
Cholesterol Activates Cyclic AMP Signaling in Metaplastic Acinar Cells


 ,
,  , , and
, , and 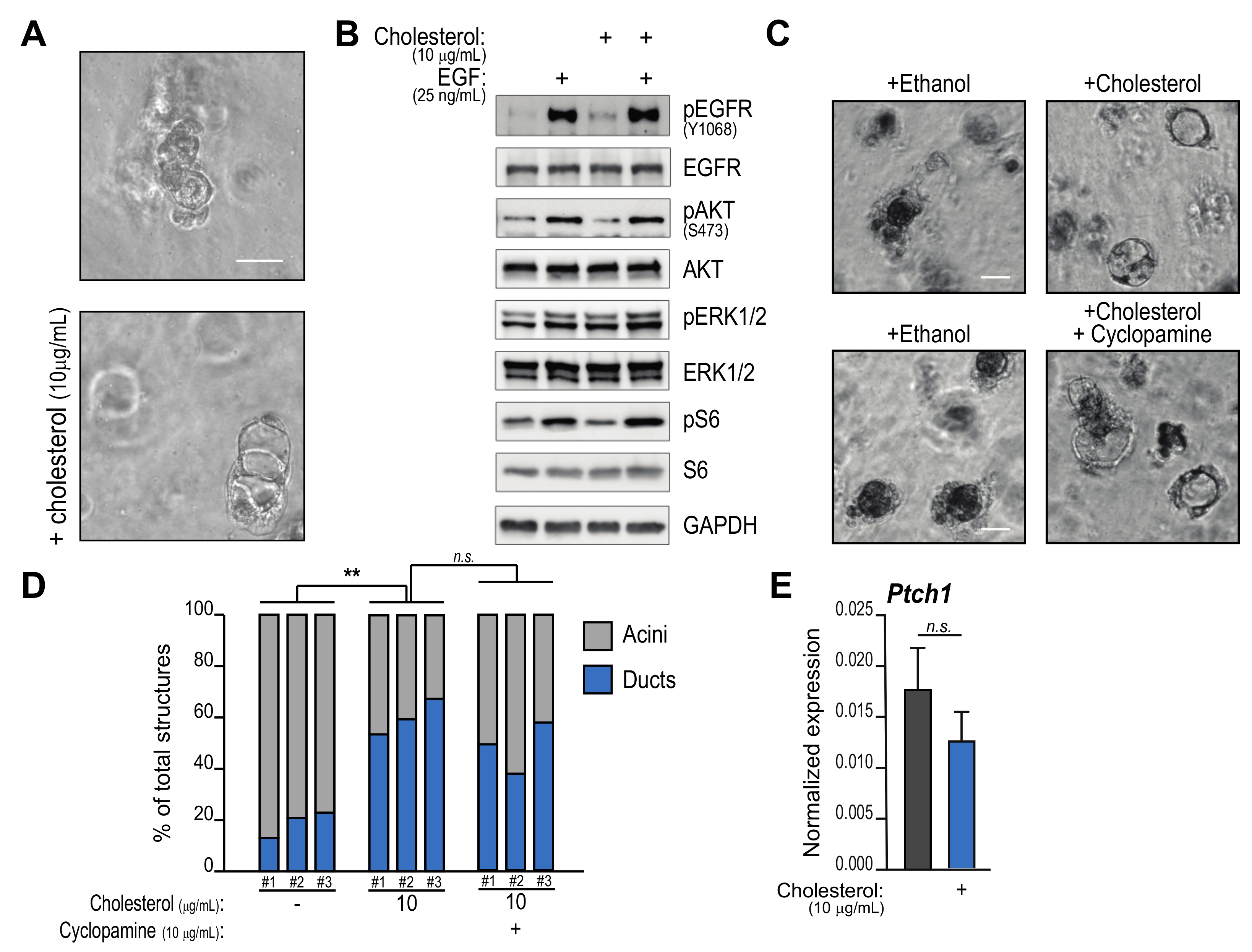{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
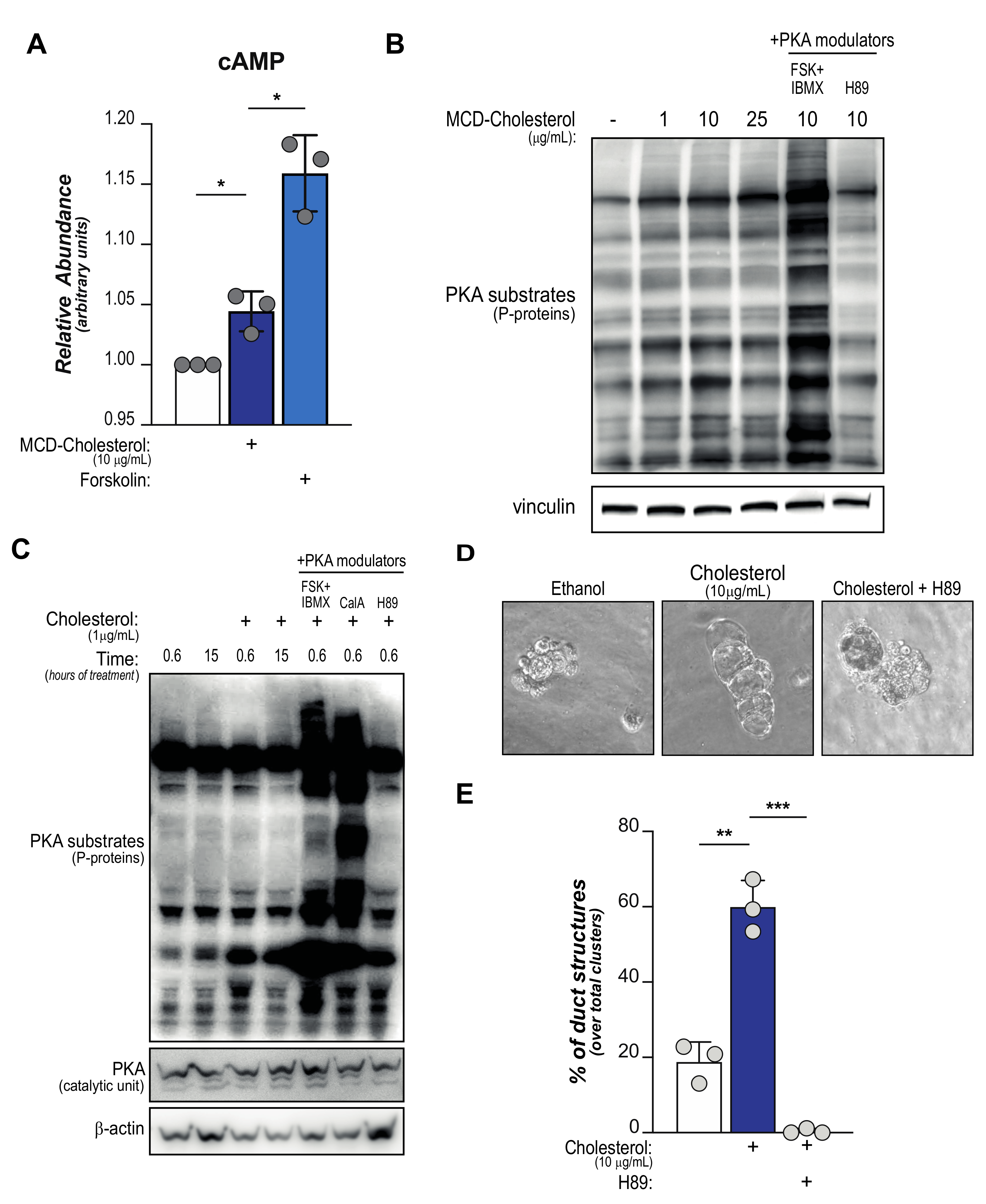
2.1. Cholesterol Activates cAMP-PKA Signaling
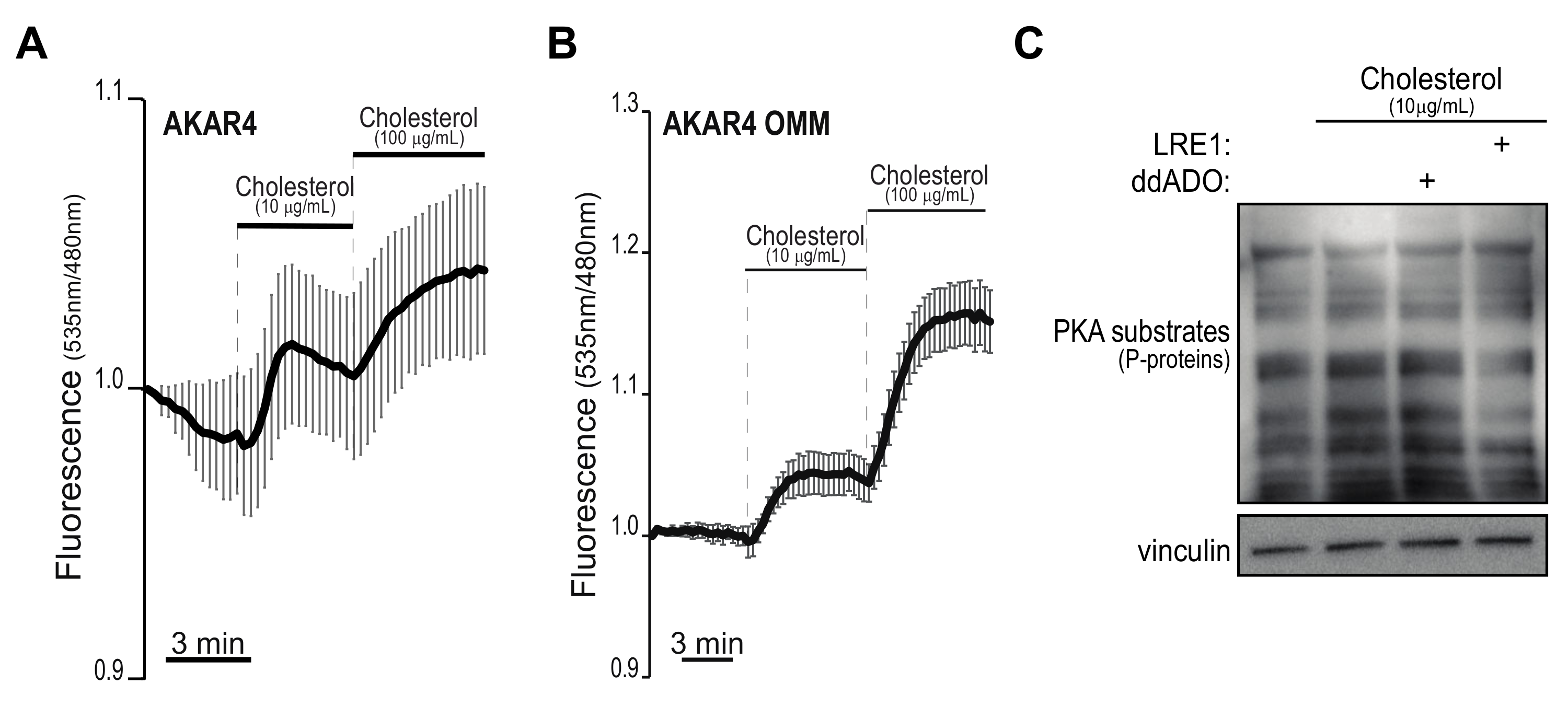
2.2. Dynamics of Cholesterol-Induced PKA Activation
2.3. PKA Is Not Sensitive to Cholesterol Stimulation in PDA Neoplastic Cells
2.4. Baseline PKA Activity Evolves during Pancreatic Carcinogenesis
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Acinar Cell Isolation, Culture and Treatment
4.3. Quantitative PCR (qPCR)
4.4. Cell Lines and Culture
4.5. Protein Lysate Preparation for Western Blotting
4.6. FRET Analysis of PKA Activity
4.7. Immunoassay (ELISA) for cAMP
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef] [Green Version]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081. [Google Scholar] [CrossRef]
- Paoli, C.; Carrer, A. Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation. Cancers 2020, 12, 2606. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.L.; Von Figura, G.; Mayes, E.; Liu, F.; Dubois, C.L.; Iv, J.P.M.; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derle, A.; De Santis, M.C.; Gozzelino, L.; Ratto, E.; Martini, M. The role of metabolic adaptation to nutrient stress in pancreatic cancer. Cell Stress 2018, 2, 332–339. [Google Scholar] [CrossRef]
- Chandwani, R.; Allen, P.J. Cystic Neoplasms of the Pancreas. Annu. Rev. Med. 2016, 67, 45–57. [Google Scholar] [CrossRef]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and Repair of the Exocrine Pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef] [Green Version]
- Neinast, M.D.; Jang, C.; Hui, S.; Anthony, T.G.; Rabinowitz, J.D.; Arany, Z.; Neinast, M.D.; Jang, C.; Hui, S.; Murashige, D.S.; et al. Quantitative Analysis of the Whole-Body Metabolic Fate of Branched-Chain Amino Acids Article Quantitative Analysis of the Whole-Body Metabolic Fate of Branched-Chain Amino Acids. Cell Metab. 2019, 29, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, L.; Fagman, J.B.; Kim, J.Y.; Todoric, J.; Gukovsky, I.; Mackey, M.; Ellisman, M.H.; Karin, M. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6166–E6174. [Google Scholar] [CrossRef] [Green Version]
- Gerasimenko, J.V.; Gerasimenko, O.V.; Petersen, O.H. The role of Ca2+ in the pathophysiology of pancreatitis. J. Physiol. 2014, 592, 269–280. [Google Scholar] [CrossRef]
- Lu, Z.; Kolodecik, T.R.; Karne, S.; Nyce, M.; Gorelick, F. Effect of ligands that increase cAMP on caerulein-induced zymogen activation in pancreatic acini. Am. J. Physiol. Liver Physiol. 2003, 285, G822–G828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanger, B.Z.; Hebrok, M. Control of cell identity in pancreas development and regeneration. Gastroenterology 2013, 144, 1170–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziv, O.; Glaser, B.; Dor, Y. The plastic pancreas. Dev. Cell 2013, 26, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrer, A.; Trefely, S.; Zhao, S.; Campbell, S.L.; Norgard, R.J.; Schultz, K.C.; Sidoli, S.; Parris, J.L.D.; Affronti, H.C.; Sivanand, S.; et al. Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 2019, 9, 416–435. [Google Scholar] [CrossRef] [Green Version]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef]
- Lardon, J.; Bouwens, L. Metaplasia in the pancreas. Differentiation 2005, 73, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, D.S.; Shinozuka, H.; Dekker, A. Focal acinar cell dysplasia in human pancreas. Cancer 1980, 45, 534–540. [Google Scholar] [CrossRef]
- Wu, C.-Y.C.; Carpenter, E.S.; Takeuchi, K.K.; Halbrook, C.J.; Peverley, L.V.; Bien, H.; Hall, J.C.; DelGiorno, K.E.; Pal, D.; Song, Y.; et al. PI3K regulation of RAC1 is required for KRAS-induced pancreatic tumorigenesis in mice. Gastroenterology 2014, 147, 1405–1416. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.-M.; Singh, G.; Koenig, A.; Liou, G.-Y.; Storz, P.; Zhang, J.-S.; Regul, L.; Nagarajan, S.; Kühnemuth, B.; Johnsen, S.A.; et al. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar–Ductal Transdifferentiation in the Pancreas. Gastroenterology 2015, 148, 1024–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbrook, C.J.; Wen, H.-J.; Ruggeri, J.M.; Takeuchi, K.K.; Zhang, Y.; di Magliano, M.P.; Crawford, H.C. Mitogen-activated Protein Kinase Kinase Activity Maintains Acinar-to-Ductal Metaplasia and Is Required for Organ Regeneration in Pancreatitis. Cell. Mol. Gastroenterol. Hepatol. 2016, 3, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrer, A.; Wellen, K.E. Metabolism and epigenetics: A link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, U.C.; Watanabe, T.; Bendtsen, F. The relationship between the use of statins and mortality, severity, and pancreatic cancer in Danish patients with chronic pancreatitis. Eur. J. Gastroenterol. Hepatol. 2018, 30, 346–351. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- Hamada, T.; Khalaf, N.; Yuan, C.; Babic, A.; Morales-Oyarvide, V.; Qian, Z.R.; Nowak, J.A.; Ng, K.; Kraft, P.; Rubinson, D.A.; et al. Statin use and pancreatic cancer risk in two prospective cohort studies. J. Gastroenterol. 2018. [Google Scholar] [CrossRef]
- Chen, M.-J.; Tsan, Y.-T.; Liou, J.-M.; Lee, Y.-C.; Wu, M.-S.; Chiu, H.-M.; Wang, H.-P.; Chen, P.-C. Statins and the risk of pancreatic cancer in Type 2 diabetic patients-A population-based cohort study. Int. J. Cancer 2016, 138, 594–603. [Google Scholar] [CrossRef]
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986. [Google Scholar] [CrossRef]
- Wiggins, S.V.; Steegborn, C.; Levin, L.R.; Buck, J. Pharmacological modulation of the CO2/HCO3−/pH-, calcium-, and ATP-sensing soluble adenylyl cyclase. Pharmacol. Ther. 2018, 190, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.B.; Agarwal, S.R.; Harvey, R.D.; Ostrom, R.S. cAMP Signaling Compartmentation: Adenylyl Cyclases as Anchors of Dynamic Signaling Complexes. Mol. Pharmacol. 2018, 93, 270–276. [Google Scholar] [CrossRef] [Green Version]
- Lefkimmiatis, K.; Zaccolo, M. cAMP signaling in subcellular compartments. Pharmacol. Ther. 2014, 143, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Kolodecik, T.R.; Shugrue, C.A.; Thrower, E.C.; Levin, L.R.; Buck, J.; Gorelick, F.S. Activation of Soluble Adenylyl Cyclase Protects against Secretagogue Stimulated Zymogen Activation in Rat Pancreaic Acinar Cells. PLoS ONE 2012, 7, e41320. [Google Scholar] [CrossRef] [Green Version]
- Innamorati, G.; Valenti, M.T.; Giacomello, L.; Dalle Carbonare, L.; Bassi, C. GNAS Mutations: Drivers or Co-Pilots? Yet, Promising Diagnostic Biomarkers. Trends Cancer 2016, 2, 282–285. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Kato, Y.; Mizukami, Y.; Widholz, S.; Boukhali, M.; Revenco, I.; Grossman, E.A.; Ji, F.; Sadreyev, R.I.; Liss, A.S.; et al. Mutant GNAS drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat. Cell Biol. 2018, 20, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Ideno, N.; Yamaguchi, H.; Ghosh, B.; Gupta, S.; Okumura, T.; Steffen, D.J.; Fisher, C.G.; Wood, L.D.; Singhi, A.D.; Nakamura, M.; et al. GNASR201C Induces Pancreatic Cystic Neoplasms in Mice That Express Activated KRAS by Inhibiting YAP1 Signaling. Gastroenterology 2018, 155, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, S.; Tissier, F.; Ragazzon, B.; Rebours, V.; Saloustros, E.; Perlemoine, K.; Vincent-Dejean, C.; Meurette, G.; Cassagnau, E.; Dousset, B.; et al. Pancreatic Ductal and Acinar Cell Neoplasms in Carney Complex: A Possible New Association. J. Clin. Endocrinol. Metab. 2011, 96, E1888–E1895. [Google Scholar] [CrossRef] [Green Version]
- Taki, K.; Ohmuraya, M.; Tanji, E.; Komatsu, H.; Hashimoto, D.; Semba, K.; Araki, K.; Kawaguchi, Y.; Baba, H.; Furukawa, T. GNASR201H and KrasG12D cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm. Oncogene 2016, 35, 2407–2412. [Google Scholar] [CrossRef]
- Wang, K.; Wong, Y.H. G protein signaling controls the differentiation of multiple cell lineages. BioFactors 2009, 35, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Clements, W.K.; Traver, D. Signalling pathways that control vertebrate haematopoietic stem cell specification. Nat. Rev. Immunol. 2013, 13, 336–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, M.G.; Gonen, T.; Scott, J.D. Local cAMP signaling in disease at a glance. J. Cell Sci. 2013, 126, 4537–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddappa, R.; Martens, A.; Doorn, J.; Leusink, A.; Olivo, C.; Licht, R.; van Rijn, L.; Gaspar, C.; Fodde, R.; Janssen, F.; et al. cAMP/PKA pathway activation in human mesenchymal stem cells in vitro results in robust bone formation in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 7281–7286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubeseder-Martellato, C.; Alexandrow, K.; Hidalgo-Sastre, A.; Heid, I.; Boos, S.L.; Briel, T.; Schmid, R.M.; Siveke, J.T. Oncogenic KRas-induced Increase in Fluid-phase Endocytosis is Dependent on N-WASP and is Required for the Formation of Pancreatic Preneoplastic Lesions. EBioMedicine 2017, 15, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Riobo, N.A. Cholesterol and its derivatives in Sonic Hedgehog signaling and cancer. Curr. Opin. Pharmacol. 2012, 12, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pitarresi, J.R.; Cuitiño, M.C.; Kladney, R.D.; Woelke, S.A.; Sizemore, G.M.; Nayak, S.G.; Egriboz, O.; Schweickert, P.G.; Yu, L.; et al. Genetic ablation of smoothened in pancreatic fibroblasts increases acinar-ductal metaplasia. Genes Dev. 2016, 30, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Peruzzo, R.; Bachmann, M.; Montà, G.D.; Vicario, M.; Santinon, G.; Mattarei, A.; Moro, E.; Quintana-Cabrera, R.; Scorrano, L.; et al. Impaired Mitochondrial ATP Production Downregulates Wnt Signaling via ER Stress Induction. Cell Rep. 2019, 28, 1949–1960. [Google Scholar] [CrossRef] [Green Version]
- Pinto, C.; Papa, D.; Hübner, M.; Mou, T.-C.; Lushington, G.H.; Seifert, R. Activation and Inhibition of Adenylyl Cyclase Isoforms by Forskolin Analogs. J. Pharmacol. Exp. Ther. 2008, 325, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaumond, F.; Bidaut, G.; Ouaissi, M.; Servais, S.; Gouirand, V.; Olivares, O.; Lac, S.; Borge, L.; Roques, J.; Gayet, O.; et al. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 2473–2478. [Google Scholar] [CrossRef] [Green Version]
- Pendin, D.; Greotti, E.; Lefkimmiatis, K.; Pozzan, T. Exploring cells with targeted biosensors. J. Gen. Physiol. 2016, 149, 1–36. [Google Scholar] [CrossRef]
- Burdyga, A.; Surdo, N.C.; Monterisi, S.; Di Benedetto, G.; Grisan, F.; Penna, E.; Pellegrini, L.; Zaccolo, M.; Bortolozzi, M.; Swietach, P.; et al. Phosphatases control PKA-dependent functional microdomains at the outer mitochondrial membrane. Proc. Natl. Acad. Sci. USA 2018, 115, E6497–E6506. [Google Scholar] [CrossRef] [Green Version]
- Grisan, F.; Burdyga, A.; Iannucci, L.F.; Surdo, N.C.; Pozzan, T.; Di Benedetto, G.; Lefkimmiatis, K. Studying β1 and β2 adrenergic receptor signals in cardiac cells using FRET-based sensors. Prog. Biophys. Mol. Biol. 2020, 154, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Espiritu, L.; Kleinboelting, S.; Navarrete, F.A.; Alvau, A.; Visconti, P.E.; Valsecchi, F.; Starkov, A.; Manfredi, G.; Buck, H.; Adura, C.; et al. Discovery of LRE1 as a specific and allosteric inhibitor of soluble adenylyl cyclase. Nat. Chem. Biol. 2016, 12, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Ono, Y.; Tanino, M.; Karasaki, H.; Yamaguchi, H.; Furukawa, T.; Enomoto, K.; Ueda, J.; Sumi, A.; Katayama, J.; et al. Pathways of Progression From Intraductal Papillary Mucinous Neoplasm to Pancreatic Ductal Adenocarcinoma Based on Molecular Features. Gastroenterology 2019, 156, 647–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Song, B.; Xu, C. Cholesterol metabolism in cancer: Mechanisms and therapeutic opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.-Z.; Cai, Z.; Shi, S.; Jiang, H.; Shang, Y.-R.; Ma, N.; Wang, J.-J.; Guan, D.-X.; Chen, T.-W.; Rong, Y.-F.; et al. Cilia loss sensitizes cells to transformation by activating the mevalonate pathway. J. Exp. Med. 2017, 215, 177–195. [Google Scholar] [CrossRef] [Green Version]
- Cobo, I.; Martinelli, P.; Flández, M.; Bakiri, L.; Zhang, M.; Carrillo-De-Santa-Pau, E.; Jia, J.; Lobo, V.J.S.A.; Megías, D.; Felipe, I.; et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature 2018, 554, 533–537. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The role of cholesterol in cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Hulce, J.J.; Cognetta, A.B.; Niphakis, M.J.; Tully, S.E.; Cravatt, B.F. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat. Methods 2013, 10, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Gabitova, L.; Gorin, A.; Astsaturov, I. Molecular Pathways: Sterols and Receptor Signaling in Cancer. Clin. Cancer Res. 2014, 20, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Gabitova-Cornell, L.; Surumbayeva, A.; Peri, S.; Franco-Barraza, J.; Restifo, D.; Weitz, N.; Ogier, C.; Goldman, A.R.; Hartman, T.R.; Francescone, R.; et al. Cholesterol Pathway Inhibition Induces TGF-β Signaling to Promote Basal Differentiation in Pancreatic Cancer. Cancer Cell 2020, 38, 567–583. [Google Scholar] [CrossRef]
- Nöthlings, U.; Wilkens, L.R.; Murphy, S.P.; Hankin, J.H.; Henderson, B.E.; Kolonel, L.N. Meat and Fat Intake as Risk Factors for Pancreatic Cancer: The Multiethnic Cohort Study. JNCI J. Natl. Cancer Inst. 2005, 97, 1458–1465. [Google Scholar] [CrossRef]
- Gong, J.; Sachdev, E.; Robbins, L.A.; Lin, E.; Hendifar, A.E.; Mita, M.M. Statins and pancreatic cancer. Oncol. Lett. 2017, 13, 1035–1040. [Google Scholar] [CrossRef] [Green Version]
- Ringerike, T.; Blystad, F.D.; Levy, F.O.; Madshus, I.H.; Stang, E. Cholesterol is important in control of EGF receptor kinase activity but EGF receptors are not concentrated in caveolae. J. Cell Sci. 2002, 115, 1331–1340. [Google Scholar]
- Irwin, M.E.; Mueller, K.L.; Bohin, N.; Ge, Y.; Boerner, J.L. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell. Physiol. 2011, 226, 2316–2328. [Google Scholar] [CrossRef] [Green Version]
- Lim, C.-Y.; Davis, O.B.; Shin, H.R.; Zhang, J.; Berdan, C.A.; Jiang, X.; Counihan, J.L.; Ory, D.S.; Nomura, D.K.; Zoncu, R. ER–lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann–Pick type C. Nat. Cell Biol. 2019, 21, 1206–1218. [Google Scholar] [CrossRef]
- Alonso-Curbelo, D.; Ho, Y.-J.; Burdziak, C.; Maag, J.L.V.; Morris, J.P.; Chandwani, R.; Chen, H.-A.; Tsanov, K.M.; Barriga, F.M.; Luan, W.; et al. A gene–environment-induced epigenetic program initiates tumorigenesis. Nature 2021. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.F.; Lim, H.W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell Histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grisan, F.; Spacci, M.; Paoli, C.; Costamagna, A.; Fantuz, M.; Martini, M.; Lefkimmiatis, K.; Carrer, A. Cholesterol Activates Cyclic AMP Signaling in Metaplastic Acinar Cells. Metabolites 2021, 11, 141. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11030141
Grisan F, Spacci M, Paoli C, Costamagna A, Fantuz M, Martini M, Lefkimmiatis K, Carrer A. Cholesterol Activates Cyclic AMP Signaling in Metaplastic Acinar Cells. Metabolites. 2021; 11(3):141. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11030141
Chicago/Turabian StyleGrisan, Francesca, Martina Spacci, Carlotta Paoli, Andrea Costamagna, Marco Fantuz, Miriam Martini, Konstantinos Lefkimmiatis, and Alessandro Carrer. 2021. "Cholesterol Activates Cyclic AMP Signaling in Metaplastic Acinar Cells" Metabolites 11, no. 3: 141. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11030141