
Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry

 , ,
, ,
Abstract
:1. Introduction
2. Results
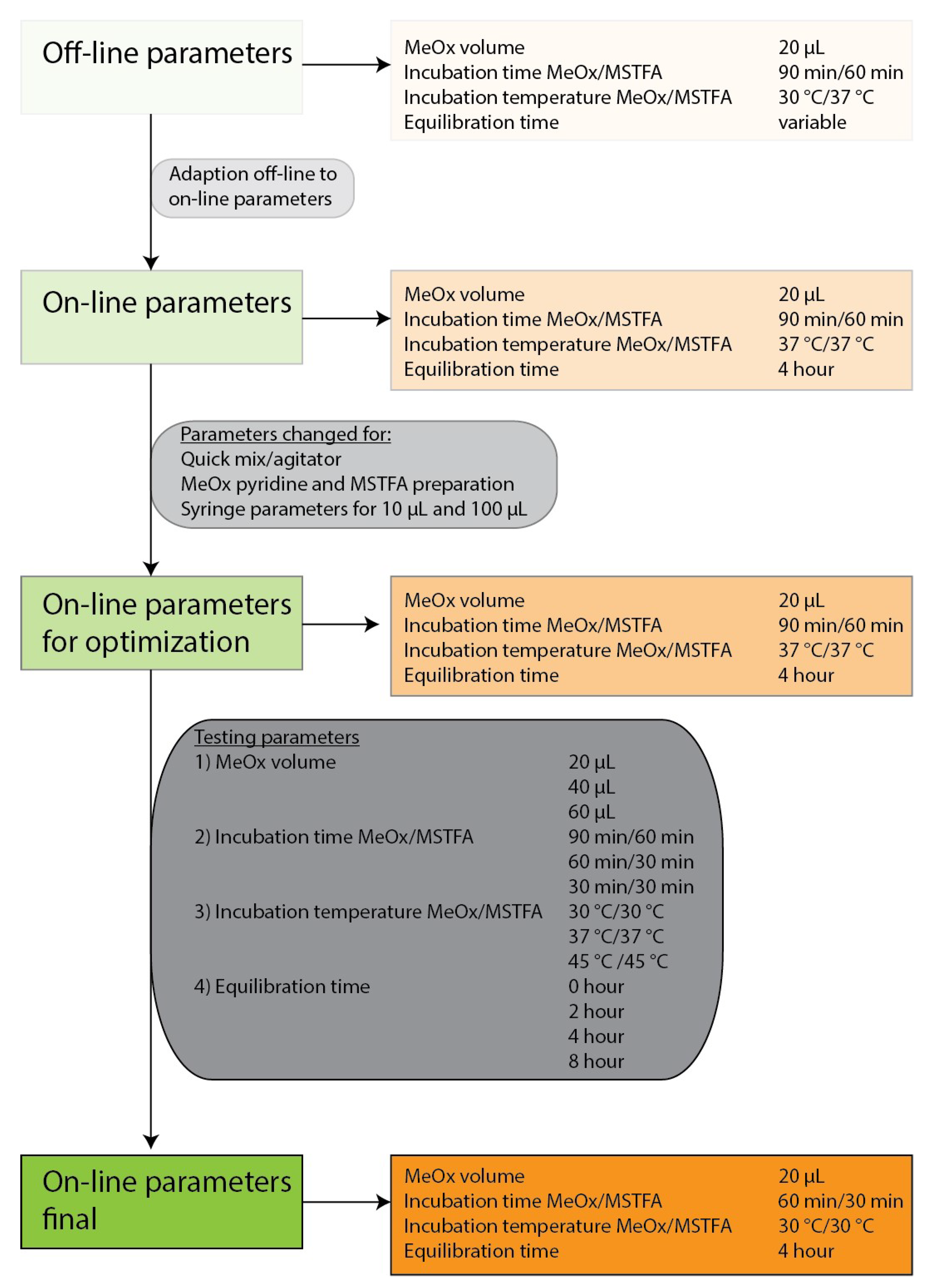
2.1. Optimization of the Derivatization Parameters
2.1.1. MeOx Volume
2.1.2. Incubation Time
2.1.3. Incubation Temperature
2.1.4. Equilibration Time
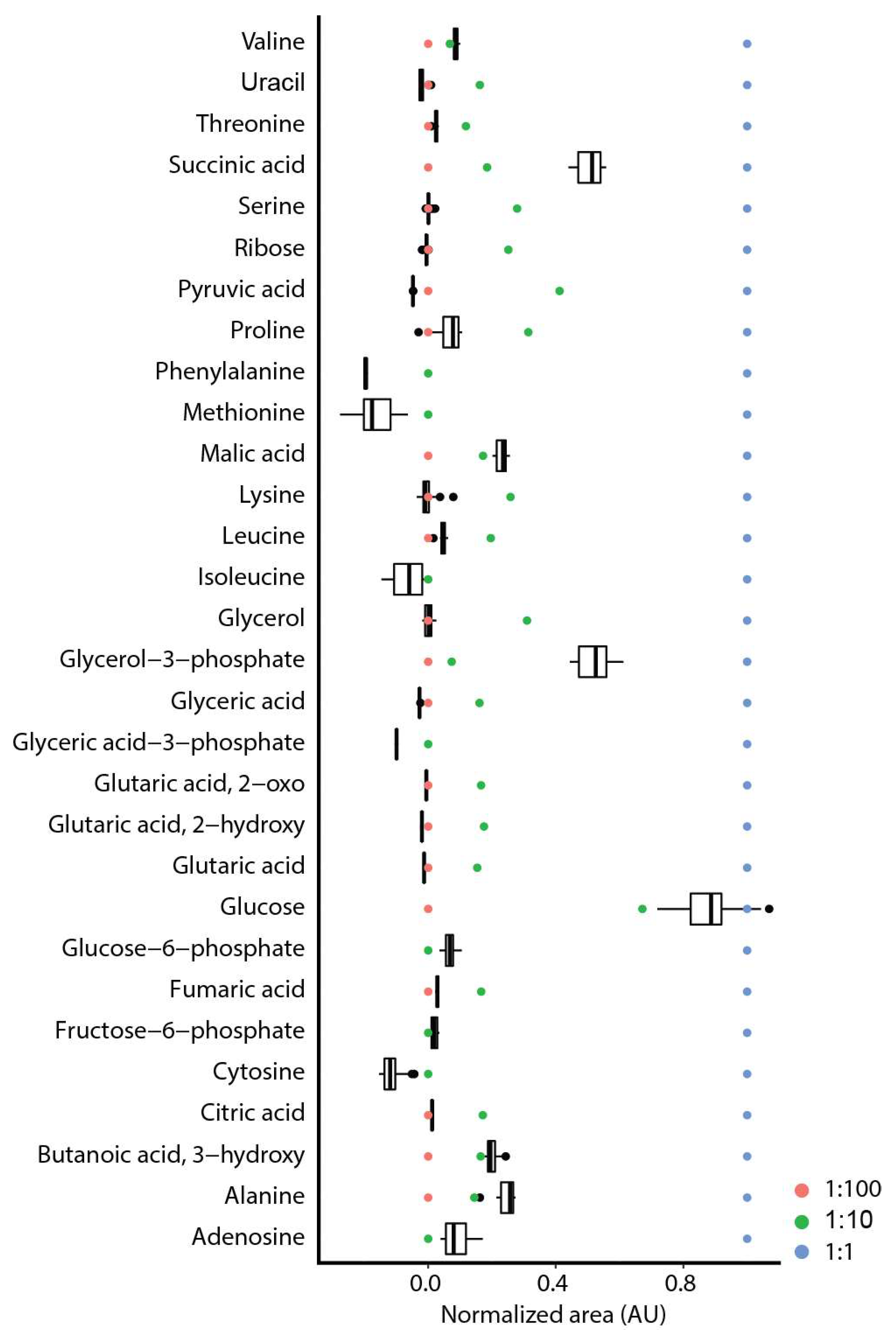
2.2. Repeatability and Reproducibility
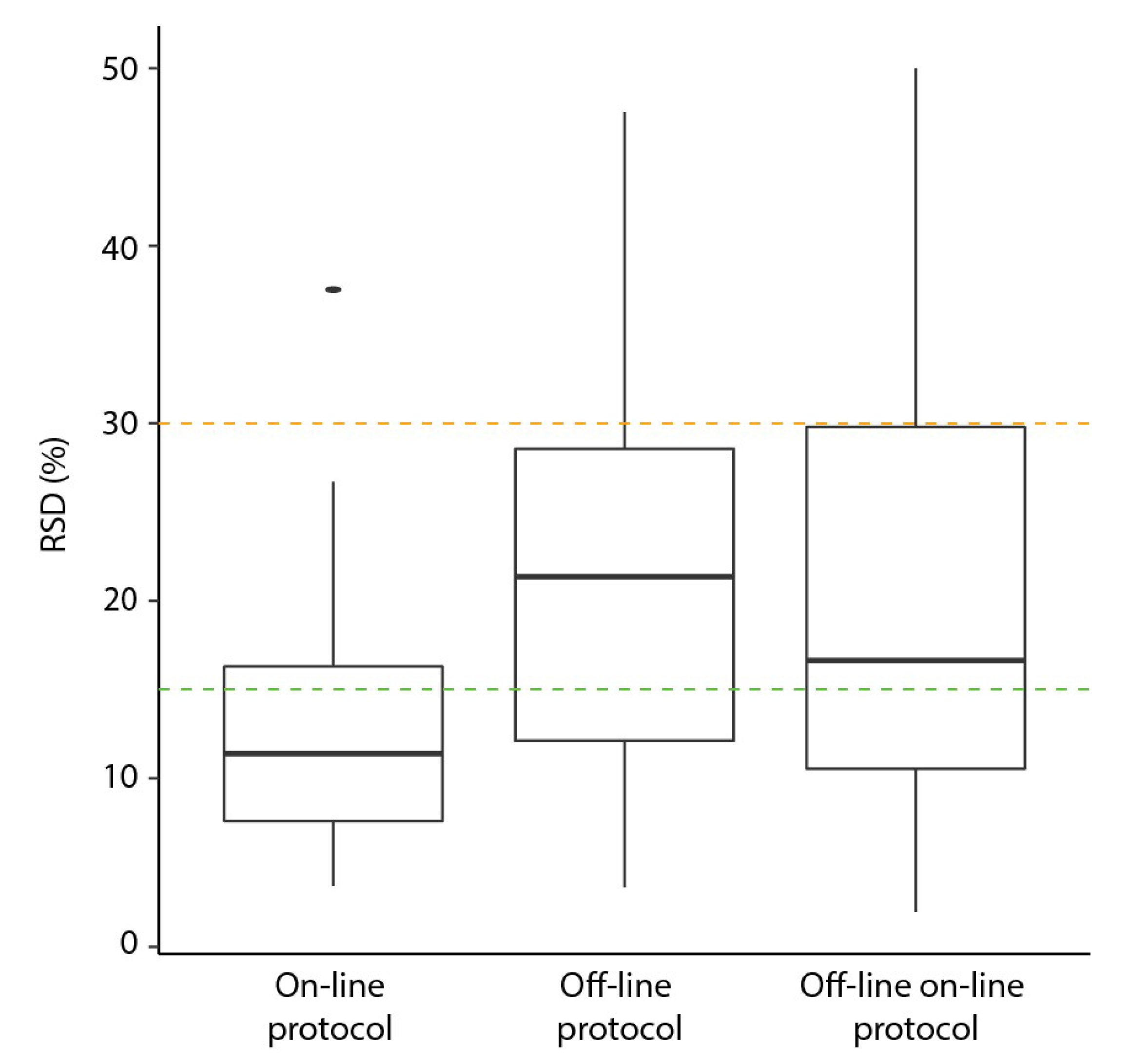
2.3. Comparison of on-Line to off-Line Derivatization
3. Discussion
4. Materials and Methods
4.1. Extraction of Calibration Standards
4.2. Plasma and Serum Extraction
4.3. Liver Extraction
4.4. GC-MS Metabolomics Measurement of Key Central Carbon Pathway Metabolites
4.4.1. On-line Derivatization
4.4.2. Off-line Derivatization
4.4.3. Instrumentation
4.4.4. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halket, J.M.; Waterman, D.; Przyborowska, A.M.; Patel, R.K.; Fraser, P.D.; Bramley, P.M. Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J. Exp. Bot. 2005, 56, 219–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villas-Boas, S.G.; Smart, K.F.; Sivakumaran, S.; Lane, G.A. Alkylation or Silylation for Analysis of Amino and Non-Amino Organic Acids by GC-MS? Metabolites 2011, 1, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; David, F.; Lynen, F.; Rumpel, K.; Dugardeyn, J.; Van Der Straeten, D.; Xu, G.; Sandra, P. Evaluation of automated sample preparation, retention time locked gas chromatography-mass spectrometry and data analysis methods for the metabolomic study of Arabidopsis species. J. Chromatogr. A 2011, 1218, 3247–3254. [Google Scholar] [CrossRef] [PubMed]
- Abbiss, H.; Rawlinson, C.; Maker, G.L.; Trengove, R. Assessment of automated trimethylsilyl derivatization protocols for GC–MS-based untargeted metabolomic analysis of urine. Metabolomics 2015, 11, 1908–1921. [Google Scholar] [CrossRef]
- Fritsche-Guenther, R.; Gloaguen, Y.; Kirchner, M.; Mertins, P.; Tunn, P.U.; Kirwan, J.A. Progression-Dependent Altered Metabolism in Osteosarcoma Resulting in Different Nutrient Source Dependencies. Cancers 2020, 12, 1371. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Blaschke, B.; Benn, A.; Hammer, E.; Witt, E.; Kirwan, J.; Fritsche-Guenther, R.; Gloaguen, Y.; Bartsch, C.; Vietzke, A.; et al. Sex-specific metabolic and functional differences in human umbilical vein endothelial cells from twin pairs. Atherosclerosis 2019, 291, 99–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritsche-Guenther, R.; Zasada, C.; Mastrobuoni, G.; Royla, N.; Rainer, R.; Rossner, F.; Pietzke, M.; Klipp, E.; Sers, C.; Kempa, S. Alterations of mTOR signaling impact metabolic stress resistance in colorectal carcinomas with BRAF and KRAS mutations. Sci. Rep. 2018, 8, 9204. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Broadhurst, D.; Begley, P.; Zelena, E.; Francis-McIntyre, S.; Anderson, N.; Brown, M.; Knowles, J.D.; Halsall, A.; Haselden, J.N.; et al. Procedures for large-scale metabolic profiling of serum and plasma using gas chromatography and liquid chromatography coupled to mass spectrometry. Nat. Protoc. 2011, 6, 1060–1083. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics 2011, 7, 307–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Shi, S.; Yi, Z.; He, R.; Lu, H.; Liang, Y. MeOx-TMS derivatization for GC-MS metabolicprofiling of urine and application in thediscrimination between normal C57BL/6J and type2 diabetic KK-Ay mice. AnalyticalMethods 2014, 6, 4380. [Google Scholar] [CrossRef]
- Gullberg, J.; Jonsson, P.; Nordstrom, A.; Sjostrom, M.; Moritz, T. Design of experiments: An efficient strategy to identify factors influencing extraction and derivatization of Arabidopsis thaliana samples in metabolomic studies with gas chromatography/mass spectrometry. Anal. Biochem. 2004, 331, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Pietzke, M.; Zasada, C.; Mudrich, S.; Kempa, S. Decoding the dynamics of cellular metabolism and the action of 3-bromopyruvate and 2-deoxyglucose using pulsed stable isotope-resolved metabolomics. Cancer Metab. 2014, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opialla, T.; Kempa, S.; Pietzke, M. Towards a More Reliable Identification of Isomeric Metabolites Using Pattern Guided Retention Validation. Metabolites 2020, 10, 457. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter- | MeOx Volume | Time | Temperature | Equilibration |
|---|---|---|---|---|
| Analyzed replicates | 20 µL: 5 40 µL: 5 60 µL: 4 | 30/30 min: 4 60/30 min: 3 90/60 min: 5 | 30 °C: 4 37 °C: 3 45 °C: 3 | 0 h: 4 2 h: 4 4 h: 5 8 h: 4 |
| Detected compounds | 20 µL: 32 40 µL: 33 60 µL: 32 | 30/30 min: 33 60/30 min: 34 90/60 min: 31 | 30 °C: 38 37 °C: 37 45 °C: 36 | 0 h: 34 2 h: 36 4 h: 36 8 h: 34 |
| Median RSD (%) | 20 µL: 17 40 µL: 27 60 µL: 33 | 30/30 min: 23 60/30 min: 14 90/60 min: 18 | 30 °C: 10 37 °C: 10 45 °C: 21 | 0 h: 11 2 h: 21 4 h: 15 8 h: 15 |
| Parameter | Plasma | Liver | Batch 1 | Batch 2 | Batch 3 |
|---|---|---|---|---|---|
| Number of metabolites | 0.5 | 0 | 1.9 | 2.4 | 2.2 |
| Number of missing values | 11 (0.8%) | 0 (0%) | 24 (11%) | 25 (12%) | 25 (12%) |
| Median RSD | 16% | 10% | 21% | 20% | 19% |
| RSD range | 11–28% | 2–56% | 3–42% | 12–69% | 13–39% |
| Parameter | On-Line | Off-Line (Original) | Off-Line with On-Line Settings (OLOLP) |
|---|---|---|---|
| Replicates | 9 | 9 | 8 |
| Number metabolites | 0.73 | 0.83 | 0.71 |
| Number of missing values | 14 (6%) | 7 (3%) | 5 (3%) |
| Median RSD | 11% | 21% | 17% |
| RSD range | 4–38% | 4–48% | 2–50% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritsche-Guenther, R.; Gloaguen, Y.; Bauer, A.; Opialla, T.; Kempa, S.; Fleming, C.A.; Redmond, H.P.; Kirwan, J.A. Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry. Metabolites 2021, 11, 888. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11120888
Fritsche-Guenther R, Gloaguen Y, Bauer A, Opialla T, Kempa S, Fleming CA, Redmond HP, Kirwan JA. Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry. Metabolites. 2021; 11(12):888. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11120888
Chicago/Turabian StyleFritsche-Guenther, Raphaela, Yoann Gloaguen, Anna Bauer, Tobias Opialla, Stefan Kempa, Christina A. Fleming, Henry Paul Redmond, and Jennifer A. Kirwan. 2021. "Optimized Workflow for On-Line Derivatization for Targeted Metabolomics Approach by Gas Chromatography-Mass Spectrometry" Metabolites 11, no. 12: 888. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11120888