
Docking-Based Virtual Screening and Molecular Dynamics Simulations of Quercetin Analogs as Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors of Mycobacterium tuberculosis

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Structure Preparation
2.2. Preparation of Small Molecule Library
2.3. Docking-Based Virtual Screening
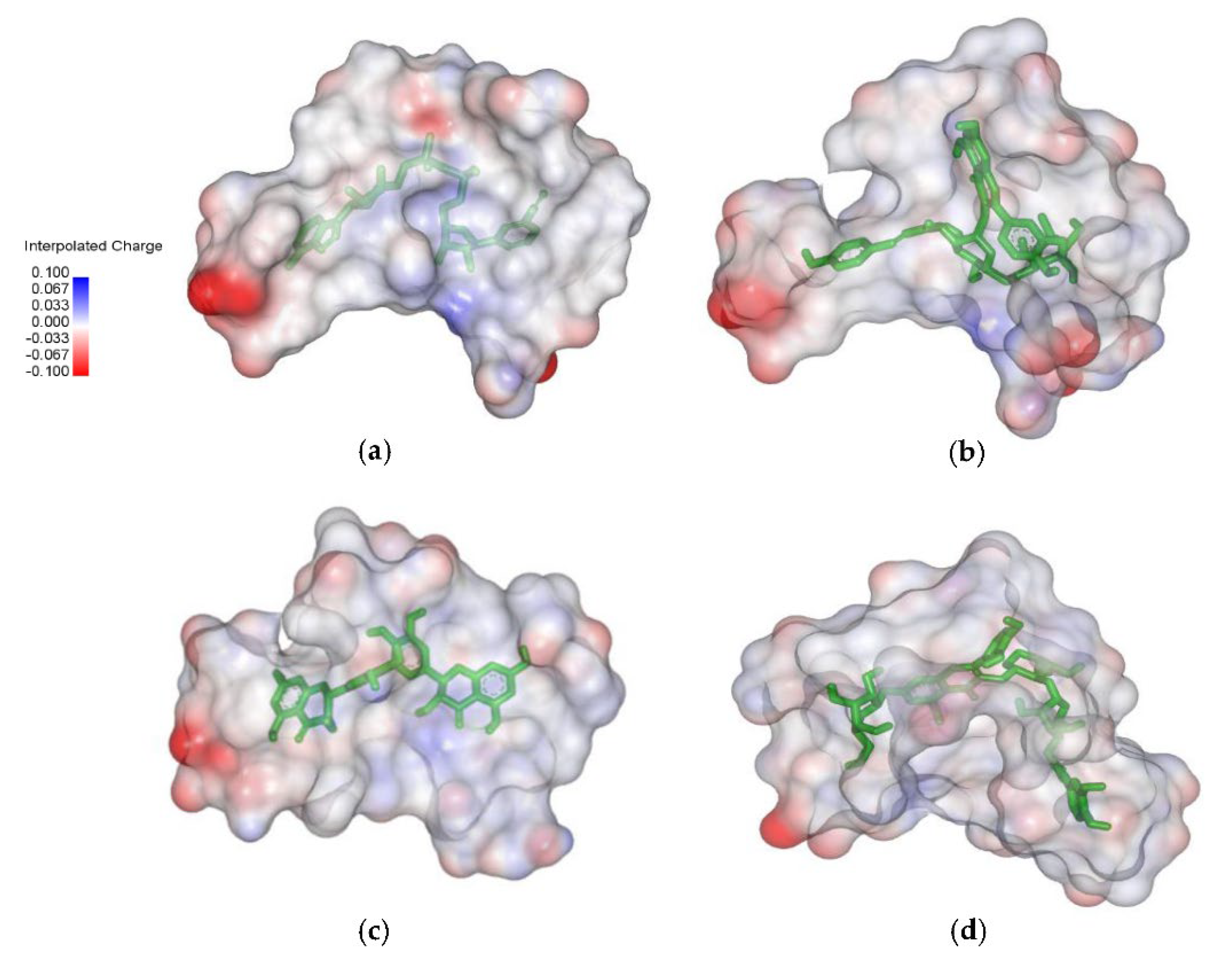
2.4. Analysis of the Protein–Ligand Complex
2.5. Molecular Dynamic (MD) Simulation
2.6. Binding Free Energy (MM-PBSA) Analysis
3. Results
3.1. Docking-Based Virtual Screening
3.2. Structural Analysis of Systems
3.2.1. Root-Mean-Square Deviation (RMSD) and Root-Mean-Square Fluctuation (RMSF)
3.2.2. Radius Gyration (Rg) and Solvent Accessible Surface Area (SASA)
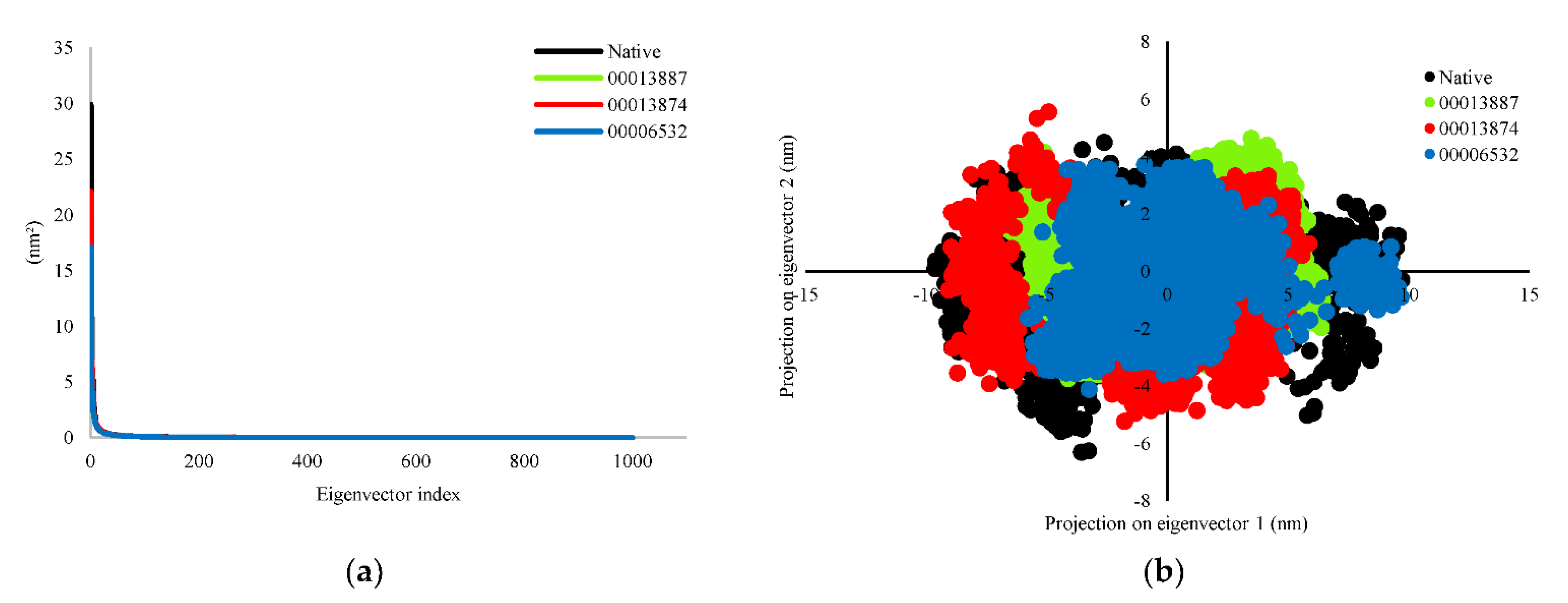
3.2.3. Principal Component Analysis (PCA)
3.3. Binding Free Energy InhA-Ligands
3.4. Hydrogen Bonds Established between Receptor-Ligands
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sotgiu, G.; Centis, R.; D’ambrosio, L.; Migliori, G.B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmins, G.S.; Deretic, V. Mechanisms of action of isoniazid. Mol. Microbiol. 2006, 5, 1220–1227. [Google Scholar] [CrossRef] [PubMed]
- Marrakchi, H.; Lanéelle, G.; Quémard, A. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 2000, 2, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to Synthesis and Processing of Mycolic Acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2005, 1, 81–101. [Google Scholar] [CrossRef] [Green Version]
- Rotta, M.; Pissinate, K.; Villela, A.D.; Back, D.F.; Timmers, L.F.S.M.; Bachega, J.F.R.; de Souza, O.N.; Santos, D.S.; Basso, L.A.; Machado, P. Piperazine derivatives: Synthesis, inhibition of the Mycobacterium tuberculosis enoyl-acyl carrier protein reductase and SAR studies. Eur. J. Med. Chem. 2015, 1, 436–447. [Google Scholar] [CrossRef]
- Lei, B.; Wei, C.J.; Tu, S.C. Action Mechanism of Antitubercular Isoniazid. J. Biol Chem. 2000, 4, 2520–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants—rich sources for a new group of multi-potent plant extracts. Molecules 2009, 6, 2016–2031. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Jiang, X.; Gao, F.; Song, J.; Sun, J.; Wang, L.; Sun, X.; Lu, Z.; Zhang, H. Identification of plant-derived natural products as potential inhibitors of the Mycobacterium tuberculosis proteasome. BMC Complement. Altern. Med. 2014, 14, 400. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Teskey, G.; Islamoglu, H.; Gutierrez, M.; Salaiz, O.; Munjal, S.; Fraix, M.P.; Sathananthan, A.; Nieman, D.C.; Venketaraman, V. Flavonoid Mixture Inhibits Mycobacterium tuberculosis Survival and Infectivity. Molecules 2019, 5, 851. [Google Scholar] [CrossRef] [Green Version]
- Sasikumar, K.; Ghosh, A.R.; Dusthackeer, A. Antimycobacterial potentials of quercetin and rutin against Mycobacterium tuberculosis H37Rv. Biotech 2018, 10, 427. [Google Scholar] [CrossRef]
- Pawar, A.; Jha, P.; Konwar, C.; Chaudhry, U.; Chopra, M.; Saluja, D. Ethambutol targets the glutamate racemase of Mycobacterium tuberculosis—an enzyme involved in peptidoglycan biosynthesis. Appl. Microbiol. Biotechnol. 2019, 2, 843–851. [Google Scholar] [CrossRef]
- Premalatha, E.; Dineshraj, R.; Iyanar, K.; Bhaarath, K.S.; Sharavanan, T.K.V. Molecular docking study on quercetin derivatives as inhibitors of PantothenateSynthetase (PanC) of Mycobacterium tuberculosis. Int. J. Res. Pharm. Sci. 2020, 3, 3684–3690. [Google Scholar] [CrossRef]
- Pitaloka, D.A.E.; Damayanti, S.; Artarini, A.A.; Sukandar, E.Y. Molecular Docking, Dynamics Simulation, and Scanning Electron Microscopy (SEM) Examination of Clinically Isolated Mycobacterium tuberculosis by Ursolic Acid: A Pentacyclic Triterpenes. Indones J. Chem. 2019, 2, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Olson, A.J. Using AutoDock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leung, K.S.; Wong, M.H. idock: A multithreaded virtual screening tool for flexible ligand docking. IEEE Symp. Comput. Intell. Bioinform. Comput. Biol. CIBCB 2012, 77–84. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 22, 3219–3228. [Google Scholar] [CrossRef]
- Petrov, D.; Zagrovic, B. Are Current Atomistic Force Fields Accurate Enough to Study Proteins in Crowded Environments? PLoS Comput. Biol. 2014, 5, e1003638. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.W.S.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Gao, X.; Fang, J. Multiple Staggered Mesh Ewald: Boosting the Accuracy of the Smooth Particle Mesh Ewald Method. J. Chem. Theory Comput. 2016, 11, 5596–5608. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 7, 1951–1962. [Google Scholar] [CrossRef]
- Tabrizi, A.M.; Goossens, S.; Rahimi, A.M.; Knepley, M.; Bardhan, J.P. Predicting solvation free energies and thermodynamics in polar solvents and mixtures using a solvation-layer interface condition. J. Chem Phys. 2017, 9, 094103. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, J.; Okimoto, N.; Morimoto, G.; Taiji, M. A new set of atomic radii for accurate estimation of solvation free energy by Poisson–Boltzmann solvent model. J. Comput. Chem. 2014, 29, 2132. [Google Scholar] [CrossRef] [Green Version]
- Shoichet, B.K.; McGovern, S.L.; Wei, B.; Irwin, J.J. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 2002, 4, 439–446. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Vilchèze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 1999, 22, 15582–15589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhasini, M.; Sailatha, E.; Gunasekaran, S.; Ramkumaar, G.R. Vibrational and electronic investigations, thermodynamic parameters, HOMO and LUMO analysis on Lornoxicam by density functional theory. J. Mol. Struct. 2015, 1100, 116–128. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys. 2016, 32, 22129–22139. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 3, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, M.; Catanzaro, D.; Catanzaro, A.; Rodwell, T.C. Genetic mutations associated with isoniazid resistance in Mycobacterium tuberculosis: A systematic review. PLoS ONE 2015, 3, e0119628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dessen, A.; Quemard, A.; Blanchard, J.S.; Jacobs, W.R., Jr.; Sacchettini, J.C. Cyrstal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 1995, 5204, 1638–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
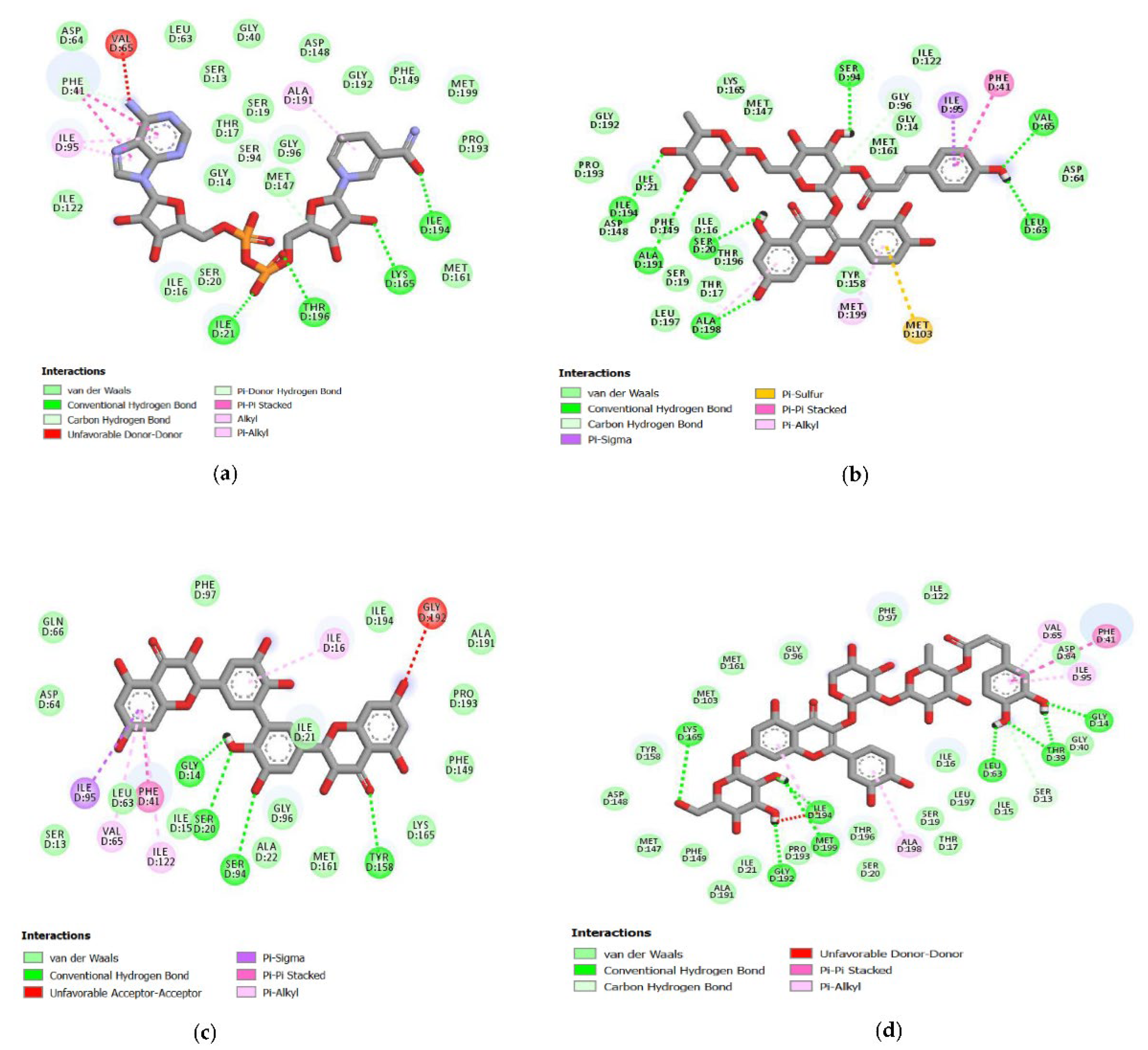



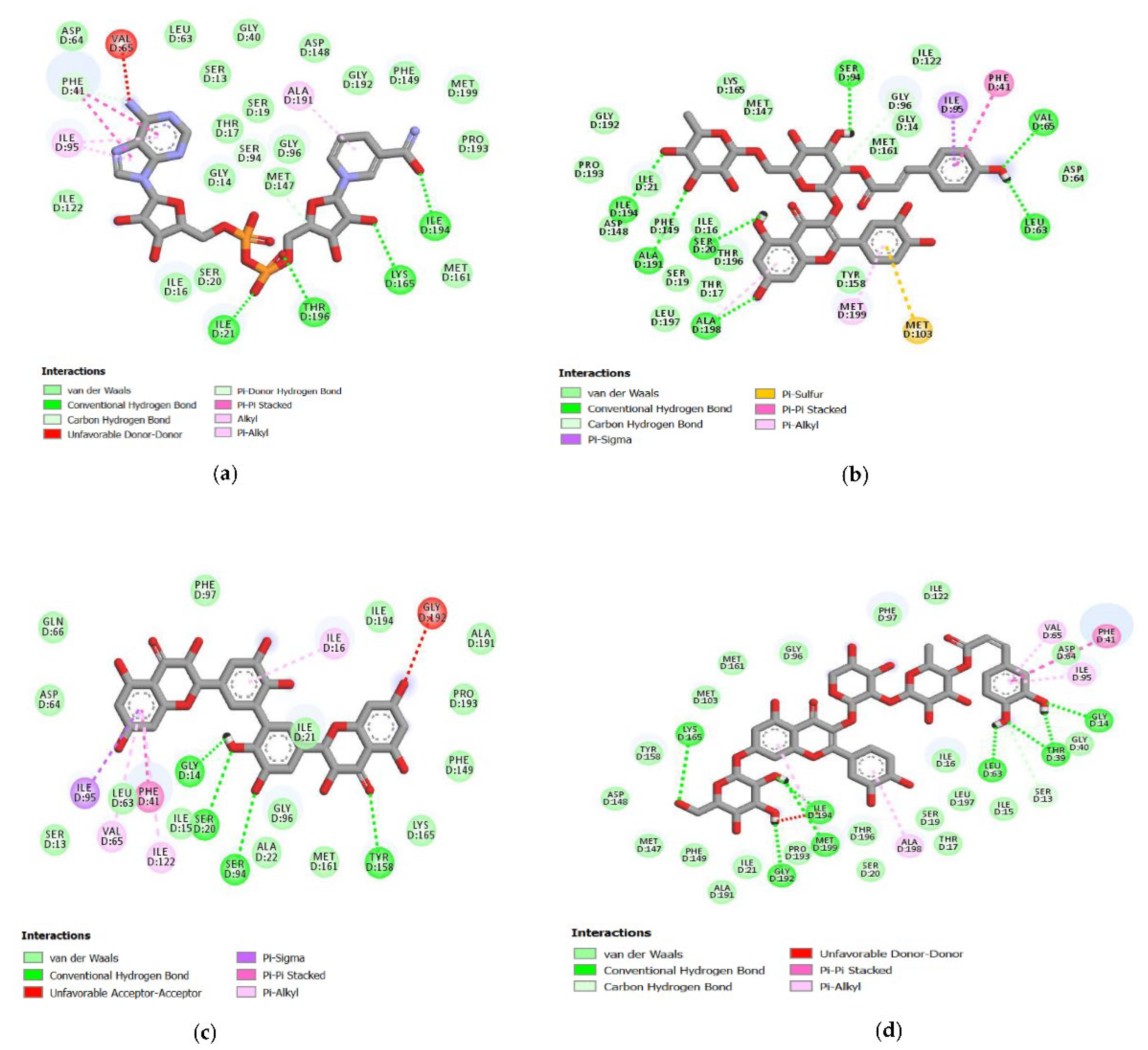{kind=link}
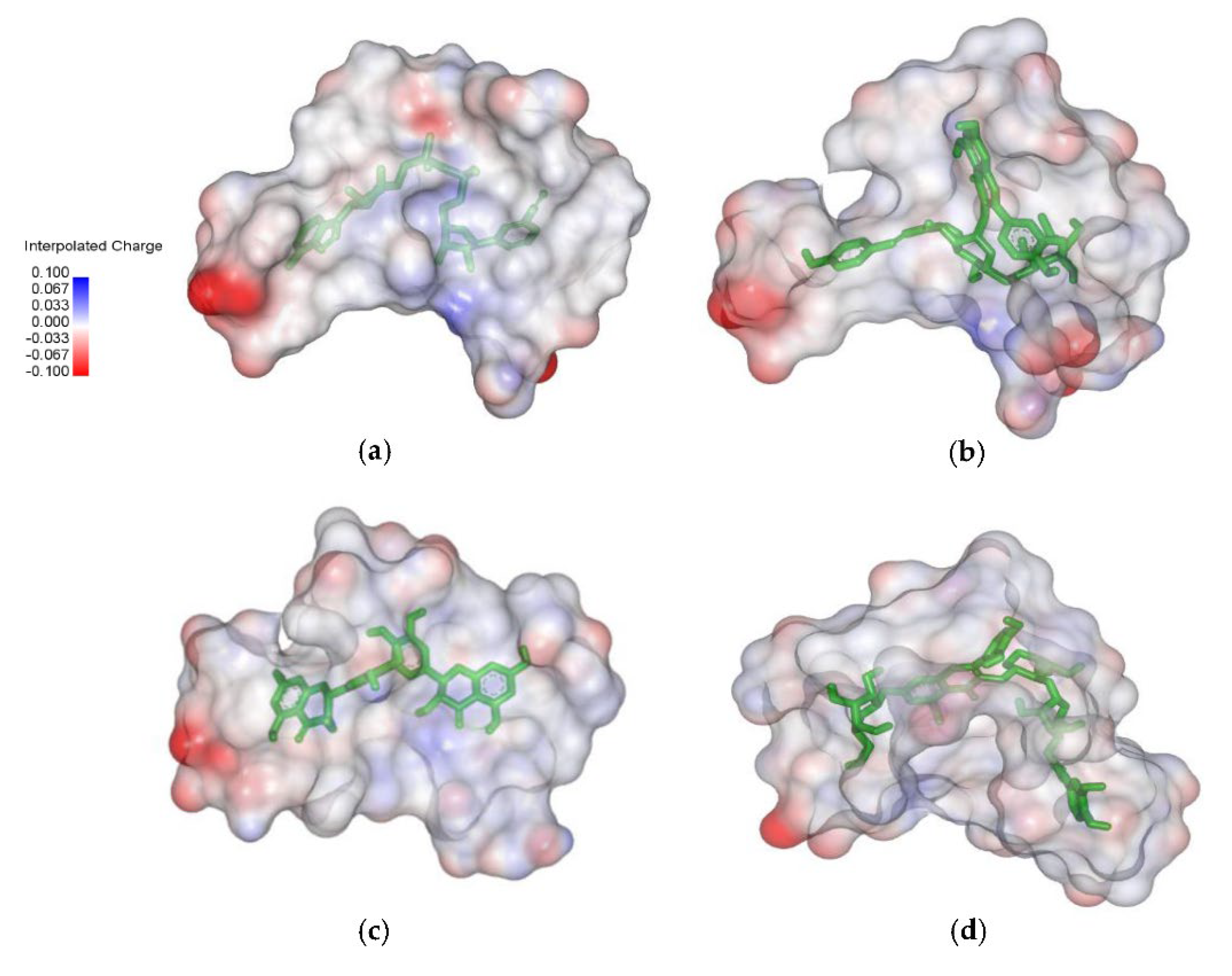{kind=link}
{kind=link}
{kind=link}
| Molecule | Van der Waals Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Solvation Energy (kJ/mol) | S.A.S.A. Energy (kJ/mol) | Bond Prediction Energy (kJ/mol) |
|---|---|---|---|---|---|
| NAD | −286.465 ± 21.372 | −147.213 ± 25.396 | 372.266 ± 33.929 | −26.158 ± 1.823 | −87.570 ± 40.255 |
| C00006532 | −274.790 ± 18.218 | −52.562 ± 26.593 | 257.072 ± 38.295 | −26.968 ± 1.197 | −97.248 ± 24.281 |
| C00013874 | −355.187 ± 20.678 | −124.388 ± 21.176 | 362.838 ± 29.135 | −31.914 ± 1.180 | −148.651 ± 23.604 |
| C00013887 | −335.165 ± 35.626 | −135.347 ± 56.100 | 392.283 ± 45.968 | −36.080 ± 1.507 | −114.310 ± 29.345 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pitaloka, D.A.E.; Ramadhan, D.S.F.; Arfan; Chaidir, L.; Fakih, T.M. Docking-Based Virtual Screening and Molecular Dynamics Simulations of Quercetin Analogs as Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors of Mycobacterium tuberculosis. Sci. Pharm. 2021, 89, 20. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm89020020
Pitaloka DAE, Ramadhan DSF, Arfan, Chaidir L, Fakih TM. Docking-Based Virtual Screening and Molecular Dynamics Simulations of Quercetin Analogs as Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors of Mycobacterium tuberculosis. Scientia Pharmaceutica. 2021; 89(2):20. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm89020020
Chicago/Turabian StylePitaloka, Dian Ayu Eka, Dwi Syah Fitra Ramadhan, Arfan, Lidya Chaidir, and Taufik Muhammad Fakih. 2021. "Docking-Based Virtual Screening and Molecular Dynamics Simulations of Quercetin Analogs as Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors of Mycobacterium tuberculosis" Scientia Pharmaceutica 89, no. 2: 20. https://0-doi-org.brum.beds.ac.uk/10.3390/scipharm89020020