administration. Promising in vitro results were obtained with a number of compounds such as verapamil (see Section 2.4.3 and Figure 9) and ciclosporin analogs and other natural products [68,69], but these and numerous synthetic inhibitors of the second and third generations (elacridar, tariquidar, zosuquidar, Figure 23) failed in clinical trials. Recently, the bisbenzylisoquinoline alkaloid tetrandrine (CBT-01®, Figure 23) in combination with paclitaxel has advanced clinical trials [70].
7. Pharmacokinetic Enhancers for Levodopa
7.1. Parkinson’s Disease and Levodopa
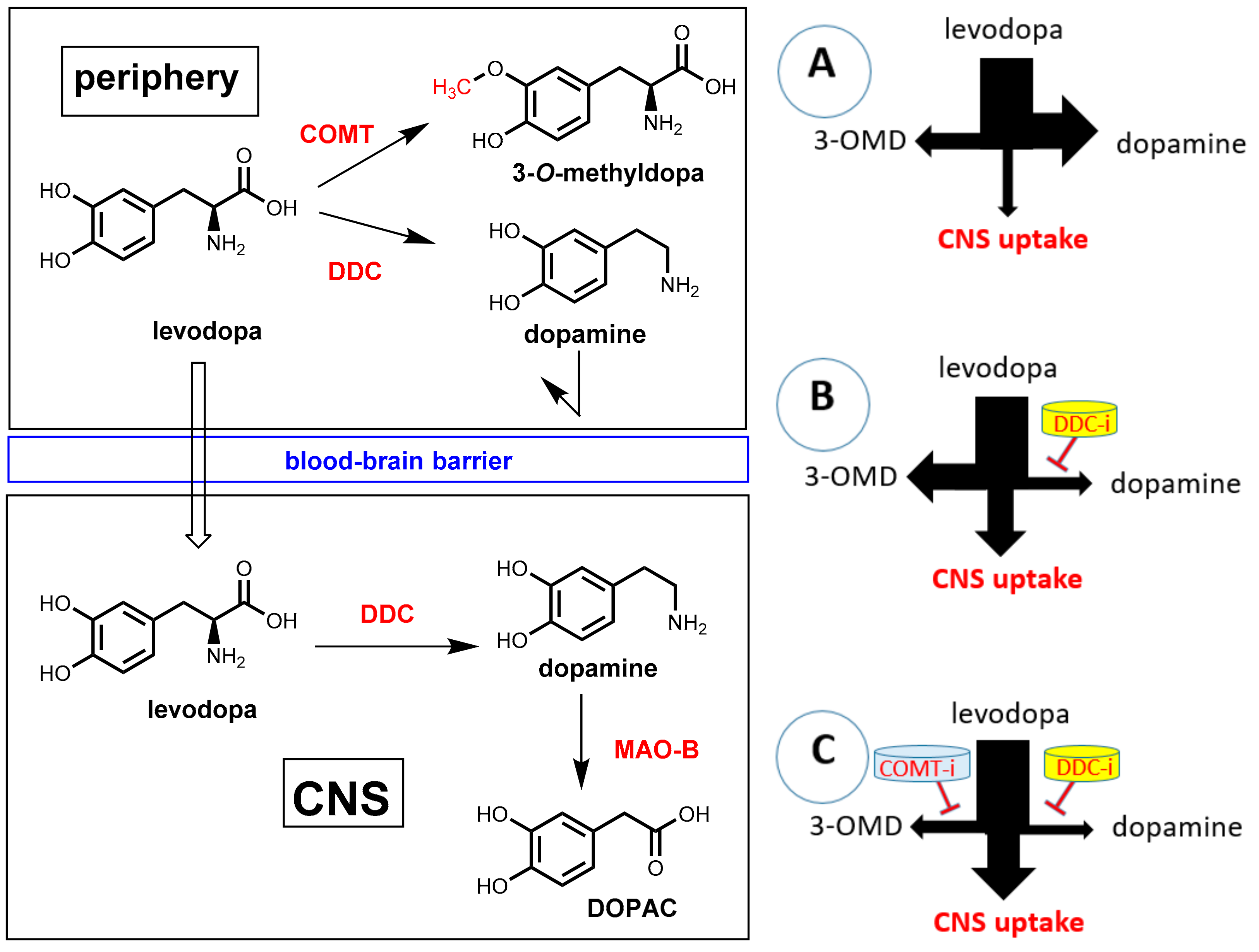
Parkinson’s disease (PD) is a neurodegenerative disease characterized by a progressive loss of dopamine-producing neurons in the substantia nigra and striatum. The shortage of dopaminergic input results in the typical motor symptoms (rigidity, akinesia, and tremors). Direct supplementation of dopamine is ineffective since this compound is not able to pass the blood-brain barrier. One of the most important treatments for PD patients is oral administration of the amino acid levodopa (L-DOPA, L-3,4-dihydroxyphenylalanine; Figure 24), which represents a biochemical precursor of dopamine. In contrast to dopamine, levodopa is taken up into the brain by utilizing active transport systems. Subsequent decarboxylation by means of the enzyme DOPA decarboxylase (DDC, synonym: aromatic L-amino acid decarboxylase, AADC) leads to dopamine in the brain [71,72]. Significant amounts of orally applied levodopa can, however, be lost by undesired metabolism before transport into the brain. The enzyme DOPA decarboxylase, which is essential for bioactivation in brain, is also significantly expressed in peripheral tissues. During intestinal absorption and peripheral circulation, about 70% of levodopa is lost by enzymatic decarboxylation and, consequently, significant levodopa in the brain cannot be achieved by levodopa monotherapy. By co-administration of a DOPA decarboxylase inhibitor (see Section 7.2), this premature decarboxylation in the periphery can be suppressed significantly and uptake of therapeutically relevant amounts of levodopa is achieved. However, even under co-administration of a peripheral DOPA decarboxylase inhibitor, less than 10% of the orally administered levodopa reaches the brain. Furthermore, under suppression of peripheral decarboxylation of levodopa, a second metabolic pathway becomes dominating. O-methylation of one phenolic group catalyzed by the enzyme catechol-O-methyltransferase (COMT) gives 3-O-methyldopa (3-OMD) as the main metabolite (Figure 24). 3-OMD is pharmacologically inactive, but it further reduces the bioavailability of levodopa by competing with it for active transport across the blood-brain barrier. Co-administration of a second pharmacokinetic enhancer, which is a COMT inhibitor, ends up in a suppression of both enzymatic reactions, which lead to peripheral loss of dopamine, and a therapeutic effect on Parkinson’s disease can be achieved with a minimum dosage of levodopa. Furthermore, side effects that are caused by peripheral levodopa metabolites (mainly dopamine) can be minimized this way [73].
7.2. DOPA Decarboxylase Inhibitors: Carbidopa and Benserazide
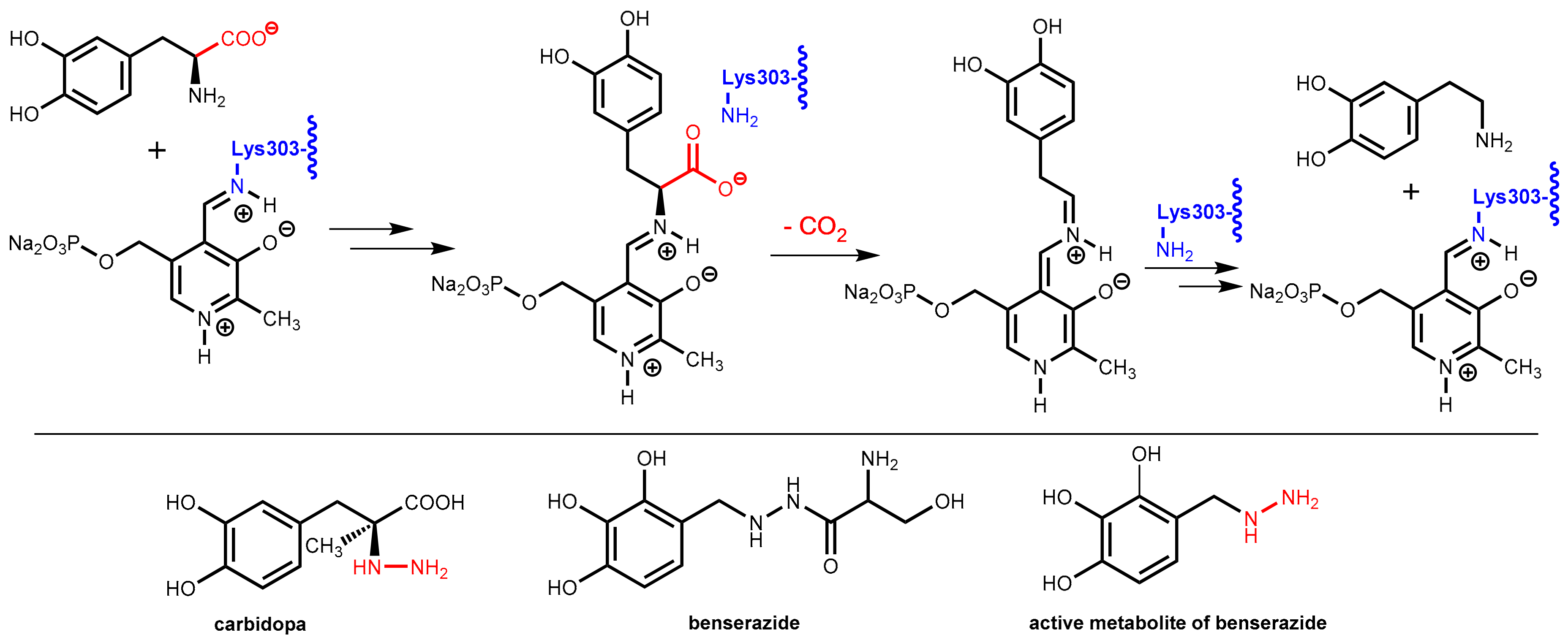
The enzyme DOPA decarboxylase contains a pyridoxal phosphate co-factor in its active site. This co-factor is bound to the enzyme by formation of an imine with the primary amino group in the side-chain of Lys303. Decarboxylation of levodopa starts with an imine exchange reaction and the levodopa bound to the co-factor is decarboxylated in the following. In the final step, Lys303 forms an imine with the co-factor again and releases dopamine from the enzyme (Figure 25).
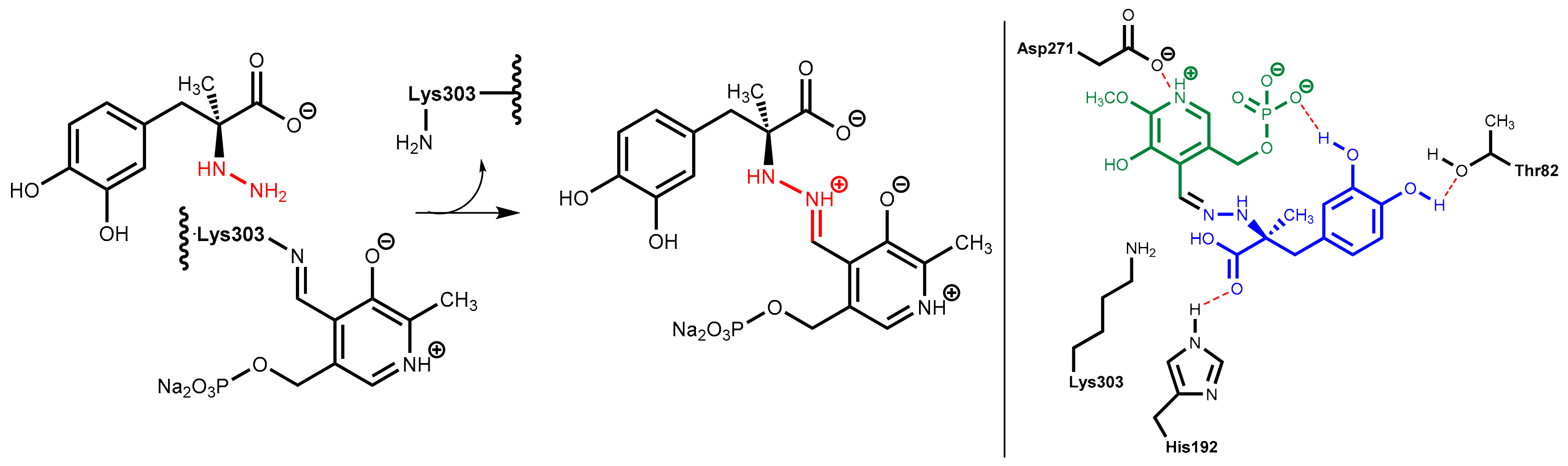
DOPA decarboxylase inhibitors were designed for inhibiting exclusively the decarboxylation of levodopa in the periphery. Consequently, these inhibitors typically have physico-chemical properties that guarantee that they do not pass the blood-brain barrier. Presently, only carbidopa and benserazide are used in therapy. Carbidopa is a hydrazine analogue of levodopa and, like levodopa, it has an (S)-configuration. In benserazide, 2,3,4-trihydroxybenzylhydrazine is connected to racemic serine via an amide bond and in vivo amide hydrolysis gives the free benzylhydrazine metabolite (Figure 25). In both active compounds, the free hydrazine group is essential for inhibiting the target enzyme since hydrazines form very stable condensation products (hydrazones) with aromatic aldehydes. The active hydrazines react with the enzyme-bound pyridoxal phosphate to give the corresponding hydrazones (Figure 26, left). These hydrazines are rather stable and are fixed in the active site of the enzyme by a network of polar interactions (Figure 26, right), which results in an enduring inhibition of the enzyme. This is demonstrated by crystal structure analyses [74]. However, because the active hydrazines can also react with free pyridoxal phosphate and other pyridoxal phosphate-containing enzymes, these inhibitors are not absolutely selective for DOPA decarboxylase, which results in a number of adverse side effects [72].
7.3. COMT Inhibitors
7.3.1. Catechol-O-Methyltransferase (COMT)
As pointed out above, COMT inhibitors block the unwanted O-methylation of levodopa to 3-O-methyldopa (3-OMD) (Figure 24) in peripheral tissues, which complements the effect of DOPA decarboxylase inhibitors and prolongs the pharmacological effect of levodopa as well as diminishes its therapeutic dose [75]. The combination of levodopa/DOPA decarboxylase inhibitors with a COMT inhibitor is frequently named “adjunctive therapy” in literature. This therapy is typically applied in patients with end-of-dose motor fluctuations whose symptoms cannot be stabilized with a levodopa/DOPA decarboxylase inhibitor combination alone.
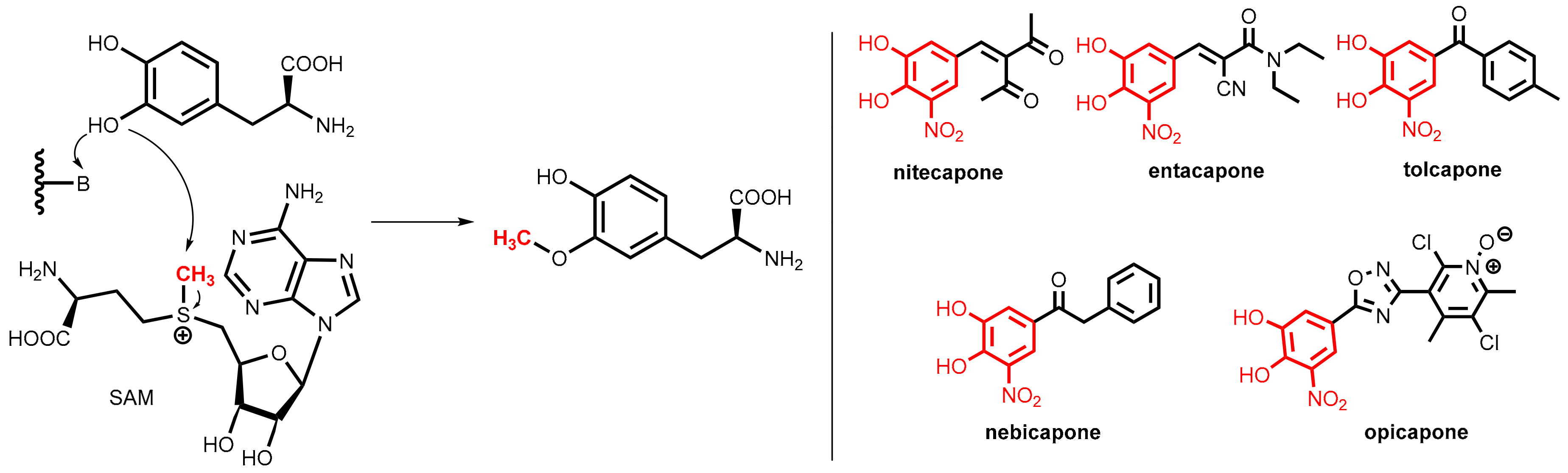
COMT is a magnesium-dependent intracellular enzyme that transfers a methyl group from the methyl donor S-adenosylmethionine (SAM) regioselectively to the 3-hydroxy group of levodopa and related substrates bearing an ortho-dihydroxybenzene (catechol) motif (Figure 27, left).
All of the clinically relevant second-generation COMT inhibitors for oral application (nitecapone, entacapone, tolcapone, nebicapone, and opicapone, Figure 27, right) are synthetic catechol derivatives, but subtle differences exist in the molecular modes of COMT inhibition.
7.3.2. COMT Inhibitors
Polyhydroxylated arenes were long known for their inhibitory activity on COMT, but the breakthrough was achieved by finding that the inhibitory activity of catechols can be significantly improved by the introduction of a strongly electron-withdrawing group (nitro > cyano or carbonyl groups) at the ortho-position to one of the hydroxyl groups. All of the relevant COMT inhibitors shown in Figure 27 bear this structural motif. Compared to older inhibitors, these compounds bind more tightly to the active site of the enzyme (for this reason they are named “tight-binding inhibitors”) but conversely do not undergo extensive O-methylation by COMT. The additional electron-withdrawing group renders the adjacent hydroxyl group significantly more acidic and, most likely, the delocalization of the negative charge of the resulting phenolate anion. This, thereby, decreases the reactivity toward O-methylation [75].
The styryl derivatives nitecapone and entacapone [76] are rather polar compounds with poor penetration of the blood-brain barrier and so they are active exclusively in the periphery. Tolcapone [73] shares with nitecapone and entacapone the same nitrocatechol pharmacophore, but it displays a different pharmacokinetic profile. Due to its lipophilic 4-methylbenzoyl side chain, it can cross the blood-brain barrier and was demonstrated to be an equipotent inhibitor of both brain and peripheral COMT. Due to liver toxicity concerns, the application of tolcapone is restricted in many countries. Nebicapone, albeit having strong structural similarity to tolcapone, displays weaker brain COMT inhibition.
Opicapone is the first peripheral COMT inhibitor of the third generation and was launched in 2016 [77,78]. Instead of the vinyl and acyl residues of the older inhibitors, this chemotype contains a heterocyclic ring (here an 1,2,4-oxadiazole) in meta-position to the nitro group of the nitrocatechol pharmacophore. Replacement of the phenyl substituent (see tolcapone and nebicapone) by a polar pyridine-N-oxide resulted in very high binding affinity, which includes a slow complex dissociation rate constant and a long duration of action on exclusively peripheral COMT in vivo.
Note: A further therapeutic option for the treatment of Parkinson’s disease are monoamine oxidase B (MAO-B) inhibitors such as selegiline, rasagiline, and safinamide [79]. These drugs reduce oxidative metabolism of physiological dopamine in the brain (see Figure 24). However, these drugs cannot be classified as pharmacokinetic enhancers in the strict sense since they were not designed to have an impact on the pharmacokinetics of a co-administered drug.
8. Dextromethorphan Plus Quinidine
Pseudobulbar affect (also known as pathological laughing and crying, affective lability, and expressive emotional incontinence) is characterized by uncontrollable, inappropriate laughing and/or crying. It occurs in patients with neurological disorders such as amyotrophic lateral sclerosis, multiple sclerosis, or traumatic brain injury. No causative therapy is available at the moment.
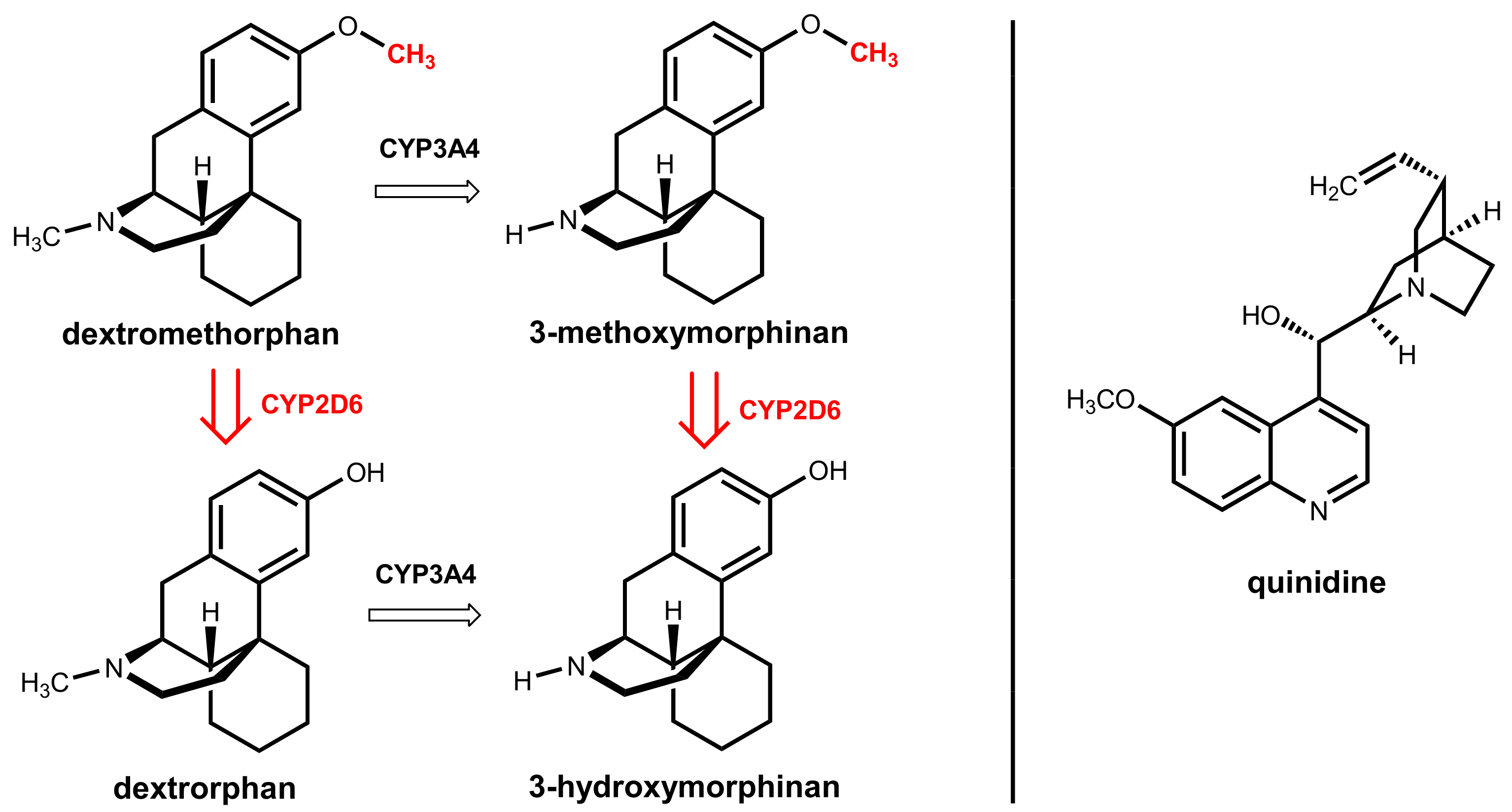
A drug combination of dextromethorphan and quinidine has been approved in the USA as Nuedexta® for treatment. The active agent in this combination is dextromethorphan, which is a well-established antitussive drug that can cross the blood-brain barrier. This compound is, among other molecular effects, a sigma-1 receptor agonist and an uncompetitive NMDA receptor antagonist, but the precise mechanism of action in patients with a pseudobulbar affect is still unknown [80].
Dextromethorphan underlies extensive oxidative hepatic first-pass metabolism by CYP2D6, which catalyzes O-demethylation to give the main metabolite dextrorphan. An alternative, less relevant pathway is catalyzed by CYP3A4 and gives 3-methoxymorphinan under oxidative N-demethylation (Figure 28) [81]. Quinidine, which is the other component in the combination, was historically used as a class I antiarrhythmic drug. Here, it acts as a metabolic enhancer since it is a potent and specific inhibitor of hepatic CYP2D6 [82]. A dose of 2 × 30 mg/day is sufficient for the complete inhibition of the first-pass metabolism and reaches therapeutic dextromethorphan concentrations. This dose is significantly lower than the 600 to 1600 mg/day dose of quinidine routinely used to treat cardiac arrhythmia [81].
9. Summary
Pharmacokinetic enhancement (boosting) is a concept where alterations in metabolic rates of a drug (or its precursor) through inhibitory effects of another, co-administered compound (“booster”) lead to increases in available concentrations of the active agent and prolongation of its half-life [33].
This purposefully induced interaction of two xenobiotics can, when carefully managed, provide the following benefits in therapy:
- it allows for the administration of lower doses of the active agent while maintaining therapeutic levels at the relevant compartment
- it can eliminate variability in systemic exposure
- it can enable overcoming resistance problems in anti-infective and cancer therapy
- it reduces the extent of toxic side effects since lower daily doses of the primary therapeutic agent (drug) are administered
- it reduces the therapy costs since lower amounts of drugs are needed
- it can reduce the pill burden or dosing frequency and improves the medication adherence of patients
Meanwhile, pharmacokinetic enhancers have been developed and launched for a large number of drugs for various therapeutic indications. This progress has been fostered by new insights from fundamental and applied research such as genome analysis, protein crystallography, and molecular modeling. Since the knowledge about biochemical and physiological processes and the options for their targeted modulation is ever increasing, further progress in the development of pharmacokinetic enhancers to the advantage of patients is to be expected.
Author Contributions
Conceptualization, F.B.; Data Curation, J.K. and F.B.; Writing-Original Draft Preparation, F.B.; Writing-Review & Editing, J.K. and F.B.; Visualization, J.K. and F.B.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- González-Bello, C. Antibiotic Adjuvants–A Strategy to Unlock Bacterial Resistance to Antibiotics. Bioorg. Med. Chem. Lett. 2017, 27, 4221–4228. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Abboud, M.I.; Markoulides, M.S.; Brem, J.; Schofield, C.J. The Road to Avibactam: The First Clinically Useful Non-β-Lactam Working Somewhat Like a β-Lactam. Future Med. Chem. 2016, 8, 1063–1084. [Google Scholar] [CrossRef] [PubMed]
- Padayatti, P.S.; Helfand, M.S.; Totir, M.A.; Carey, M.P.; Carey, P.R.; Bonomo, R.A.; van den Akker, F. High Resolution Crystal Structures of the Trans-Enamine Intermediates Formed by Sulbactam and Clavulanic Acid and E166a Shv-1 β-Lactamase. J. Biol. Chem. 2005, 280, 34900–34907. [Google Scholar] [CrossRef] [PubMed]
- Padayatti, P.S.; Helfand, M.S.; Totir, M.A.; Carey, M.P.; Hujer, A.M.; Carey, P.R.; Bonomo, R.A.; van den Akker, F. Tazobactam Forms a Stoichiometric Trans-Enamine Intermediate in the E166a Variant of Shv-1 β-Lactamase: 1.63 Å Crystal Structure. Biochemistry 2004, 43, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.; Hampel, B.; Bruckner, G.; Koeppe, P. The Pharmacokinetics of Sultamicillin. APMIS Suppl. 1989, 5, 17–22. [Google Scholar]
- Shirley, M. Ceftazidime-Avibactam: A Review in the Treatment of Serious Gram-Negative Bacterial Infections. Drugs 2018, 78, 675–692. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (Rpx7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef] [PubMed]
- Kahan, F.M.; Kropp, H.; Sundelof, J.G.; Birnbaum, J. Thienamycin: Development of Imipenem-Cilastatin. J. Antimicrob. Chemother. 1983, 12, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.J.; Forrester, L.J.; Zahler, W.L.; Burks, M. β-Lactamase Activity of Purified and Partially Characterized Human Renal Dipeptidase. J. Biol. Chem. 1984, 259, 14586–14590. [Google Scholar] [PubMed]
- Keynan, S.; Hooper, N.M.; Felici, A.; Amicosante, G.; Turner, A.J. The Renal Membrane Dipeptidase (Dehydropeptidase I) Inhibitor, Cilastatin, Inhibits the Bacterial Metallo-β-Lactamase Enzyme Cpha. Antimicrob. Agents Chemother. 1995, 39, 1629–1631. [Google Scholar] [CrossRef] [PubMed]
- Drusano, G.L. An Overview of the Pharmacology of Imipenem/Cilastatin. J. Antimicrob. Chemother. 1986, 18 (Suppl. E), 79–92. [Google Scholar] [CrossRef] [PubMed]
- Mainz, D.; Borner, K.; Koeppe, P.; Kotwas, J.; Lode, H. Pharmacokinetics of Lansoprazole, Amoxicillin and Clarithromycin after Simultaneous and Single Administration. J. Antimicrob. Chemother. 2002, 50, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Ushiama, H.; Echizen, H.; Nachi, S.; Ohnishi, A. Dose-Dependent Inhibition of Cyp3a Activity by Clarithromycin During Helicobacter Pylori Eradication Therapy Assessed by Changes in Plasma Lansoprazole Levels and Partial Cortisol Clearance to 6β-Hydroxycortisol. Clin. Pharmacol. Ther. 2002, 72, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Yasui-Furukori, N.; Uno, T.; Takahata, T.; Sugawara, K.; Munakata, A.; Tateishi, T. Effects of Clarithromycin on Lansoprazole Pharmacokinetics between Cyp2c19 Genotypes. Br. J. Clin. Pharmacol. 2005, 59, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Ohashi, K.; Kobayashi, K.; Iida, I.; Yoshida, H.; Shirai, N.; Takashima, M.; Kosuge, K.; Hanai, H.; Chiba, K.; et al. Effects of Clarithromycin on the Metabolism of Omeprazole in Relation to Cyp2c19 Genotype Status in Humans. Clin. Pharmacol. Ther. 1999, 66, 265–274. [Google Scholar] [CrossRef]
- Meyer, U.A. Metabolic Interactions of the Proton-Pump Inhibitors Lansoprazole, Omeprazole and Pantoprazole with Other Drugs. Eur. J. Gastroenterol. Hepatol. 1996, 8 (Suppl. 1), S21–S25. [Google Scholar] [CrossRef] [PubMed]
- Shirasaka, Y.; Sager, J.E.; Lutz, J.D.; Davis, C.; Isoherranen, N. Inhibition of Cyp2c19 and Cyp3a4 by Omeprazole Metabolites and Their Contribution to Drug-Drug Interactions. Drug Metab. Dispos. 2013, 41, 1414–1424. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, L.; Pazzucconi, F.; Ferrara, S.; Di Paolo, A.; Tacca, M.D.; Sirtori, C. Pharmacokinetic Interactions between Omeprazole/Pantoprazole and Clarithromycin in Health Volunteers. Pharmacol. Res. 2004, 49, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.H.; Woodward, R.; Peters, L.; Verwey, W.F.; Mattis, P.A. The Prolongation of Penicillin Retention in the Body by Means of Para-Aminohippuric Acid. Science 1944, 100, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.; Koch, S.E.; Tranter, M.; Rubinstein, J. The History and Future of Probenecid. Cardiovasc. Toxicol. 2012, 12, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.F.; Israili, Z.H.; Dayton, P.G. Clinical Pharmacokinetics of Probenecid. Clin. Pharmacokinet. 1981, 6, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Landersdorfer, C.B.; Kirkpatrick, C.M.; Kinzig, M.; Bulitta, J.B.; Holzgrabe, U.; Jaehde, U.; Reiter, A.; Naber, K.G.; Rodamer, M.; Sorgel, F. Competitive Inhibition of Renal Tubular Secretion of Ciprofloxacin and Metabolite by Probenecid. Br. J. Clin. Pharmacol. 2010, 69, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Bostian, K.A. Practical Applications and Feasibility of Efflux Pump Inhibitors in the Clinic-A Vision for Applied Use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Spengler, G.; Kincses, A.; Gajdacs, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Venter, H.; Ma, S. Efflux Pump Inhibitors: A Novel Approach to Combat Efflux-Mediated Drug Resistance in Bacteria. Curr. Drug Targets 2016, 17, 702–719. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Cohen, K.A.; Winglee, K.; Maiga, M.; Diarra, B.; Bishai, W.R. Efflux Inhibition with Verapamil Potentiates Bedaquiline in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Pule, C.M.; Sampson, S.L.; Warren, R.M.; Black, P.A.; van Helden, P.D.; Victor, T.C.; Louw, G.E. Efflux Pump Inhibitors: Targeting Mycobacterial Efflux Systems to Enhance Tb Therapy. J. Antimicrob. Chemother. 2016, 71, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tasneen, R.; Peloquin, C.A.; Almeida, D.V.; Li, S.Y.; Barnes-Boyle, K.; Lu, Y.; Nuermberger, E. Verapamil Increases the Bioavailability and Efficacy of Bedaquiline but Not Clofazimine in a Murine Model of Tuberculosis. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Larson, K.B.; Wang, K.; Delille, C.; Otofokun, I.; Acosta, E.P. Pharmacokinetic Enhancers in HIV Therapeutics. Clin. Pharmacokinet. 2014, 53, 865–872. [Google Scholar] [CrossRef] [PubMed]
- Croxtall, J.D.; Perry, C.M. Lopinavir/Ritonavir: A Review of Its Use in the Management of HIV-1 Infection. Drugs 2010, 70, 1885–1915. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Chu, Y.; Wang, Y. HIV Protease Inhibitors: A Review of Molecular Selectivity and Toxicity. HIV AIDS (Auckl.) 2015, 7, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Von Richter, O.; Burk, O.; Fromm, M.F.; Thon, K.P.; Eichelbaum, M.; Kivisto, K.T. Cytochrome P450 3a4 and P-Glycoprotein Expression in Human Small Intestinal Enterocytes and Hepatocytes: A Comparative Analysis in Paired Tissue Specimens. Clin. Pharmacol. Ther. 2004, 75, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Renjifo, B.; van Wyk, J.; Salem, A.H.; Bow, D.; Ng, J.; Norton, M. Pharmacokinetic Enhancement in HIV Antiretroviral Therapy: A Comparison of Ritonavir and Cobicistat. AIDS Rev. 2015, 17, 37–46. [Google Scholar] [PubMed]
- Tseng, A.; Hughes, C.A.; Wu, J.; Seet, J.; Phillips, E.J. Cobicistat Versus Ritonavir: Similar Pharmacokinetic Enhancers but Some Important Differences. Ann. Pharmacother. 2017, 51, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Atta, M.G.; De Seigneux, S.; Lucas, G.M. Clinical Pharmacology in HIV Therapy. Clin. J. Am. Soc. Nephrol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hussaini, T. Paritaprevir/Ritonavir-Ombitasvir and Dasabuvir, the 3d Regimen for the Treatment of Chronic Hepatitis C Virus Infection: A Concise Review. Hepat. Med. 2016, 8, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Tran, T.; Chen, T.; Mikus, G.; Greenblatt, D.J. Inhibition of Human Cytochromes P450 in Vitro by Ritonavir and Cobicistat. J. Pharm. Pharmacol. 2017, 69, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Lepist, E.-I.; Phan, T.K.; Roy, A.; Tong, L.; MacLennan, K.; Murray, B.; Ray, A.S. Cobicistat Boosts the Intestinal Absorption of Transport Substrates, Including HIV Protease Inhibitors and Gs-7340, in Vitro. Antimicrob. Agents Chemother. 2012, 56, 5409–5413. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Desai, M.C. Cobicistat and Ritonavir as Pharmacoenhancers for Antiviral Drugs. In Successful Strategies for the Discovery of Antiviral Drugs; Desai, M.C., Meanwell, N.A., Eds.; The Royal Society of Chemistry: London, UK, 2013; pp. 451–481. ISBN 978-1-84973-657-2. [Google Scholar]
- Harbeson, S.L.; Tung, R.D. Preparation of Azapeptide Derivatives as HIV Protease Inhibitors. U.S. Patent 815,880,5B2, 17 April 2012. [Google Scholar]
- Hezode, C.; Asselah, T.; Reddy, K.R.; Hassanein, T.; Berenguer, M.; Fleischer-Stepniewska, K.; Marcellin, P.; Hall, C.; Schnell, G.; Pilot-Matias, T.; et al. Ombitasvir Plus Paritaprevir Plus Ritonavir with or without Ribavirin in Treatment-Naive and Treatment-Experienced Patients with Genotype 4 Chronic Hepatitis C Virus Infection (Pearl-I): A Randomised, Open-Label Trial. Lancet 2015, 385, 2502–2509. [Google Scholar] [CrossRef]
- Keating, G. Ombitasvir/Paritaprevir/Ritonavir: A Review in Chronic HCV Genotype 4 Infection. Drugs 2016, 76, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, O.M.; Gale, S.E.; Santevecchi, B. Ombitasvir/Paritaprevir/Ritonavir and Dasabuvir Tablets for Hepatitis C Virus Genotype 1 Infection. Ann. Pharmacother. 2015, 49, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Felmlee, D.J. Mechanisms of Hepatitis C Viral Resistance to Direct Acting Antivirals. Viruses 2015, 7, 6716–6729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petta, S.; Marzioni, M.; Russo, P.; Aghemo, A.; Alberti, A.; Ascione, A.; Antinori, A.; Bruno, R.; Bruno, S.; Chirianni, A.; et al. Plus Ribavirin for Patients with Hepatitis C Virus Genotype 1 or 4 Infection with Cirrhosis (Abacus): A Prospective Observational Study. Lancet Gastroenterol. Hepatol. 2017, 2, 427–434. [Google Scholar] [CrossRef]
- Ouwerkerk-Mahadevan, S.; Snoeys, J.; Peeters, M.; Beumont-Mauviel, M.; Simion, A. Drug-Drug Interactions with the NS3/4a Protease Inhibitor Simeprevir. Clin. Pharmacokinet. 2016, 55, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Petty, B.G.; Flaherty, J.; Fisher, P.E.; Polis, M.A.; Wachsman, M.; Lietman, P.S.; Lalezari, J.P.; Hitchcock, M.J.; Jaffe, H.S. Clinical Pharmacokinetics of Cidofovir in Human Immunodeficiency Virus-Infected Patients. Antimicrob. Agents Chemother. 1995, 39, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Unadkat, J.D.; Kirby, B.J.; Endres, C.J.; Zolnerciks, J.K. The Impact and in Vitro to in Vivo Prediction of Transporter-Based Drug–Drug Interactions in Humans. In Enzyme- and Transporter-Based Drug-Drug Interactions; Pang, K.S., Rodrigues, A.D., Eds.; Springer: New York, NY, USA, 2010; pp. 517–553. [Google Scholar] [CrossRef]
- Uwai, Y.; Ida, H.; Tsuji, Y.; Katsura, T.; Inui, K.-I. Renal Transport of Adefovir, Cidofovir, and Tenofovir by Slc22a Family Members (Hoat1, Hoat3, and Hoct2). Pharm. Res. 2007, 24, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.L.; Rodriguez, C.A.; Mucci, M.; Ingrosso, A.; Duncan, B.A.; Nickens, D.J. Pharmacokinetics and Renal Effects of Cidofovir with a Reduced Dose of Probenecid in HIV-Infected Patients with Cytomegalovirus Retinitis. J. Clin. Pharmacol. 2003, 43, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Butler, D. Wartime Tactic Doubles Power of Scarce Bird-Flu Drug. Nature 2005, 438, 6. [Google Scholar] [CrossRef] [PubMed]
- Hill, G.; Cihlar, T.; Oo, C.; Ho, E.S.; Prior, K.; Wiltshire, H.; Barrett, J.; Liu, B.; Ward, P. The Anti-Influenza Drug Oseltamivir Exhibits Low Potential to Induce Pharmacokinetic Drug Interactions Via Renal Secretion—Correlation of in Vivo and in Vitro Studies. Drug Metab. Disposition 2002, 30, 13–19. [Google Scholar] [CrossRef]
- Schilsky, R.L.; Kindler, H.L. Eniluracil: An Irreversible Inhibitor of Dihydropyrimidine Dehydrogenase. Expert Opin. Investig. Drugs 2000, 9, 1635–1649. [Google Scholar] [CrossRef] [PubMed]
- Paff, M.T.; Baccanari, D.P.; Davis, S.T.; Cao, S.; Tansik, R.L.; Rustum, Y.M.; Spector, T. Preclinical Development of Eniluracil: Enhancing the Therapeutic Index and Dosing Convenience of 5-Fluorouracil. Invest. New Drugs 2000, 18, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Sanford, M. S-1 (Teysuno(R)): A Review of Its Use in Advanced Gastric Cancer in Non-Asian Populations. Drugs 2013, 73, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Nadal, J.C.; Van Groeningen, C.J.; Pinedo, H.M.; Peters, G.J. In Vivo Potentiation of 5-Fluorouracil by Leucovorin in Murine Colon Carcinoma. Biomed. Pharmacother. 1988, 42, 387–393. [Google Scholar] [PubMed]
- Diasio, R.B. Oral DPD-Inhibitory Fluoropyrimidine Drugs. Oncology (Williston Park) 2000, 14, 19–23. [Google Scholar] [PubMed]
- Peters, G.J.; Noordhuis, P.; Van Kuilenburg, A.B.; Schornagel, J.H.; Gall, H.; Turner, S.L.; Swart, M.S.; Voorn, D.; Van Gennip, A.H.; Wanders, J.; et al. Pharmacokinetics of S-1, an Oral Formulation of Ftorafur, Oxonic Acid and 5-Chloro-2,4-Dihydroxypyridine (Molar Ratio 1:0.4:1) in Patients with Solid Tumors. Cancer Chemother. Pharmacol. 2003, 52, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sugimachi, K.; Maehara, Y. A Phase Ii Trial of a New 5-Fluorouracil Derivative, Bof-A2 (Emitefur), for Patients with Advanced Gastric Cancer. Surg. Today 2000, 30, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.J.; Chestnut, W.G.; Merrill, B.M.; Spector, T. Mechanism-Based Inactivation of Dihydropyrimidine Dehydrogenase by 5-Ethynyluracil. J. Biol. Chem. 1992, 267, 5236–5242. [Google Scholar] [PubMed]
- Shirasaka, T.; Shimamoto, Y.; Fukushima, M. Inhibition by Oxonic Acid of Gastrointestinal Toxicity of 5-Fluorouracil without Loss of Its Antitumor Activity in Rats. Cancer Res. 1993, 53, 4004–4009. [Google Scholar] [PubMed]
- Kish, T.; Uppal, P. Trifluridine/Tipiracil (Lonsurf) for the Treatment of Metastatic Colorectal Cancer. P&T 2016, 41, 314–325. [Google Scholar]
- Puthiamadathil, J.M.; Weinberg, B.A. Emerging Combination Therapies for Metastatic Colorectal Cancer-Impact of Trifluridine/Tipiracil. Cancer Manag. Res. 2017, 9, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, M.; Suzuki, N.; Emura, T.; Yano, S.; Kazuno, H.; Tada, Y.; Yamada, Y.; Asao, T. Structure and Activity of Specific Inhibitors of Thymidine Phosphorylase to Potentiate the Function of Antitumor 2′-Deoxyribonucleosides. Biochem. Pharmacol. 2000, 59, 1227–1236. [Google Scholar] [CrossRef]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural Alkaloids as P-Gp Inhibitors for Multidrug Resistance Reversal in Cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.; Zia-Ul-Haq, M. Natural Products as Alternative Choices for P-Glycoprotein (P-Gp) Inhibition. Molecules 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Qing, Z.X.; Huang, J.L.; Yang, X.Y.; Liu, J.H.; Cao, H.L.; Xiang, F.; Cheng, P.; Zeng, J.G. Anticancer and Reversing Multidrug Resistance Activities of Natural Isoquinoline Alkaloids and Their Structure-Activity Relationship. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Robey, R.W.; Chen, C.C.; Draper, D.; Luchenko, V.; Barnett, D.; Oldham, R.K.; Caluag, Z.; Frye, A.R.; Steinberg, S.M.; et al. A Pharmacodynamic Study of the P-Glycoprotein Antagonist Cbt-1® in Combination with Paclitaxel in Solid Tumors. Oncologist 2012, 17, e512–e523. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, H.; Nishikawa, N.; Nagai, M.; Tsujii, T.; Yabe, H.; Kubo, M.; Ieiri, I.; Nomoto, M. Pharmacokinetics of Levodopa/Benserazide Versus Levodopa/Carbidopa in Healthy Subjects and Patients with Parkinson’s Disease. Neurol. Clin. Neurosci. 2015, 3, 68–73. [Google Scholar] [CrossRef]
- Daidone, F.; Montioli, R.; Paiardini, A.; Cellini, B.; Macchiarulo, A.; Giardina, G.; Bossa, F.; Borri Voltattorni, C. Identification by Virtual Screening and in Vitro Testing of Human Dopa Decarboxylase Inhibitors. PLoS ONE 2012, 7, e31610. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Lyseng-Williamson, K.A. Tolcapone: A Review of Its Use in the Management of Parkinson’s Disease. CNS Drugs 2005, 19, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, P.; Dominici, P.; Borri-Voltattorni, C.; Jansonius, J.N.; Malashkevich, V.N. Structural Insight into Parkinson’s Disease Treatment from Drug-Inhibited Dopa Decarboxylase. Nat. Struct. Biol. 2001, 8, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.E.; Soares-da-Silva, P. Medicinal Chemistry of Catechol O-Methyltransferase (Comt) Inhibitors and Their Therapeutic Utility. J. Med. Chem. 2014, 57, 8692–8717. [Google Scholar] [CrossRef] [PubMed]
- Schrag, A. Entacapone in the Treatment of Parkinson’s Disease. Lancet Neurol. 2005, 4, 366–370. [Google Scholar] [CrossRef]
- Bonifacio, M.J.; Torrao, L.; Loureiro, A.I.; Palma, P.N.; Wright, L.C.; Soares-da-Silva, P. Pharmacological Profile of Opicapone, a Third-Generation Nitrocatechol Catechol-O-Methyl Transferase Inhibitor, in the Rat. Br. J. Pharmacol. 2015, 172, 1739–1752. [Google Scholar] [CrossRef] [PubMed]
- Annus, A.; Vecsei, L. Spotlight on Opicapone as an Adjunct to Levodopa in Parkinson’s Disease: Design, Development and Potential Place in Therapy. Drug Des. Devel. Ther. 2017, 11, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Gordin, A.; Kaakkola, S.; Teravainen, H. Clinical Advantages of COMT Inhibition with Entacapone-A Review. J. Neural. Transm. (Vienna) 2004, 111, 1343–1363. [Google Scholar] [CrossRef] [PubMed]
- Garnock-Jones, K.P. Dextromethorphan/Quinidine: In Pseudobulbar Affect. CNS Drugs 2011, 25, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A. Dextromethorphan/Quinidine: A Novel Dextromethorphan Product for the Treatment of Emotional Lability. Expert Opin. Pharmacother. 2006, 7, 2581–2598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Britto, M.R.; Valderhaug, K.L.; Wedlund, P.J.; Smith, R.A. Dextromethorphan: Enhancing Its Systemic Availability by Way of Low-Dose Quinidine-Mediated Inhibition of Cytochrome P4502D6. Clin. Pharmacol. Ther. 1992, 51, 647–655. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A): Formation of a stable O-acyl-enzyme complex by reaction of the β-lactam antibiotic with the bacterial target enzyme (in red) is the molecular basis of the antibacterial activity. (B): Hydrolysis of the β-lactam antibiotic by bacterial β-lactamases leads to a loss of antibiotic activity. (C): Active site view from a crystal structure of a penicillin-binding protein (PBP) from Thermosynechococcus elongates acylated by benzylpenicillin (in blue) (PDB ID: 2J8Y).
Figure 1.
(A): Formation of a stable O-acyl-enzyme complex by reaction of the β-lactam antibiotic with the bacterial target enzyme (in red) is the molecular basis of the antibacterial activity. (B): Hydrolysis of the β-lactam antibiotic by bacterial β-lactamases leads to a loss of antibiotic activity. (C): Active site view from a crystal structure of a penicillin-binding protein (PBP) from Thermosynechococcus elongates acylated by benzylpenicillin (in blue) (PDB ID: 2J8Y).

Figure 2.
Structures of the β-lactam-type β-lactamase inhibitors clavulanic acid, sulbactam, and tazobactam.
Figure 2.
Structures of the β-lactam-type β-lactamase inhibitors clavulanic acid, sulbactam, and tazobactam.

Figure 3.
(A): Inactivation of a β-lactamase enzyme (relevant serine residues are marked in red and blue) by clavulanic acid. The C3 unit from clavulanic acid, which is bonded covalently to the enzyme, is highlighted with balls in magenta. (B) Inactivation of a β-lactamase enzyme by tazobactam. (C) Active site view from a crystal structure of the intermediate covalent adduct formed between a β-lactamase (in red) from Mycobacterium tuberculosis and clavulanic acid (binding fragment highlighted in blue) (PDB ID: 3CG5).
Figure 3.
(A): Inactivation of a β-lactamase enzyme (relevant serine residues are marked in red and blue) by clavulanic acid. The C3 unit from clavulanic acid, which is bonded covalently to the enzyme, is highlighted with balls in magenta. (B) Inactivation of a β-lactamase enzyme by tazobactam. (C) Active site view from a crystal structure of the intermediate covalent adduct formed between a β-lactamase (in red) from Mycobacterium tuberculosis and clavulanic acid (binding fragment highlighted in blue) (PDB ID: 3CG5).

Figure 4.
In the “mutual prodrug” sultamicillin, the β-lactamase-labile antibiotic ampicillin (blue) and the β-lactamase inhibitor sulbactam (green) are connected with an esterase-labile di-ester moiety. Enzymatic ester hydrolysis releases both active compounds as well as one equivalent formaldehyde.
Figure 4.
In the “mutual prodrug” sultamicillin, the β-lactamase-labile antibiotic ampicillin (blue) and the β-lactamase inhibitor sulbactam (green) are connected with an esterase-labile di-ester moiety. Enzymatic ester hydrolysis releases both active compounds as well as one equivalent formaldehyde.

Figure 5.
(Left): Structures of the 1,6-diazabicyclo [3.2.1] octane-type β-lactamase inhibitors avibactam, relebactam, zidebactam, and NXL-105. (Right) Reversible binding of avibactam (in blue) to the Ser69 (in red) residue of a β-lactamase from Klebsiella pneumoniae (from PDB ID: 4ZBE).
Figure 5.
(Left): Structures of the 1,6-diazabicyclo [3.2.1] octane-type β-lactamase inhibitors avibactam, relebactam, zidebactam, and NXL-105. (Right) Reversible binding of avibactam (in blue) to the Ser69 (in red) residue of a β-lactamase from Klebsiella pneumoniae (from PDB ID: 4ZBE).

Figure 6.
(Left): Formation of a tetrahedral, negatively charged intermediate as the first step of the reaction of the β-lactamase inhibitor sulbactam with a serine residue in the active site of a β-lactamase. (Right): In an analogous manner, boronic acid derivatives like vaborbactam can reversibly form negatively charged adducts with a serine residue in the active site of β-lactamases.
Figure 6.
(Left): Formation of a tetrahedral, negatively charged intermediate as the first step of the reaction of the β-lactamase inhibitor sulbactam with a serine residue in the active site of a β-lactamase. (Right): In an analogous manner, boronic acid derivatives like vaborbactam can reversibly form negatively charged adducts with a serine residue in the active site of β-lactamases.

Figure 7.
Structures of the antibiotic imipenem and the dehydropeptidase-I inhibitor cilastatin. Both compounds contain a dehydropeptide structural motif (highlighted in blue) responsible for affinity for the enzyme dehydropeptidase-I.
Figure 7.
Structures of the antibiotic imipenem and the dehydropeptidase-I inhibitor cilastatin. Both compounds contain a dehydropeptide structural motif (highlighted in blue) responsible for affinity for the enzyme dehydropeptidase-I.

Figure 8.
Structures of the macrolide antibiotic clarithromycin and the proton pump inhibitors omeprazole, lansoprazole, and pantoprazole.
Figure 8.
Structures of the macrolide antibiotic clarithromycin and the proton pump inhibitors omeprazole, lansoprazole, and pantoprazole.

Figure 9.
Structures of probenecid, verapamil, and bedaquiline.

Figure 10.
Structures of the pharmacokinetic enhancers ritonavir and cobicistat (differences in structure highlighted in red) as well as antiretroviral drugs boosted by these compounds: the HIV protease inhibitors lopinavir, atazanavir, darunavir, and the integrase inhibitor elvitegravir.
Figure 10.
Structures of the pharmacokinetic enhancers ritonavir and cobicistat (differences in structure highlighted in red) as well as antiretroviral drugs boosted by these compounds: the HIV protease inhibitors lopinavir, atazanavir, darunavir, and the integrase inhibitor elvitegravir.

Figure 11.
Structures of novel pharmacokinetic enhancers for HIV protease inhibitors in the development.
Figure 11.
Structures of novel pharmacokinetic enhancers for HIV protease inhibitors in the development.

Figure 12.
The HCV polyprotein is a single protein (around 3000 amino acids), which undergoes cleavage catalyzed by host endopeptidases (for structural proteins) and autoproteolytically (indicated by blue arrows) by its own non-structural protein NS3/4A with protease activity. This results in the formation of structural proteins (core and envelope proteins) and non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). Direct-acting antiviral drugs interact with diverse non-structural proteins (indicated by red bars) [44].
Figure 12.
The HCV polyprotein is a single protein (around 3000 amino acids), which undergoes cleavage catalyzed by host endopeptidases (for structural proteins) and autoproteolytically (indicated by blue arrows) by its own non-structural protein NS3/4A with protease activity. This results in the formation of structural proteins (core and envelope proteins) and non-structural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). Direct-acting antiviral drugs interact with diverse non-structural proteins (indicated by red bars) [44].

Figure 13.
Structures of novel direct-acting antiviral drugs paritaprevir (NS3/4A protease inhibitor), ombitasvir (inhibitor of the non-structural protein NS5A involved in viral replication, assembly, and release of HCV particles), and dasabuvir (inhibitor of the RNA polymerase NS5B).
Figure 13.
Structures of novel direct-acting antiviral drugs paritaprevir (NS3/4A protease inhibitor), ombitasvir (inhibitor of the non-structural protein NS5A involved in viral replication, assembly, and release of HCV particles), and dasabuvir (inhibitor of the RNA polymerase NS5B).

Figure 14.
Structures of the antiviral drugs cidofovir und oseltamivir.

Figure 15.
Structures of 5-fluorouracil (5-FU) and its pro-drug tegafur.

Figure 16.
Bioactivation of tegafur to give 5-FU and subsequent metabolism of 5-FU (black arrows). The structures of the main toxic metabolites are shown in the box. Relevant enzymes are highlighted in blue. The biological effects of the toxic metabolites FUTP and FdUTP are indicated by red arrows. The effects of pharmacokinetic enhancers gimeracil and oteracil (both in Teysuno®) and of the active 5-FU metabolite FdUMP on enzymes are indicated by red bars. Abbreviations: 5-FU 5-fluorouracil, CH2THF methylene tetrahydrofolate, DHFU dihydrofluorouracil, DPD dihydropyrimidine dehydrogenase, FBAL fluoro-β-alanine, FdUDP fluorodeoxyuridine diphosphate, FdUMP fluorodeoxyuridine monophosphate, FdUTP fluorodeoxyuridine triphosphate, FUMP fluorouridine monophosphate, FUDP fluorouridine diphosphate, FUDR fluorodeoxyuridine, FUTP fluorouridine triphosphate, OPRT orotate phosphoribosyltransferase, PRPP phosphoribosyl pyrophosphate, TK thymidine kinase, and TP thymidine phosphorylase [56].
Figure 16.
Bioactivation of tegafur to give 5-FU and subsequent metabolism of 5-FU (black arrows). The structures of the main toxic metabolites are shown in the box. Relevant enzymes are highlighted in blue. The biological effects of the toxic metabolites FUTP and FdUTP are indicated by red arrows. The effects of pharmacokinetic enhancers gimeracil and oteracil (both in Teysuno®) and of the active 5-FU metabolite FdUMP on enzymes are indicated by red bars. Abbreviations: 5-FU 5-fluorouracil, CH2THF methylene tetrahydrofolate, DHFU dihydrofluorouracil, DPD dihydropyrimidine dehydrogenase, FBAL fluoro-β-alanine, FdUDP fluorodeoxyuridine diphosphate, FdUMP fluorodeoxyuridine monophosphate, FdUTP fluorodeoxyuridine triphosphate, FUMP fluorouridine monophosphate, FUDP fluorouridine diphosphate, FUDR fluorodeoxyuridine, FUTP fluorouridine triphosphate, OPRT orotate phosphoribosyltransferase, PRPP phosphoribosyl pyrophosphate, TK thymidine kinase, and TP thymidine phosphorylase [56].

Figure 17.
(A) Reduction of 5-FU to dihydrofluorouracil (DHFU) catalyzed by the enzyme dihydropyrimidine dehydrogenase (DPD), which is followed by hydrolysis to give fluoro-β-alanine (FBAL). (B) Structures of the competitive DPD inhibitors uracil, gimeracil, and CNDP.
Figure 17.
(A) Reduction of 5-FU to dihydrofluorouracil (DHFU) catalyzed by the enzyme dihydropyrimidine dehydrogenase (DPD), which is followed by hydrolysis to give fluoro-β-alanine (FBAL). (B) Structures of the competitive DPD inhibitors uracil, gimeracil, and CNDP.

Figure 18.
Covalent binding of eniluracil to DPD after reductive bioactivation by the target enzyme DPD itself [62].
Figure 18.
Covalent binding of eniluracil to DPD after reductive bioactivation by the target enzyme DPD itself [62].

Figure 19.
Emitefur (BOF-A2) is a mutual pro-drug in which a 5-FU (marked in red) derivative is covalently coupled (using an inert isophthalic acid unit) with a CNDP (marked in blue) derivative.
Figure 19.
Emitefur (BOF-A2) is a mutual pro-drug in which a 5-FU (marked in red) derivative is covalently coupled (using an inert isophthalic acid unit) with a CNDP (marked in blue) derivative.

Figure 20.
Function of the enzyme orotate phosphoribosyltransferase (OPRT) and its inhibition by oteracil (in red).
Figure 20.
Function of the enzyme orotate phosphoribosyltransferase (OPRT) and its inhibition by oteracil (in red).

Figure 21.
(Left) Structures of the antimetabolite trifluridine and its sites of bioactivation (green) and degradation (red). (Right) The thymidine phosphorylase inhibitor tipiracil.
Figure 21.
(Left) Structures of the antimetabolite trifluridine and its sites of bioactivation (green) and degradation (red). (Right) The thymidine phosphorylase inhibitor tipiracil.

Figure 22.
Inactivation of trifluridine catalyzed by the enzyme thymidine phosphorylase.

Figure 23.
Structures of inhibitors of selected P-glycoprotein efflux pumps: elacridar, zosuquidar, and the alkaloid tetrandrine.
Figure 23.
Structures of inhibitors of selected P-glycoprotein efflux pumps: elacridar, zosuquidar, and the alkaloid tetrandrine.

Figure 24.
Transport and metabolism of levodopa. (Left): In the periphery, levodopa can undergo metabolism by both DOPA decarboxylase (DDC, AADC) or catechol-O-methyltransferase (COMT). Only unchanged levodopa is taken up into the brain. There it is decarboxylated by DDC to give the active compound dopamine. Dopamine is metabolized in the brain by monoamine oxidase B (MAO-B) as well as COMT and DDC (not shown in the graphic). (Right) (A): Application of levodopa without a pharmacokinetic enhancer results in extremely low uptake into the brain. The major part is lost by peripheral decarboxylation to give dopamine. (B): Co-administration of a DOPA decarboxylase inhibitor (inhibition indicated by a red bar) leads to significant enhancement of dopamine uptake into the brain, but by the action of peripheral COMT, 3-O-methyldopa (3-OMD) becomes the main peripheral metabolite. (C): Combination of levodopa with both a DOPA decarboxylase inhibitor and a COMT inhibitor (2 red bars) leads to a maximum uptake of levodopa into the brain.
Figure 24.
Transport and metabolism of levodopa. (Left): In the periphery, levodopa can undergo metabolism by both DOPA decarboxylase (DDC, AADC) or catechol-O-methyltransferase (COMT). Only unchanged levodopa is taken up into the brain. There it is decarboxylated by DDC to give the active compound dopamine. Dopamine is metabolized in the brain by monoamine oxidase B (MAO-B) as well as COMT and DDC (not shown in the graphic). (Right) (A): Application of levodopa without a pharmacokinetic enhancer results in extremely low uptake into the brain. The major part is lost by peripheral decarboxylation to give dopamine. (B): Co-administration of a DOPA decarboxylase inhibitor (inhibition indicated by a red bar) leads to significant enhancement of dopamine uptake into the brain, but by the action of peripheral COMT, 3-O-methyldopa (3-OMD) becomes the main peripheral metabolite. (C): Combination of levodopa with both a DOPA decarboxylase inhibitor and a COMT inhibitor (2 red bars) leads to a maximum uptake of levodopa into the brain.

Figure 25.
(Top): Molecular mechanism of the decarboxylation of levodopa by DOPA decarboxylase (relevant lysine residue highlighted in blue) aided by the co-factor pyridoxal phosphate. (Bottom): Structures of the DOPA decarboxylase inhibitors carbidopa, benserazide, and its active metabolite. The essential hydrazine moieties in the active inhibitors are highlighted in red.
Figure 25.
(Top): Molecular mechanism of the decarboxylation of levodopa by DOPA decarboxylase (relevant lysine residue highlighted in blue) aided by the co-factor pyridoxal phosphate. (Bottom): Structures of the DOPA decarboxylase inhibitors carbidopa, benserazide, and its active metabolite. The essential hydrazine moieties in the active inhibitors are highlighted in red.

Figure 26.
(Left): Carbidopa forms a hydrazine (highlighted in red) with the pyridoxal phosphate in the active site of DOPA decarboxylase. (Right): This hydrazine (carbidopa fragment highlighted in blue, pyridoxal fragment in green) is bound in the active site via numerous polar interactions.
Figure 26.
(Left): Carbidopa forms a hydrazine (highlighted in red) with the pyridoxal phosphate in the active site of DOPA decarboxylase. (Right): This hydrazine (carbidopa fragment highlighted in blue, pyridoxal fragment in green) is bound in the active site via numerous polar interactions.

Figure 27.
(Left): Regioselective O-methylation of levodopa catalyzed by COMT, the methyl group (in red) is provided by S-adenosylmethionine (SAM). (Right): Clinically relevant COMT inhibitors: nitecapone, entacapone, tolcapone, nebicapone, and opicapone. All of them share the nitrocatechol pharmacophore (highlighted in red).
Figure 27.
(Left): Regioselective O-methylation of levodopa catalyzed by COMT, the methyl group (in red) is provided by S-adenosylmethionine (SAM). (Right): Clinically relevant COMT inhibitors: nitecapone, entacapone, tolcapone, nebicapone, and opicapone. All of them share the nitrocatechol pharmacophore (highlighted in red).

Figure 28.
(Left): The active component dextromethorphan is mainly metabolized by CYP2D6 (highlighted in red). (Right): Structure of the CYP2D6 inhibitor quinidine, which is a pharmacokinetic enhancer for dextromethorphan.
Figure 28.
(Left): The active component dextromethorphan is mainly metabolized by CYP2D6 (highlighted in red). (Right): Structure of the CYP2D6 inhibitor quinidine, which is a pharmacokinetic enhancer for dextromethorphan.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Krauß, J.; Bracher, F. Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond. Sci. Pharm. 2018, 86, 43. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm86040043
AMA Style
Krauß J, Bracher F. Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond. Scientia Pharmaceutica. 2018; 86(4):43. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm86040043
Chicago/Turabian StyleKrauß, Jürgen, and Franz Bracher. 2018. "Pharmacokinetic Enhancers (Boosters)—Escort for Drugs against Degrading Enzymes and Beyond" Scientia Pharmaceutica 86, no. 4: 43. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm86040043