
Molecular Mechanisms of the SLC13A5 Gene Transcription


Department of Pharmaceutical Sciences, University of Maryland School of Pharmacy, 20 Penn Street, Baltimore, MD 21201, USA
*
Author to whom correspondence should be addressed.
Metabolites 2021, 11(10), 706; https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11100706
Submission received: 9 September 2021
/
Revised: 12 October 2021
/
Accepted: 13 October 2021
/
Published: 15 October 2021
(This article belongs to the Special Issue I'm Not Dead Yet in Metabolic Regulation)
Abstract
:Citrate is a crucial energy sensor that plays a central role in cellular metabolic homeostasis. The solute carrier family 13 member 5 (SLC13A5), a sodium-coupled citrate transporter highly expressed in the mammalian liver with relatively low levels in the testis and brain, imports citrate from extracellular spaces into the cells. The perturbation of SLC13A5 expression and/or activity is associated with non-alcoholic fatty liver disease, obesity, insulin resistance, cell proliferation, and early infantile epileptic encephalopathy. SLC13A5 has been proposed as a promising therapeutic target for the treatment of these metabolic disorders. In the liver, the inductive expression of SLC13A5 has been linked to several xenobiotic receptors such as the pregnane X receptor and the aryl hydrocarbon receptor as well as certain hormonal and nutritional stimuli. Nevertheless, in comparison to the heightened interest in understanding the biological function and clinical relevance of SLC13A5, studies focusing on the regulatory mechanisms of SLC13A5 expression are relatively limited. In this review, we discuss the current advances in our understanding of the molecular mechanisms by which the expression of SLC13A5 is regulated. We expect this review will provide greater insights into the regulation of the SLC13A5 gene transcription and the signaling pathways involved therein.
1. Introduction
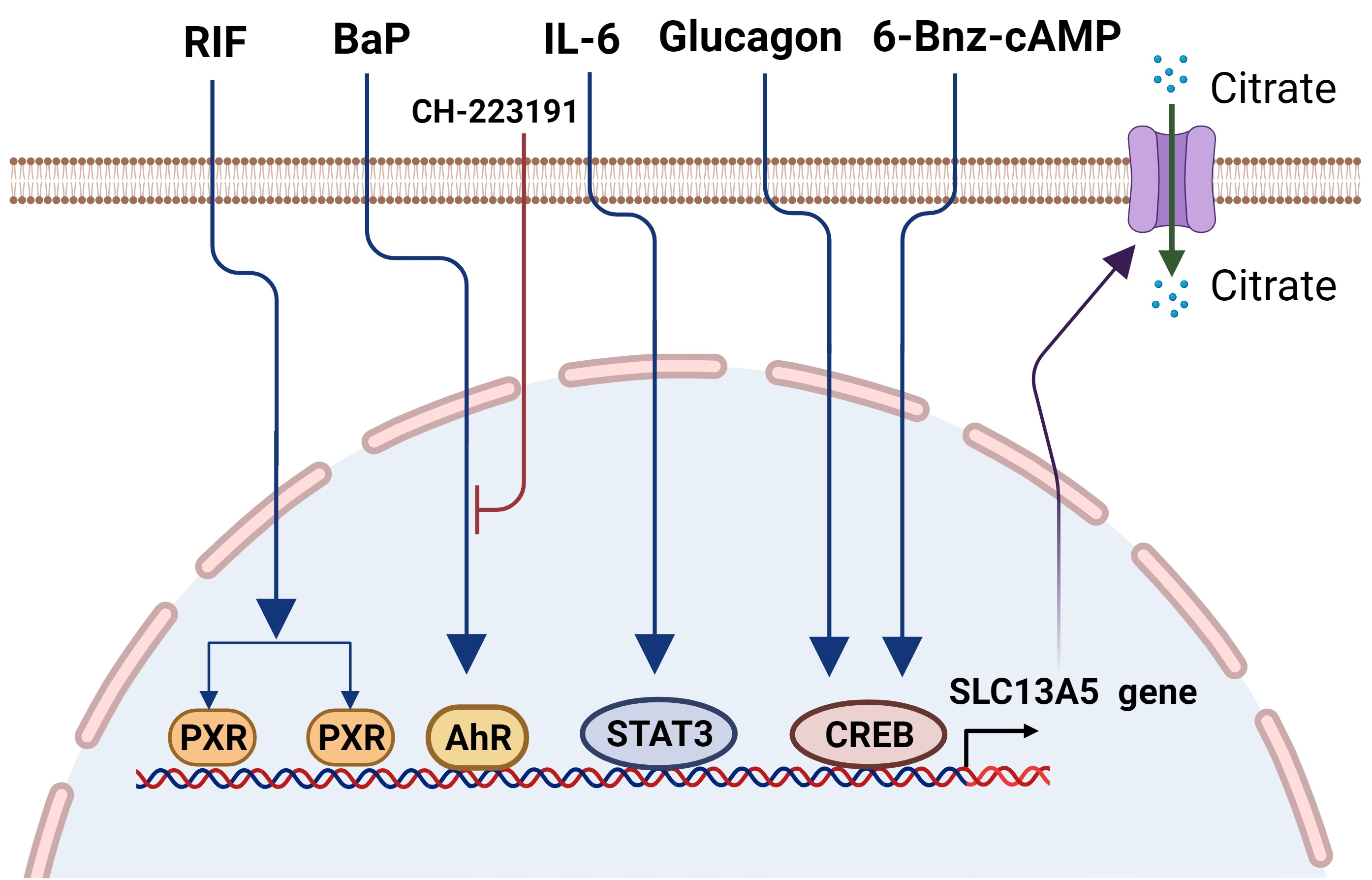
The solute carrier family 13 member 5 (SLC13A5), also known as the Na+/citrate cotransporter (NaCT), is a member of the sodium dicarboxylate/sulfate cotransporter family. It recognizes and transports various dicarboxylate and tricarboxylate intermediates of the tricarboxylic acid (TCA) cycle with citrate as the preferred substrate [1,2]. Intracellular citrate functions as an energy sensor by influencing glycolysis, the TCA cycle, gluconeogenesis, and fatty acid synthesis, which are pivotal for the energy homeostasis of the cells [3,4]. The level of the cytosolic citrate is well-maintained by endogenous mitochondrial biosynthesis and exogenous uptake from the circulation. Mitochondrial citrate generated from the TCA cycle is transported outside the mitochondria by the citrate carrier (CIC), which is encoded by the SLC25A1 gene [5]. Alternatively, cytosolic citrate can be imported from extracellular spaces via SLC13A5/NaCT expressed on the cell plasma membrane [6]. Cytosolic citrate is subsequently cleaved into acetyl-CoA and oxaloacetate by ATP citrate lyase (ACLY) [7]. Both acetyl-CoA and oxaloacetate are precursors of multiple anabolic reactions important for the de novo biosynthesis of fatty acids and steroids that are the building blocks for new cells [8]. On the other hand, elevated cytosolic citrate can repress glycolysis and the TCA cycle by inhibiting phosphofructokinase 1 (PFK1), PFK2, and pyruvate kinase (PK), key enzymes in the glycolysis pathway (Figure 1) [3,9]. In humans, the normal blood concentration of citrate is within the range of ~100–150 µM, which is several times higher than that of all other TCA cycle intermediates combined [10]. Thus, SLC13A5, being the primary uptake transporter of citrate, may play a key physiologic role in the generation of metabolic energy by facilitating the utilization of the circulating citrate. Specifically, in comparison to the ubiquitous biosynthesis of mitochondrial citrate and the broad expression of CIC/SLC25A1, SLC13A5 is preferentially expressed in tissues such as the liver, testis, and brain, which would influence physiological and pathophysiological conditions such as lipid metabolism, gluconeogenesis, obesity, type 2 diabetes, and epilepsy in an organ-specific pattern [1,11].
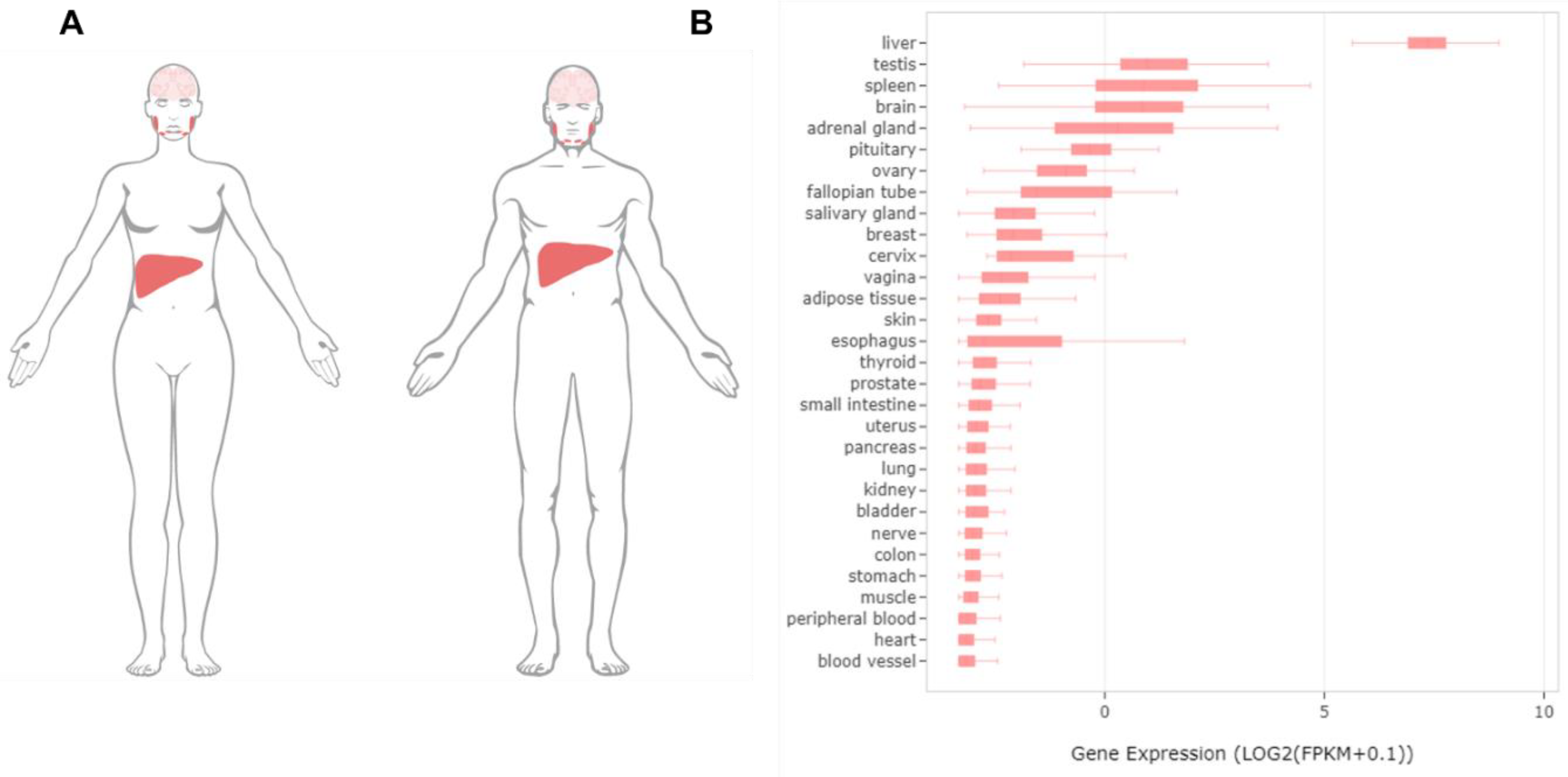
The cloning of the mammalian SLC13A5 was triggered directly by its structural and functional similarity with a Drosophila gene termed INDY, an acronym for “I’m Not Dead Yet”. The name of INDY was given by Rogina et al. based on the finding that the mutation of INDY was associated with a significant extension of the lifespan of fruit flies without affecting their energy intake [12]. Such a beneficial effect on the lifespan was also observed in Caenorhabditis elegans when the expression of an Na+-coupled citrate transporter (the C. elegans version of INDY) was repressed [13]. The subsequent structural-based functional postulation and experimental validation established INDY as a transporter protein that has a substrate preference for tricarboxylate citrate rather than for dicarboxylates [2]. SLC13A5/NaCT, the mammalian ortholog of Drosophila INDY (mINDY), was initially identified by screening a rat brain cDNA library with an EST (established sequence tag) probe, which led to the cloning of the full-length cDNA of the transporter as the newest member of the SLC13A family [2]. Subsequent studies revealed that, in humans, SLC13A5 mRNA is predominantly expressed in the liver followed by the testis, brain, spleen, bone marrow, and adrenal glands at much lower levels [14,15,16] (Figure 2). The human SLC13A5 gene locates in chromosome 17 with a size of approximately 30 kb containing 12 exons, exhibiting a preferential uptake of tricarboxylate citrate that differs significantly from that of its sister transporters SLC13A2 (NaDC1) and SLC13A3 (NaDC3) [1,17]. In line with its predominance in the liver, a high expression of SLC13A5 has been found primarily localized in the sinusoidal membrane of hepatocytes, echoing its capacity of uptake citrate from the blood circulation [18,19]. Accumulating evidence has revealed that SLC13A5 plays an important role in controlling the hepatic citrate level and is linked to various metabolic disorders. The pharmacological inhibition and genetic silencing of SLC13A5 in the liver was reported to reduce hepatic lipid accumulation, improve hepatic insulin sensitivity, and repress liver tumor cell proliferation [20,21,22]. SLC13A5 knockout (−/−) mice were protected from high-fat diet (HFD)-induced obesity, a fatty liver, and insulin resistance [15,23]. On the other hand, the induction of SLC13A5 expressions in rat and human primary hepatocytes by benzo[a]pyrene (BaP) and rifampicin (RIF), respectively, resulted in an increased citrate uptake and/or hepatic lipid accumulation [24,25,26]. Notably, in addition to the perturbation of the SLC13A5 expression by xenobiotic and endobiotic chemicals, inherited genetic variants of SLC13A5 (particularly non-synonymous single nucleotide variants (SNVs) in the coding region) can directly affect the structure, activity, and expression of this transporter. To date, numerous naturally occurring mutations have been identified in the SLC13A5 gene [27,28,29]. A few of these variants are associated with the disruption of the primary and secondary structure of the transporter, leading to protein internalization from the plasma membrane and a loss of the uptake function [29,30]. Most recently, human SLC13A5 protein structures in a complex with citrate or PF-06649298 (a known SLC13A5 inhibitor) were determined by Sauer et al., which provided structural insights for potential drug developments and the understanding of how various mutations affect the structure activity relationship of SLC13A5 [27].
Collectively, SLC13A5 has been established as a key transmembrane protein regulating cytosolic citrate levels in several energy-sensitive organs such as the liver and brain. The alteration of the SLC13A5 expression and function in these tissues is closely associated with obesity, type 2 diabetes, non-alcoholic fatty liver disease (NAFLD), inflammation, cancer, and neurological disorders. The purpose of this review is to highlight the recent progress in our understanding of the molecular mechanisms underlying the genetic and epigenetic regulation of the SLC13A5 gene expression in a tissue-specific manner, with a specific focus centering on the hepatic regulation of the SLC13A5 transcription.
2. Upregulation of the SLC13A5 Expression
As an uptake transporter sensing intracellular citrate levels, the expression of SLC13A5 can be altered in response to various metabolic stresses and chemical challenges. An increased expression of SLC13A5 in the liver has been observed in patients with obesity, NAFLD, and type 2 diabetes as well as in cultured primary hepatocytes treated with glucagon, BaP, or RIF. Our understanding of the mechanisms underlying SLC13A5 upregulation has advanced significantly in the past ten years or so.
2.1. PXR in the SLC13A5 Transcription
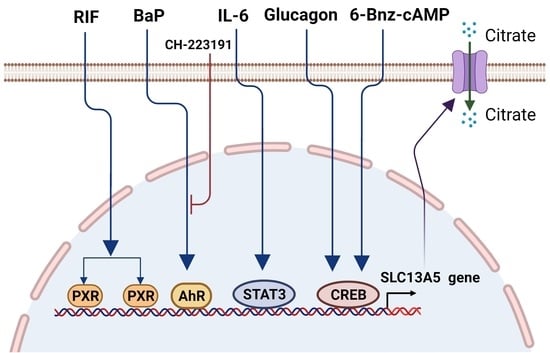
The pregnane X receptor (PXR, NR1I2), also known as the steroid X receptor (SXR), has been characterized as a master regulator of the expression of numerous phase I and phase II drug-metabolizing enzymes as well as drug transporters [31,32]. The perturbation of the PXR expression and activity can alter the absorption, distribution, metabolism, and excretion (ADME) profile of many drugs and results in clinically significant drug–drug interactions [33,34,35,36]. Hence, the screening of the PXR activity of drug candidates has been a common practice in numerous pharmaceutical companies. In addition to a xenobiotic disposition, mounting evidence suggests that PXR coordinates various physiological and pathophysiological processes involving energy homeostasis, inflammation, and cell proliferation [37,38,39,40,41,42,43]. Using wild-type, PXR knockout, and human PXR transgenic mice, several studies have revealed that the activation of PXR enhances lipogenesis whilst reducing lipid oxidation, resulting in a fatty liver phenotype [39,44,45]. In an attempt to decipher the transcriptional profile of the human PXR activation, a microarray analysis was conducted in human primary hepatocytes treated with RIF, the prototypical agonist of human PXR. Of note, SLC13A5 was identified as one of the top genes that was robustly induced by RIF along with other known PXR target genes such as CYP3A4. This finding was subsequently confirmed by RT-PCR and Western blotting analyses in hepatocytes obtained from multiple human liver donors [25]. Notably, the RIF-mediated induction of SLC13A5 was markedly repressed by sulforaphane, a selective PXR deactivator [46], or the knockdown of the human PXR expression via lentiviral small hairpin RNA. Recently, the induction of the mRNA expression of a mouse Slc13a5 was also reported in mice treated with the prototypical mouse PXR activator, pregnenolone 16α-carbonitrile (PCN), albeit to a lesser extent [47].
Mechanistically, two clusters of potential PXR binding sites were identified at approximately −22 and −1.7 kb upstream of the SLC13A5 transcription start site, as shown in Figure 2. Utilizing cell-based luciferase reporter assays, electrophoretic mobility shift assays (EMSA), and chromatin immunoprecipitation (ChIP) assays, two distal enhancers containing the core site of AGGTCA spaced by 4 nucleotides (DR4) were functionally characterized as key elements involving the PXR-mediated transcription of the SLC13A5 gene [25]. It is worth noting that whilst the induction of the SLC13A5 gene by RIF in human primary hepatocytes is robust, the activation of the reporter construct of SLC13A5 containing the new DR4s by RIF is relatively moderate. Most recently, a comprehensive in silico analysis of the −30kb upstream and the intron regions of SLC13A5 was carried out to search for potential PXR response elements, which has led to the identification of two DR4s located in the introns of SLC13A5 with high raw scores (unpublished data). Although more functional experiments are expected, an initial EMSA analysis indicated that the new intron DR4s exhibit a potent binding capacity to the PXR/RXR heterodimer. Additionally, we noticed that the RIF-mediated activation of the SLC13A5 reporter gene in HepG2 cells cotransfected with PXR was markedly lower than that in human primary hepatocytes, which contained a full spectrum of liver-enriched transcription factors. Together, these findings suggest that additional novel response elements in the SLC13A5 gene, as well as hepatic transcription factors other than PXR, might collectively contribute to the optimal induction of SLC13A5 in the liver.
Phenobarbital (PB), although a prototypical activator of the constitutive androstane receptor (CAR, NR1I3) in rodents, is a dual activator of both human CAR and PXR, and can induce a large array of genes associated with drug metabolism and clearance [48,49]. Our recent results indicate that a PB treatment markedly induced the expression of SLC13A5 in both the mRNA and protein levels in human primary hepatocytes (unpublished data). Interestingly, the selective human CAR agonist, 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime (CITCO), did not induce SLC13A5 expression either in human primary hepatocytes (unpublished data) or in HepaRG cells based on the analysis of our published RNA-seq data (GSE71446) [48]. Further studies using specific inhibitors of hCAR and hPXR as well as the HepaRG wild-type, CAR−/−, and PXR−/− cell lines revealed that PB induces the expression of SLC13A5 in liver cells via a PXR-mediated signaling pathway independent of CAR (unpublished data). Collectively, these findings indicate that although PXR and CAR share many target genes that encode drug-metabolizing enzymes and drug transporters, the transcription of SLC13A5 is upregulated by the activation of PXR but not CAR.
2.2. AhR in the SLC13A5 Transcription
The aryl hydrocarbon receptor (AhR) is a ligand-activated transcription factor that belongs to the basic helix-loop-helix PER-ARNT-SIM (PAS) gene family [50]. Inactivated AhR is expressed primarily in the cytoplasm as a protein complex with several chaperones including two heat shock protein 90 (HSP90), a co-chaperone p23, and a XAP-2 molecule [51,52]. The activation of AhR involves ligand binding, nuclear translocation, and interaction with the promoter of the target genes after forming a heterodimer with its partner, AhR nuclear translocator (ARNT) [36,53]. As with PXR and CAR, AhR has been referred to as an important xenobiotic sensor dictating the inductive expression of many drug-metabolizing enzymes and transporters including CYP1A1, CYP1A2, CYP1B1, UGT1A1, UGT1A3, UGT1A4, UGT1A6, and BCRP [54,55,56,57,58,59,60]. The key biological functions of AhR also include the regulation of energy metabolism and immune/inflammatory responses [61,62]. The activation of AhR was reported to induce spontaneous hepatic steatosis characterized by the accumulation of triglycerides likely via the upregulation of fatty acid uptake transporters whilst suppressing fatty acid oxidation and export [63]. A strong correlation between an exposure to AhR-activating chemicals such as polychlorinated biphenyls (PCBs) and increased metabolic disorders has been experimentally established [64,65]. At around the same time that PXR was recognized as a modulator of SLC13A5 expression, Neuschafer-Rube et al. reported that the mRNA expression of SLC13A5 was induced by a number of AhR activators including BaP, 2,3,7,8-tetrachlorodibenzodioxin (TCDD), and 3-methylcholanthrene (3MC) in cultured rat primary hepatocytes with BaP demonstrating the most robust induction (8-fold) [24]. The BaP-mediated induction of SLC13A5 was markedly attenuated by CH223191, a known AhR antagonist. A putative AhR binding site in the promoter of SLC13A5 was subsequently identified and evaluated as important for AhR/ARNT binding and the transactivation of the SLC13A5 gene (Figure 3). Functionally, the activation of AhR signaling by BaP increased the hepatic citrate uptake and resulted in an increased incorporation of radioactively-labelled citrate into lipids [24].
It is worth mentioning that the magnitude of SLC13A5 induction in rat primary hepatocytes by BaP (8-fold) at 20 µM is significantly higher than the approximately 2-fold increase achieved by the treatment of either TCDD (10 nM) or 3MC (1 µM), two prototypical AhR agonists. It is well-known that TCDD and 3MC at the concentrations of 10 nM and 1 µM, respectively, elicit a robust activation of AhR and the induction of its prototypical target genes CYP1A1 and CYP1A2 [66,67]. The obviously moderate induction of SLC13A5 achieved by TCDD and 3MC in comparison to BaP suggests the potential involvement of molecular mechanisms other than AhR. Indeed, additional experiments demonstrated a dose-dependent induction of SLC13A5 by BaP through concentrations up to 100 µM. Higher concentrations of BaP at and above 20 µM have previously been indicated to be toxic to hepatocytes [68,69]. Whether BaP causes cellular toxic responses and also contributes to its upregulation of SLC13A5 remains elusive.
2.3. CREB in the SLC13A5 Transcription
The cAMP-responsive element-binding protein (CREB) is a member of the basic leucine zipper (bZIP) class of transcription factors, regulating a variety of genes associated with glucose sensing, lipid metabolism, and fibrosis in response to the elevated second messenger cAMP [70]. Phosphorylated CREB binds to the cAMP response element (CRE) in the promoter region of its target genes, provoking the glucagon-PKA-cAMP axis as one of the primary CREB-activating signaling cascades [71].
In addition to a xenobiotic stimulation, an endogenous hormonal and/or nutritional status has been linked to the expression of SLC13A5 in mammals. In a cohort of human liver tissue samples, von Loeffelholz et al. found that the hepatic expression of SLC13A5 was positively associated with body mass index (BMI), waist circumference, body fat, and a histologically assessed liver fat content [26]. An experimental perturbation of SLC13A5 expression was also observed in mice fed with a HFD or under fasting conditions. In rat primary hepatocytes, the physiological concentration of glucagon (10 nM) significantly induced the mRNA expression of SLC13A5 and increased the hepatic uptake of [14C]-citrate [72]. It is well-known that glucagon interacts with its cell-surface receptor to increase the intracellular cAMP levels, which can in turn phosphorylate and activate the transcription factor CREB [73]. Subsequent mechanistic studies uncovered a functional CRE core site located near -324 in the promoter of SLC13A5; the mutation of the core sequence disrupted the CREB binding and abolished the glucagon-dependent induction of the SLC13A5 luciferase reporter activity. Notably, the expression of SLC13A5 was also induced in the liver of overnight-fasting and type 2 diabetic rats where both conditions elevated the CREB activity [72,74]. Of clinical importance, these hormonally and/or nutritionally enhanced uptakes of citrate via SLC13A5 upregulation can be converted by ACLY to acetyl-CoA and oxaloacetate, critical steps influencing lipogenesis and gluconeogenesis in the liver (Figure 1). Glucagon treatment in rat primary hepatocytes increased the incorporation of [14C]-labelled citrate into newly synthesized glucose [72].
Recently, Kopel et al. found that the anti-diabetic drug metformin suppresses the expression of SLC13A5 in HepG2 cells [75], albeit at a concentration (10 mM) that is several magnitudes higher than its pharmacological level (~40 µM) [76]. The signaling pathway for the metformin-mediated repression of the SLC13A5 gene is linked to its activation of AMP-activated protein kinase (AMPK), which in turn inhibits the mammalian target of rapamycin complex 1 (mTORC1) and CREB. The involvement of the AMPK pathway was further evidenced with a similar observation when the cells were treated with AICAR, another AMPK activator. Together, these results suggest that the activation of CREB may represent a common mechanism shared by both hormonal and nutritional stimuli in the upregulation of a hepatic SLC13A5 expression.
2.4. STAT3 in the SLC13A5 Expression
Cytosolic citrate is a central metabolite at the crossroads of energy homeostasis, connecting carbohydrate catabolism, lipogenesis, and gluconeogenesis, which has recently been shown to be enhanced by interleukin-6 (IL-6) [77]. IL-6 is a multifunctional cytokine that not only regulates immune and inflammatory responses but also affects hematopoiesis, metabolism, and organ development [78]. It is known that IL-6 levels are increased in obesity, type 2 diabetes, NAFLD, and several metabolic states that are associated with an elevated SLC13A5 expression [26,79,80,81].
Combining in vitro cell cultures with in vivo genetically modified mouse models and human patient samples, von Loeffelholz et al. provided experimental evidence demonstrating SLC13A5 as a target gene of a signal transducer and activator of transcription 3 (STAT3) in response to IL-6 stimulation [26]. Both the treatment of human primary hepatocytes with IL-6 and the intravenous administration of IL-6 in mice markedly increased the hepatic SLC13A5 mRNA levels. In contrast, blocking the IL-6 receptor (IL-6R) using a monoclonal antibody (tocilizumab) or through a mouse liver-specific IL-6R knockout model abolished the IL-6-mediated induction of SLC13A5. Such a correlation was also observed in NAFLD patients where SLC13A5 expression was 2-fold higher in patients with plasmid IL-6 levels above 4.81 pg/mL compared with patients with IL-6 below this cutoff line. Subsequently, two responsive elements containing the consensus sequence TT(N4-6)AA of STAT3 were identified close to −376 bp and −618 bp in the promoter of SLC13A5. IL-6 treatment efficiently activated the luciferase reporter constructs containing these two STAT3 binding sites. Nevertheless, the site-directed mutation of the STAT3 binding sites did not repress the luciferase activity stimulated by IL-6. Given the lack of evidence in this article to show that STAT3 can bind to these two responsive elements, it is possible that response elements other than these two may contribute to the observed activation. Clearly, more in-depth investigations are needed to define the mechanisms of the IL-6-mediated induction of SLC13A5.
3. Downregulation of the SLC13A5 Expression
The clinical significance of a decreasing SLC13A5 expression and activity is evident. However, many studies were conducted by artificially manipulating the SLC13A5 expression/activity via a gene knockdown, knockout, or chemical inhibition. In comparison with the upregulation of SLC13A5, much less is known regarding the mechanisms underlying its downregulation. The current literature suggests that genetic polymorphisms and the epigenetic modification of the SLC13A5 gene may contribute to its observed low expression in certain individuals.
3.1. Naturally Occurring Mutations of the SLC13A5 Gene
The effects of genetic variants on gene expressions and associated disease propensity have long been recognized. Accumulating evidence reveals that several non-synonymous mutations of the SLC13A5 gene are phenotypically linked to neonatal epilepsy, developmental delay, and tooth hypoplasia [28,30,82]. Early onset epileptic encephalopathy (EOEE) is a clinically and etiologically heterogeneous subgroup of epilepsy syndromes with a developmental delay and high mortality rate [28]. A genetic analysis on three families with an autosomal-recessive inheritance pattern of epileptic encephalopathy by Thevenon et al. resulted in the identification of three missense SNVs, c.680C > T [p.Thr227Met], c.655G > A [p.Gly219Arg], and c.1463T > C [p.Leu488Pro] that are associated with patients who have a seizure onset in the early days of life [83]. Subsequent studies have discovered more than 40 SNVs in the coding region of SLC13A5 that were termed “pathogenic SNVs”, as exemplified in Figure 4, contributing to the development of EOEE, tooth hypoplasia, and osteogenesis imperfecta [27,84].
Functionally, these variants in the coding region of the SLC13A5 gene often cause a decrease or loss of the citrate uptake capacity as the result of (1) an incompletely folded or unassembled protein that is internalized from the plasma membrane, or (2) the mutated protein has a defected binding pocket preventing sodium and/or citrate from interacting with it [28]. Of importance, because energy production in the brain is heavily reliant on the citric acid cycle, the loss of SLC13A5 function/expression is expected to have a profound influence on the synthesis of neurotransmitters such as acetylcholine, glutamate, and GABA in the central nervous system.
Compared with the heightened focus on studying missense SNVs of SLC13A5, the investigation of genetic variants occurring in the non-coding regions of SLC13A5 (the upstream/downstream of 5′ and 3′ untranslated regions as well as introns) has not been reported to date. A bioinformatic analysis of the SLC13A5 mutations located in the non-coding regions from the Exome Aggregation Consortium (ExAC) dataset (https://gnomad.broadinstitute.org, accessed on 3 July 2021) revealed a total of 420 variants in the non-coding regions with 15 reaching a frequency > 1% in the population (Table 1). However, the phenotypical significance of these variants is unknown. Theoretically, variants in the non-coding regions, particularly in the promoter regions, are expected to influence the overall expression of the gene without affecting its protein structure [85,86,87]. Thus, it is highly likely that concurrent mutations in both the coding and non-coding regions of SLC13A5 may contribute cooperatively to the development of deleterious neurological phenotypes.
3.2. Epigenetic Regulation of SLC13A5
In addition to the genetic control of the gene regulation, epigenetic modifications can alter the DNA accessibility and chromatin structure, thereby regulating the gene expression and playing a pivotal role in the normal and pathophysiological development and differentiation [89]. Major epigenetic mechanisms include DNA methylation, histone modification, and regulatory non-coding RNAs such as microRNAs and circular RNAs [90].
DNA methylation at the C5 position of cytosine in CpG dinucleotides represents one of the central epigenetic mechanisms that suppresses the gene expression. A whole-genome integrative analysis of methylation and the gene expression profiles in glioblastoma and normal brain tissues reveals that SLC13A5 is one of the 13 genes with concordant CpG islands in their promoter regions and the hypermethylation of the CpG sites of SLC13A5 is inversely correlated to its low expression in glioblastomas [91]. In a separate report, Diaz et al. found that the SLC13A5 gene was hypermethylated and downregulated in the placenta of individuals born small-for-gestational-age (SGA) who have less adipose tissue and more insulin sensitivity than appropriate-for-gestational-age (AGA) infants [92]. Similar results were also observed in renal cancer cells where DNA hypermethylation on the CpG islands of the SLC13A5 gene was identified in a cluster of renal cancer patients with poor survival rates [93]. Together, these findings suggest that with multiple CpG islands located in the promoter region of the SLC13A5 gene, its expression is sensitive to the type of DNA methylation and epigenetic modulations. Tissue-specific DNA hypermethylation resulting in a low expression of SLC13A5 may be an additional molecular mechanism contributing to inheritable neurological and developmental disorders.
4. Conclusions and Perspectives
As a crucial energy sensor regulating cytosolic citrate levels, SLC13A5 plays crucial metabolic roles maintaining energy homeostasis in the liver, brain, and several other tissues. An abnormal expression and/or activity of SLC13A5 is associated with hepatic metabolic disorders, severe early onset epilepsy, and several other disease conditions. In the past decade, significant progress has been achieved in our understanding of the molecular mechanisms by which the expression of SLC13A5 is regulated. As summarized in Table 2, transcription factors including PXR, AhR, CREB, and STAT3 have been identified as positive regulators that can upregulate SLC13A5 transcription upon xenobiotic or endobiotic stimulation. Notably, these findings are based primarily on studies conducted using hepatocyte cultures in vitro or in the liver of rodents. Given that regulation of the gene expression is generally considered to be a tissue-specific event, whether SLC13A5 in extrahepatic tissues such as the brain would be regulated in the same manner is largely unknown. Thus, the identification of the tissue-specific inducers of SLC13A5, particularly in the brain and bone marrow, may hold a greater clinical importance. In contrast to upregulation, our knowledge regarding the repression of SLC13A5 transcription is limited. Intriguingly, the depletion of SLC13A5 in the liver has demonstrated several metabolic benefits. For instance, we have shown that the silencing of SLC13A5 expression leads to a reduction in the proliferation of HepG2 and Huh7 cells accompanied by decreases in de novo lipogenesis through the activation of the AMPK-mTOR signaling pathway [20]. Moreover, the knockdown of SLC13A5 promotes hepatic ketogenesis and cellular stress, which sensitizes the response of HepG2 cells to chemotherapeutic agents [94]. Thus, understanding the mechanisms for SLC13A5 downregulation and developing potent and selective SLC13A5 modulators warrants future investigation.
Several loss-of-function mutations of the SLC13A5 gene have been closely linked to the development of EOEE. Although these missense SNVs in the coding region are deemed to affect protein conformation and function, several studies have indicated a reduction of SLC13A5 expression in a few of these patients. Whether concurrent SNVs located in the non-coding region of SLC13A5 co-contribute to neural disorders remains to be investigated. It is evident now that the perturbation of the SLC13A5 expression and activity in the central nervous system vs. in the liver may have opposite clinical consequences. Although enhancing the SLC13A5 function in the brain is expected to benefit EOEE patients, repressing its hepatic expression and activity may represent a therapeutic strategy for several metabolic disorders. Therefore, an improved understanding of the signaling control of SLC13A5 transcription in a tissue-specific manner will eventually benefit the development of SLC13A5 modulators as potential drug candidates for SLC13A5-associated diseases in humans.
Author Contributions
Conceptualization, Z.L. and H.W.; methodology, Z.L.; software, Z.L.; validation, Z.L. and H.W.; formal analysis, Z.L.; investigation, Z.L. and H.W.; resources, H.W.; data curation, Z.L. and H.W.; writing-original draft preparation, Z.L.; writing-review and editing, H.W.; visualization, Z.L.; supervision, H.W.; project administration, Z.L.; funding acquisition, H.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by the National Institute of General Medical Sciences [R01GM121550] and the National Cancer Institute [R01CA262084].
Acknowledgments
We are grateful to members of the Wang laboratory for discussions and comments on the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Inoue, K.; Zhuang, L.; Ganapathy, V. Human Na+-coupled citrate transporter: Primary structure, genomic organization, and transport function. Biochem. Biophys. Res. Commun. 2002, 299, 465–471. [Google Scholar] [CrossRef]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, V.; Infantino, V. Citrate--new functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef]
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta 2012, 1825, 111–116. [Google Scholar] [CrossRef]
- Gnoni, G.V.; Priore, P.; Geelen, M.J.; Siculella, L. The mitochondrial citrate carrier: Metabolic role and regulation of its activity and expression. IUBMB Life 2009, 61, 987–994. [Google Scholar] [CrossRef]
- Rogers, R.P.; Rogina, B. The role of INDY in metabolism, health and longevity. Front. Genet. 2015, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chypre, M.; Zaidi, N.; Smans, K. ATP-citrate lyase: A mini-review. Biochem. Biophys. Res. Commun. 2012, 422, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Wang, C.; Xu, H.; Peng, G. Targeting citrate as a novel therapeutic strategy in cancer treatment. Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188332. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Plasma Citrate Homeostasis: How It Is Regulated; And Its Physiological and Clinical Implications. An Important, But Neglected, Relationship in Medicine. HSOA J. Hum. Endocrinol. 2016, 1. [Google Scholar] [CrossRef]
- Pajor, A.M. Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch. 2014, 466, 119–130. [Google Scholar] [CrossRef]
- Rogina, B.; Reenan, R.A.; Nilsen, S.P.; Helfand, S.L. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 2000, 290, 2137–2140. [Google Scholar] [CrossRef]
- Fei, Y.J.; Liu, J.C.; Inoue, K.; Zhuang, L.; Miyake, K.; Miyauchi, S.; Ganapathy, V. Relevance of NAC-2, an Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem. J. 2004, 379, 191–198. [Google Scholar] [CrossRef]
- Inoue, K.; Fei, Y.J.; Zhuang, L.; Gopal, E.; Miyauchi, S.; Ganapathy, V. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem. J. 2004, 378, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Birkenfeld, A.L.; Lee, H.Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Mantila Roosa, S.M.; Liu, Y.; Turner, C.H. Gene expression patterns in bone following mechanical loading. J. Bone Miner. Res. 2011, 26, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Willmes, D.M.; Kurzbach, A.; Henke, C.; Schumann, T.; Zahn, G.; Heifetz, A.; Jordan, J.; Helfand, S.L.; Birkenfeld, A.L. The longevity gene INDY (I’m Not Dead Yet) in metabolic control: Potential as pharmacological target. Pharmacol. Ther. 2018, 185, 1–11. [Google Scholar] [CrossRef]
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Srinivas, S.R.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G402–G408. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma Membrane Na(+)-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules 2017, 22, 378. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, D.; Choi, E.Y.; Lapidus, R.; Zhang, L.; Huang, S.M.; Shapiro, P.; Wang, H. Silencing of solute carrier family 13 member 5 disrupts energy homeostasis and inhibits proliferation of human hepatocarcinoma cells. J. Biol. Chem. 2017, 292, 13890–13901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huard, K.; Brown, J.; Jones, J.C.; Cabral, S.; Futatsugi, K.; Gorgoglione, M.; Lanba, A.; Vera, N.B.; Zhu, Y.; Yan, Q.; et al. Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci. Rep. 2015, 5, 17391. [Google Scholar] [CrossRef] [Green Version]
- Brachs, S.; Winkel, A.F.; Tang, H.; Birkenfeld, A.L.; Brunner, B.; Jahn-Hofmann, K.; Margerie, D.; Ruetten, H.; Schmoll, D.; Spranger, J. Inhibition of citrate cotransporter Slc13a5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol. Metab. 2016, 5, 1072–1082. [Google Scholar] [CrossRef]
- Pesta, D.H.; Perry, R.J.; Guebre-Egziabher, F.; Zhang, D.; Jurczak, M.; Fischer-Rosinsky, A.; Daniels, M.A.; Willmes, D.M.; Bhanot, S.; Bornstein, S.R.; et al. Prevention of diet-induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5). Aging 2015, 7, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Neuschafer-Rube, F.; Schraplau, A.; Schewe, B.; Lieske, S.; Krutzfeldt, J.M.; Ringel, S.; Henkel, J.; Birkenfeld, A.L.; Puschel, G.P. Arylhydrocarbon receptor-dependent mIndy (Slc13a5) induction as possible contributor to benzo[a]pyrene-induced lipid accumulation in hepatocytes. Toxicology 2015, 337, 1–9. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Von Loeffelholz, C.; Lieske, S.; Neuschafer-Rube, F.; Willmes, D.M.; Raschzok, N.; Sauer, I.M.; Konig, J.; Fromm, M.F.; Horn, P.; Chatzigeorgiou, A.; et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 2017, 66, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Sauer, D.B.; Song, J.; Wang, B.; Hilton, J.K.; Karpowich, N.K.; Mindell, J.A.; Rice, W.J.; Wang, D.N. Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 2021, 591, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Hardies, K.; de Kovel, C.G.; Weckhuysen, S.; Asselbergh, B.; Geuens, T.; Deconinck, T.; Azmi, A.; May, P.; Brilstra, E.; Becker, F.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain A J. Neurol. 2015, 138, 3238–3250. [Google Scholar] [CrossRef] [Green Version]
- Klotz, J.; Porter, B.E.; Colas, C.; Schlessinger, A.; Pajor, A.M. Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 2016, 22. [Google Scholar] [CrossRef] [PubMed]
- Selch, S.; Chafai, A.; Sticht, H.; Birkenfeld, A.L.; Fromm, M.F.; Konig, J. Analysis of naturally occurring mutations in the human uptake transporter NaCT important for bone and brain development and energy metabolism. Sci. Rep. 2018, 8, 11330. [Google Scholar] [CrossRef] [PubMed]
- Sueyoshi, T.; Negishi, M. Phenobarbital response elements of cytochrome P450 genes and nuclear receptors. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Moore, J.T.; Wade, L.; Staudinger, J.L.; Watson, M.A.; Jones, S.A.; McKee, D.D.; Oliver, B.B.; Willson, T.M.; Zetterstrom, R.H.; et al. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell 1998, 92, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Willson, T.M. Regulation of xenobiotic and bile acid metabolism by the nuclear pregnane X receptor. J. Lipid Res. 2002, 43, 359–364. [Google Scholar] [CrossRef]
- di Masi, A.; De Marinis, E.; Ascenzi, P.; Marino, M. Nuclear receptors CAR and PXR: Molecular, functional, and biomedical aspects. Mol. Asp. Med. 2009, 30, 297–343. [Google Scholar] [CrossRef]
- Tolson, A.H.; Wang, H. Regulation of drug-metabolizing enzymes by xenobiotic receptors: PXR and CAR. Adv. Drug Deliv. Rev. 2010, 62, 1238–1249. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis. Gastroenterology 2008, 134, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Gao, J.; Xie, W. PXR and CAR in energy metabolism. Trends Endocrinol. Metab. 2009, 20, 273–279. [Google Scholar] [CrossRef]
- Nakamura, K.; Moore, R.; Negishi, M.; Sueyoshi, T. Nuclear pregnane X receptor cross-talk with FoxA2 to mediate drug-induced regulation of lipid metabolism in fasting mouse liver. J. Biol. Chem. 2007, 282, 9768–9776. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.; Pasquel, D.; Tyagi, R.K.; Mani, S. Acetylation of pregnane X receptor protein determines selective function independent of ligand activation. Biochem. Biophys. Res. Commun. 2011, 406, 371–376. [Google Scholar] [CrossRef] [Green Version]
- Staudinger, J.L.; Xu, C.; Biswas, A.; Mani, S. Post-translational modification of pregnane x receptor. Pharmacol. Res. 2011, 64, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.; Mani, S.; Redinbo, M.R.; Krasowski, M.D.; Li, H.; Ekins, S. Elucidating the ‘Jekyll and Hyde’ nature of PXR: The case for discovering antagonists or allosteric antagonists. Pharm. Res. 2009, 26, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Gao, J.; Xu, M.; Ren, S.; Stefanovic-Racic, M.; O’Doherty, R.M.; Xie, W. PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice. Diabetes 2013, 62, 1876–1887. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Zhai, Y.; Mu, Y.; Gong, H.; Uppal, H.; Toma, D.; Ren, S.; Evans, R.M.; Xie, W. A novel pregnane X receptor-mediated and sterol regulatory element-binding protein-independent lipogenic pathway. J. Biol. Chem. 2006, 281, 15013–15020. [Google Scholar] [CrossRef] [Green Version]
- Bitter, A.; Rummele, P.; Klein, K.; Kandel, B.A.; Rieger, J.K.; Nussler, A.K.; Zanger, U.M.; Trauner, M.; Schwab, M.; Burk, O. Pregnane X receptor activation and silencing promote steatosis of human hepatic cells by distinct lipogenic mechanisms. Arch. Toxicol. 2015, 89, 2089–2103. [Google Scholar] [CrossRef]
- Zhou, C.; Poulton, E.J.; Grun, F.; Bammler, T.K.; Blumberg, B.; Thummel, K.E.; Eaton, D.L. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol. Pharmacol. 2007, 71, 220–229. [Google Scholar] [CrossRef]
- Jiang, Y.; Yao, X.; Fan, S.; Gao, Y.; Zhang, H.; Huang, M.; Bi, H. Lipidomic profiling reveals triacylglycerol accumulation in the liver during pregnane X receptor activation-induced hepatomegaly. J. Pharm. Biomed. Anal. 2021, 195, 113851. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Mackowiak, B.; Brayman, T.G.; Mitchell, M.; Zhang, L.; Huang, S.M.; Wang, H. Genome-wide analysis of human constitutive androstane receptor (CAR) transcriptome in wild-type and CAR-knockout HepaRG cells. Biochem. Pharmacol. 2015, 98, 190–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Welch, M.A.; Li, Z.; Mackowiak, B.; Heyward, S.; Swaan, P.W.; Wang, H. Mechanistic Insights of Phenobarbital-Mediated Activation of Human but Not Mouse Pregnane X Receptor. Mol. Pharmacol. 2019, 96, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.A.; Gasiewicz, T.A.; Opanashuk, L.A. Aryl hydrocarbon receptor expression and activity in cerebellar granule neuroblasts: Implications for development and dioxin neurotoxicity. Toxicol. Sci. 2005, 83, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guyot, E.; Chevallier, A.; Barouki, R.; Coumoul, X. The AhR twist: Ligand-dependent AhR signaling and pharmaco-toxicological implications. Drug Discov. Today 2013, 18, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Kudo, I.; Hosaka, M.; Haga, A.; Tsuji, N.; Nagata, Y.; Okada, H.; Fukuda, K.; Kakizaki, Y.; Okamoto, T.; Grave, E.; et al. The regulation mechanisms of AhR by molecular chaperone complex. J. Biochem. 2018, 163, 223–232. [Google Scholar] [CrossRef]
- Beischlag, T.V.; Luis Morales, J.; Hollingshead, B.D.; Perdew, G.H. The aryl hydrocarbon receptor complex and the control of gene expression. Crit. Rev. Eukaryot Gene Expr. 2008, 18, 207–250. [Google Scholar] [CrossRef] [Green Version]
- Yueh, M.F.; Huang, Y.H.; Hiller, A.; Chen, S.; Nguyen, N.; Tukey, R.H. Involvement of the xenobiotic response element (XRE) in Ah receptor-mediated induction of human UDP-glucuronosyltransferase 1A1. J. Biol. Chem. 2003, 278, 15001–15006. [Google Scholar] [CrossRef] [Green Version]
- Shelby, M.K.; Cherrington, N.J.; Vansell, N.R.; Klaassen, C.D. Tissue mRNA expression of the rat UDP-glucuronosyltransferase gene family. Drug Metab. Dispos. 2003, 31, 326–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hankinson, O.; Brooks, B.A.; Weir-Brown, K.I.; Hoffman, E.C.; Johnson, B.S.; Nanthur, J.; Reyes, H.; Watson, A.J. Genetic and molecular analysis of the Ah receptor and of Cyp1a1 gene expression. Biochimie 1991, 73, 61–66. [Google Scholar] [CrossRef]
- Gu, Y.Z.; Hogenesch, J.B.; Bradfield, C.A. The PAS superfamily: Sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 2000, 40, 519–561. [Google Scholar] [CrossRef] [Green Version]
- Emi, Y.; Ikushiro, S.; Iyanagi, T. Xenobiotic responsive element-mediated transcriptional activation in the UDP-glucuronosyltransferase family 1 gene complex. J. Biol. Chem. 1996, 271, 3952–3958. [Google Scholar] [CrossRef] [Green Version]
- Munzel, P.A.; Schmohl, S.; Buckler, F.; Jaehrling, J.; Raschko, F.T.; Kohle, C.; Bock, K.W. Contribution of the Ah receptor to the phenolic antioxidant-mediated expression of human and rat UDP-glucuronosyltransferase UGT1A6 in Caco-2 and rat hepatoma 5L cells. Biochem. Pharmacol. 2003, 66, 841–847. [Google Scholar] [CrossRef]
- Ebert, B.; Seidel, A.; Lampen, A. Identification of BCRP as transporter of benzo[a]pyrene conjugates metabolically formed in Caco-2 cells and its induction by Ah-receptor agonists. Carcinogenesis 2005, 26, 1754–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Mackowiak, B.; Wang, H. Mechanisms of xenobiotic receptor activation: Direct vs. indirect. Biochim. Biophys. Acta 2016, 1859, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cave, M.; Appana, S.; Patel, M.; Falkner, K.C.; McClain, C.J.; Brock, G. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003-2004. Environ. Health Perspect. 2010, 118, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, Q.; Li, H.; Chen, N.; Qu, F.; Guo, J. Understanding the Multiple Effects of PCBs on Lipid Metabolism. Diabetes Metab. Syndr. Obes. 2020, 13, 3691–3702. [Google Scholar] [CrossRef] [PubMed]
- Okino, S.T.; Quattrochi, L.C.; Pookot, D.; Iwahashi, M.; Dahiya, R. A dioxin-responsive enhancer 3’ of the human CYP1A2 gene. Mol. Pharmacol. 2007, 72, 1457–1465. [Google Scholar] [CrossRef] [Green Version]
- Nakata, A.; Urano, D.; Fujii-Kuriyama, Y.; Mizuno, N.; Tago, K.; Itoh, H. G-protein signalling negatively regulates the stability of aryl hydrocarbon receptor. EMBO Rep. 2009, 10, 622–628. [Google Scholar] [CrossRef]
- Schraplau, A.; Schewe, B.; Neuschafer-Rube, F.; Ringel, S.; Neuber, C.; Kleuser, B.; Puschel, G.P. Enhanced thyroid hormone breakdown in hepatocytes by mutual induction of the constitutive androstane receptor (CAR, NR1I3) and arylhydrocarbon receptor by benzo[a]pyrene and phenobarbital. Toxicology 2015, 328, 21–28. [Google Scholar] [CrossRef]
- Li, Q.; Gao, C.; Deng, H.; Song, Q.; Yuan, L. Benzo[a]pyrene induces pyroptotic and autophagic death through inhibiting PI3K/Akt signaling pathway in HL-7702 human normal liver cells. J. Toxicol. Sci. 2019, 44, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Benchoula, K.; Parhar, I.S.; Madhavan, P.; Hwa, W.E. CREB nuclear transcription activity as a targeting factor in the treatment of diabetes and diabetes complications. Biochem. Pharmacol. 2021, 188, 114531. [Google Scholar] [CrossRef]
- Altarejos, J.Y.; Montminy, M. CREB and the CRTC co-activators: Sensors for hormonal and metabolic signals. Nat. Rev. Mol. Cell Biol. 2011, 12, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Neuschafer-Rube, F.; Lieske, S.; Kuna, M.; Henkel, J.; Perry, R.J.; Erion, D.M.; Pesta, D.; Willmes, D.M.; Brachs, S.; von Loeffelholz, C.; et al. The mammalian INDY homolog is induced by CREB in a rat model of type 2 diabetes. Diabetes 2014, 63, 1048–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jitrapakdee, S. Transcription factors and coactivators controlling nutrient and hormonal regulation of hepatic gluconeogenesis. Int. J. Biochem. Cell Biol. 2012, 44, 33–45. [Google Scholar] [CrossRef]
- Zhang, E.E.; Liu, Y.; Dentin, R.; Pongsawakul, P.Y.; Liu, A.C.; Hirota, T.; Nusinow, D.A.; Sun, X.; Landais, S.; Kodama, Y.; et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nat. Med. 2010, 16, 1152–1156. [Google Scholar] [CrossRef]
- Kopel, J.; Higuchi, K.; Ristic, B.; Sato, T.; Ramachandran, S.; Ganapathy, V. The Hepatic Plasma Membrane Citrate Transporter NaCT (SLC13A5) as a Molecular Target for Metformin. Sci. Rep. 2020, 10, 8536. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin action: Concentrations matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Perry, R.J.; Camporez, J.G.; Kursawe, R.; Titchenell, P.M.; Zhang, D.; Perry, C.J.; Jurczak, M.J.; Abudukadier, A.; Han, M.S.; Zhang, X.M.; et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell 2015, 160, 745–758. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [Green Version]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senn, J.J.; Klover, P.J.; Nowak, I.A.; Zimmers, T.A.; Koniaris, L.G.; Furlanetto, R.W.; Mooney, R.A. Suppressor of cytokine signaling-3 (SOCS-3), a potential mediator of interleukin-6-dependent insulin resistance in hepatocytes. J. Biol. Chem. 2003, 278, 13740–13746. [Google Scholar] [CrossRef] [Green Version]
- Mathis, D.; Shoelson, S.E. Immunometabolism: An emerging frontier. Nat. Rev. Immunol. 2011, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.Z.; Spelbrink, E.M.; Nye, K.L.; Hsu, E.R.; Porter, B.E. Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder. Child. Neurol. Open 2020, 7. [Google Scholar] [CrossRef]
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Juge, C.; Roubertie, A.; Heron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef]
- Matricardi, S.; De Liso, P.; Freri, E.; Costa, P.; Castellotti, B.; Magri, S.; Gellera, C.; Granata, T.; Musante, L.; Lesca, G.; et al. Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia 2020, 61, 2474–2485. [Google Scholar] [CrossRef]
- Spielmann, M.; Mundlos, S. Looking beyond the genes: The role of non-coding variants in human disease. Hum. Mol. Genet. 2016, 25, R157–R165. [Google Scholar] [CrossRef] [PubMed]
- Loehlin, D.W.; Ames, J.R.; Vaccaro, K.; Carroll, S.B. A major role for noncoding regulatory mutations in the evolution of enzyme activity. Proc. Natl. Acad. Sci. USA 2019, 116, 12383–12389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrich, W.D.; Hassan, H.E.; Wang, H. Insights into CYP2B6-mediated drug-drug interactions. Acta Pharm. Sin. B 2016, 6, 413–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Schiano, C.; Benincasa, G.; Franzese, M.; Della Mura, N.; Pane, K.; Salvatore, M.; Napoli, C. Epigenetic-sensitive pathways in personalized therapy of major cardiovascular diseases. Pharmacol. Ther. 2020, 210, 107514. [Google Scholar] [CrossRef]
- Etcheverry, A.; Aubry, M.; de Tayrac, M.; Vauleon, E.; Boniface, R.; Guenot, F.; Saikali, S.; Hamlat, A.; Riffaud, L.; Menei, P.; et al. DNA methylation in glioblastoma: Impact on gene expression and clinical outcome. BMC Genom. 2010, 11, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, M.; Garcia, C.; Sebastiani, G.; de Zegher, F.; Lopez-Bermejo, A.; Ibanez, L. Placental and Cord Blood Methylation of Genes Involved in Energy Homeostasis: Association with Fetal Growth and Neonatal Body Composition. Diabetes 2017, 66, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Arai, E.; Chiku, S.; Mori, T.; Gotoh, M.; Nakagawa, T.; Fujimoto, H.; Kanai, Y. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis 2012, 33, 1487–1493. [Google Scholar] [CrossRef]
- Hu, T.; Huang, W.; Li, Z.; Kane, M.A.; Zhang, L.; Huang, S.M.; Wang, H. Comparative proteomic analysis of SLC13A5 knockdown reveals elevated ketogenesis and enhanced cellular toxic response to chemotherapeutic agents in HepG2 cells. Toxicol. Appl. Pharmacol. 2020, 402, 115117. [Google Scholar] [CrossRef]
- Ilagan, Y.; Mamillapalli, R.; Goetz, T.G.; Kayani, J.; Taylor, H.S. Bisphenol-A exposure in utero programs a sexually dimorphic estrogenic state of hepatic metabolic gene expression. Reprod. Toxicol. 2017, 71, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Mosaoa, R.; Graham, G.T.; Kasprzyk-Pawelec, A.; Gadre, S.; Parasido, E.; Catalina-Rodriguez, O.; Foley, P.; Giaccone, G.; Cheema, A.; et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death Differ. 2020, 27, 2143–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic illustration of the cytosolic citrate balance and function. Citrate is synthesized in the mitochondria through the TCA cycle and transported to the cytosol via CIC (SLC25A1). Another source of cytosolic citrate is from extracellular spaces imported by SLC13A5/NaCT. Functionally, cytosolic citrate can (1) serve as a precursor for acetyl-CoA and play a key role in lipogenesis and histone acetylation; (2) suppress the glycolysis pathway via the inhibition of PFK1, PFK2, and PK, and (3) increase the ATP/ADP ratio and repress the AMPK signaling, affecting the cell proliferation. The schematic figures were created using BioRender (biorender.com, accessed on 4 October 2021).
Figure 1.
Schematic illustration of the cytosolic citrate balance and function. Citrate is synthesized in the mitochondria through the TCA cycle and transported to the cytosol via CIC (SLC25A1). Another source of cytosolic citrate is from extracellular spaces imported by SLC13A5/NaCT. Functionally, cytosolic citrate can (1) serve as a precursor for acetyl-CoA and play a key role in lipogenesis and histone acetylation; (2) suppress the glycolysis pathway via the inhibition of PFK1, PFK2, and PK, and (3) increase the ATP/ADP ratio and repress the AMPK signaling, affecting the cell proliferation. The schematic figures were created using BioRender (biorender.com, accessed on 4 October 2021).

Figure 2.
The distribution patterns of SLC13A5 in humans. (A) The global expression profiles of PXR in the human body (data available from https://www.proteinatlas.org/ENSG00000141485-SLC13A5/tissue#cbox, accessed on 14 July 2021). (B) The expression levels of SLC13A5 in different human tissues (generated via an Ingenuity Pathway Analysis (IPA); QIAGEN).
Figure 2.
The distribution patterns of SLC13A5 in humans. (A) The global expression profiles of PXR in the human body (data available from https://www.proteinatlas.org/ENSG00000141485-SLC13A5/tissue#cbox, accessed on 14 July 2021). (B) The expression levels of SLC13A5 in different human tissues (generated via an Ingenuity Pathway Analysis (IPA); QIAGEN).

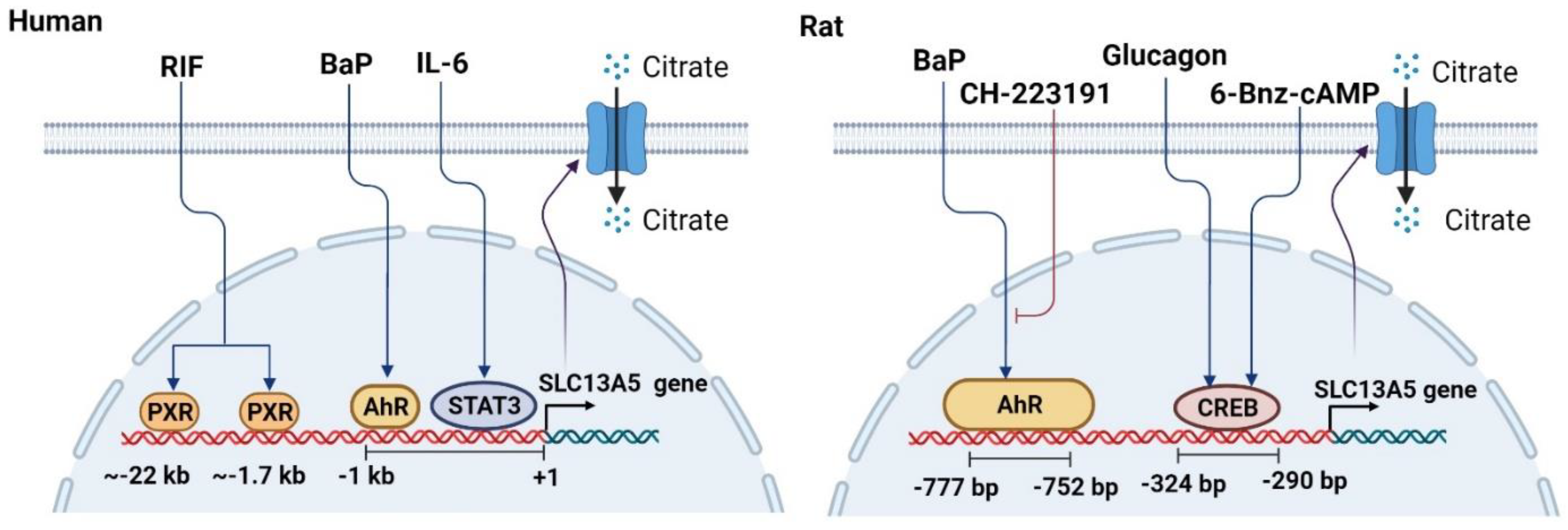
Figure 3.
Schematic illustration of the mechanisms underlying the transcriptional regulation of SLC13A5. Transcription factors including PXR, AhR, CREB, and STAT3 can positively regulate the transcription of SLC13A5 in the liver upon exposure to their activators such as RIF, BaP, glucagon, and IL-6, respectively. Specific response elements (motifs) that can bind to these transcription factors have been identified upstream of the SLC13A5 gene in human and/or rodents. CH-223191 is an antagonist of AhR.
Figure 3.
Schematic illustration of the mechanisms underlying the transcriptional regulation of SLC13A5. Transcription factors including PXR, AhR, CREB, and STAT3 can positively regulate the transcription of SLC13A5 in the liver upon exposure to their activators such as RIF, BaP, glucagon, and IL-6, respectively. Specific response elements (motifs) that can bind to these transcription factors have been identified upstream of the SLC13A5 gene in human and/or rodents. CH-223191 is an antagonist of AhR.

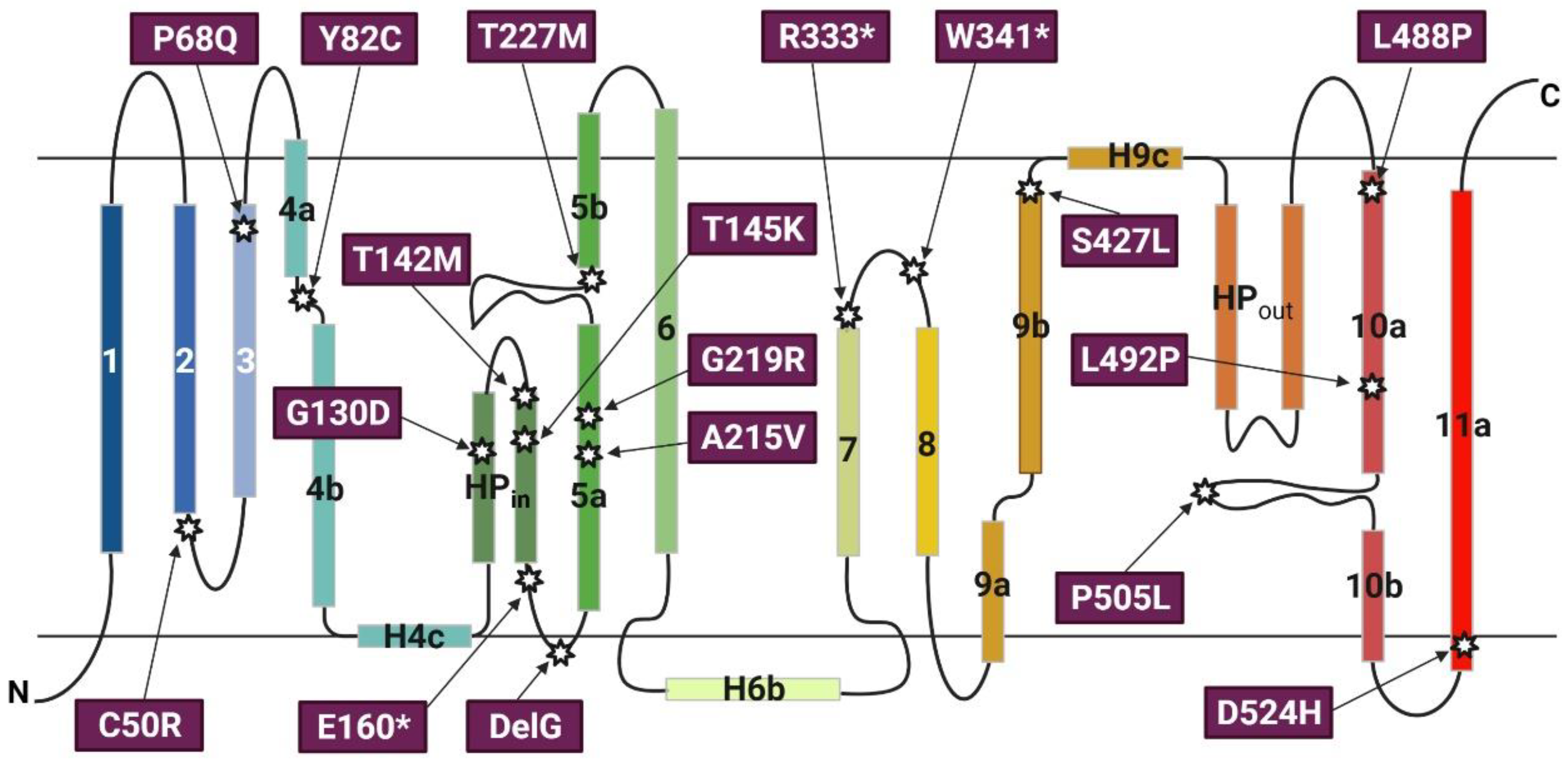
Figure 4.
Transmembrane topology of SLC13A5 and the localization of selected non-synonymous single nucleotide variants (SNVs). The transmembrane topology structure of SLC13A5 was adopted from Sauer et al. The 11 transmembrane helices are shown as numbered rectangles; the hairpin loop is labelled HP. The localization and amino acid replacement of selected SNVs previously reported as “pathogenic” are indicated. * Represents nonsense mutations.
Figure 4.
Transmembrane topology of SLC13A5 and the localization of selected non-synonymous single nucleotide variants (SNVs). The transmembrane topology structure of SLC13A5 was adopted from Sauer et al. The 11 transmembrane helices are shown as numbered rectangles; the hairpin loop is labelled HP. The localization and amino acid replacement of selected SNVs previously reported as “pathogenic” are indicated. * Represents nonsense mutations.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The variants of the SLC13A5 gene in the non-coding region (allele frequency > 1%) a.
| Chromosome | Position | rsIDs b | Reference | Alternate | Annotation | Allele Frequency (%) |
|---|---|---|---|---|---|---|
| 17 | 6616669 | rs62061545 | T | G | 5_prime_UTR_variant | 2.063 |
| 17 | 6616527 | rs62621831 | G | A | intron_variant | 8.467 |
| 17 | 6610536 | rs62061541 | A | G | intron_variant | 1.730 |
| 17 | 6610295 | rs144316437 | AAG | A | intron_variant | 4.250 |
| 17 | 6607435 | rs218678 | G | A | intron_variant | 9.240 |
| 17 | 6607389 | rs218677 | G | A | intron_variant | 2.729 |
| 17 | 6607150 | rs62061540 | G | A | intron_variant | 2.400 |
| 17 | 6599288 | rs77795940 | C | T | intron_variant | 3.760 |
| 17 | 6596497 | rs896810583 | A | AAC | intron_variant | 8.085 |
| 17 | 6594322 | rs4796540 | G | A | intron_variant | 23.338 |
| 17 | 6594291 | rs116929585 | C | T | intron_variant | 1.502 |
| 17 | 6593417 | rs218694 | A | G | intron_variant | 12.780 |
| 17 | 6593414 | rs934718226 | AC | A | intron_variant | 5.223 |
| 17 | 6591041 | rs78203589 | C | T | intron_variant | 1.335 |
| 17 | 6589495 | rs16956120 | A | G | 3_prime_UTR_variant | 1.073 |
a Gene location based on the National Center for Biotechnology Information (NCBI) Human Genome Assembly Build GRCh37/hg19. The data were collected from https://gnomad.broadinstitute.org, accessed on 3 July 2021 [88]. b Reference SNP cluster ID.
Table 2.
Transcription factors or xenobiotic chemicals that regulate SLC13A5 gene expression.
| Species | Tissue | Effect a | Reference | ||
|---|---|---|---|---|---|
| PXR | Human | Human primary hepatocytes | + | [25] | |
| AhR | Rat | Rat primary hepatocytes | + | [24] | |
| Transcription Factor | Mouse | Mouse primary hepatocytes | + | ||
| CREB | Rat | Rat primary hepatocytes | + | [72] | |
| STAT3 | Human | Human primary hepatocytes | + | [26] | |
| Glucagon | Rat | Rat primary hepatocytes | + | [72] | |
| Hormone or Cytokine | IL-6 | Human | Human primary hepatocytes | + | [26] |
| Mouse | Mouse liver | + | |||
| RIF | Human | Human primary hepatocytes | + | [25] | |
| TCDD | Rat | Rat primary hepatocytes | + | [24] | |
| 3MC | Rat | Rat primary hepatocytes | + | [24] | |
| BaP | Rat | Rat primary hepatocytes | + | [24] | |
| Mouse | Mouse primary hepatocytes | + | |||
| Chemicals or Others | Metformin | Human | HepG2 cells | − | [75] |
| AICAR | Human | HepG2 cells | − | [75] | |
| Phenobarbital | Rat | Rat primary hepatocytes | + | [24] | |
| Bisphenol-A | Mouse | Mouse liver | − | [95] | |
| CTPI-2 | Mouse | Mouse liver | + | [96] | |
| LPS | Human | Human non-parenchymal cells (including Kupffer cells) were co-cultivated with human primary hepatocytes | + | [26] | |
| 6-Bnz-cAMP | Rat | Rat primary hepatocytes | + | [72] | |
a +, induction of the SLC13A5 gene; −, repression of the SLC13A5 gene.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, Z.; Wang, H. Molecular Mechanisms of the SLC13A5 Gene Transcription. Metabolites 2021, 11, 706. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11100706
AMA Style
Li Z, Wang H. Molecular Mechanisms of the SLC13A5 Gene Transcription. Metabolites. 2021; 11(10):706. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11100706
Chicago/Turabian StyleLi, Zhihui, and Hongbing Wang. 2021. "Molecular Mechanisms of the SLC13A5 Gene Transcription" Metabolites 11, no. 10: 706. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11100706
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.