
Quantitative Assessment of Occipital Metabolic and Energetic Changes in Parkinson’s Patients, Using In Vivo 31P MRS-Based Metabolic Imaging at 7T

 , , ,
, , ,  , , ,
, , , 
Abstract
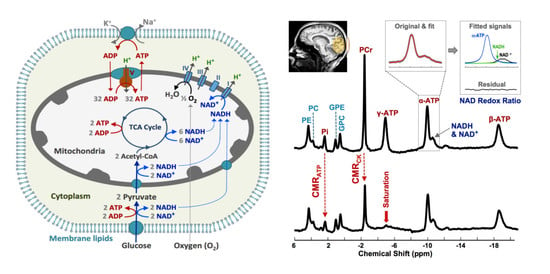
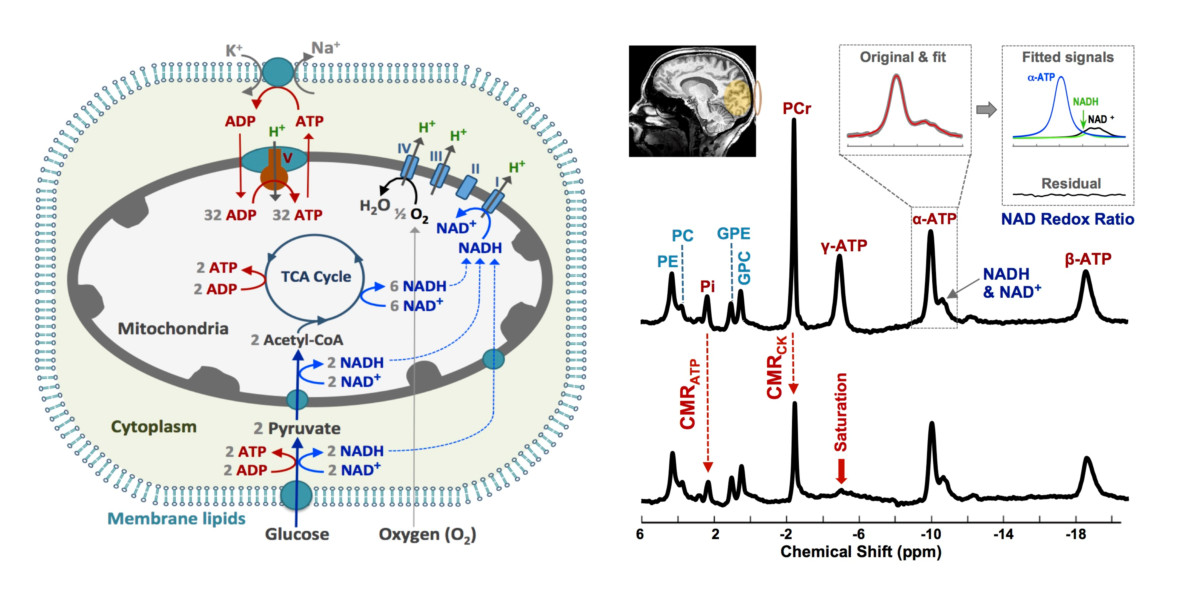
:
1. Introduction
2. Results
2.1. Characterization of the Study Participants
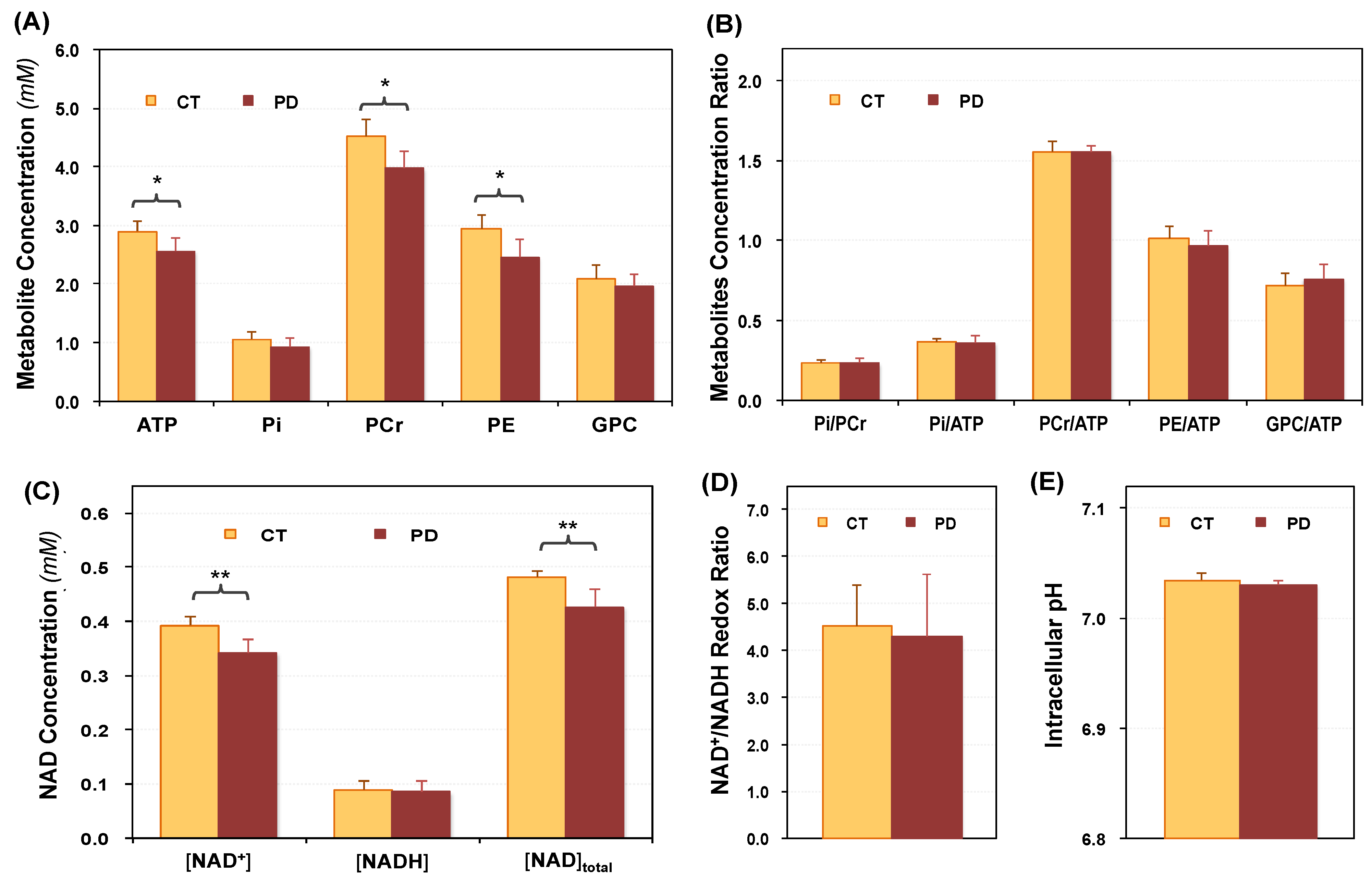
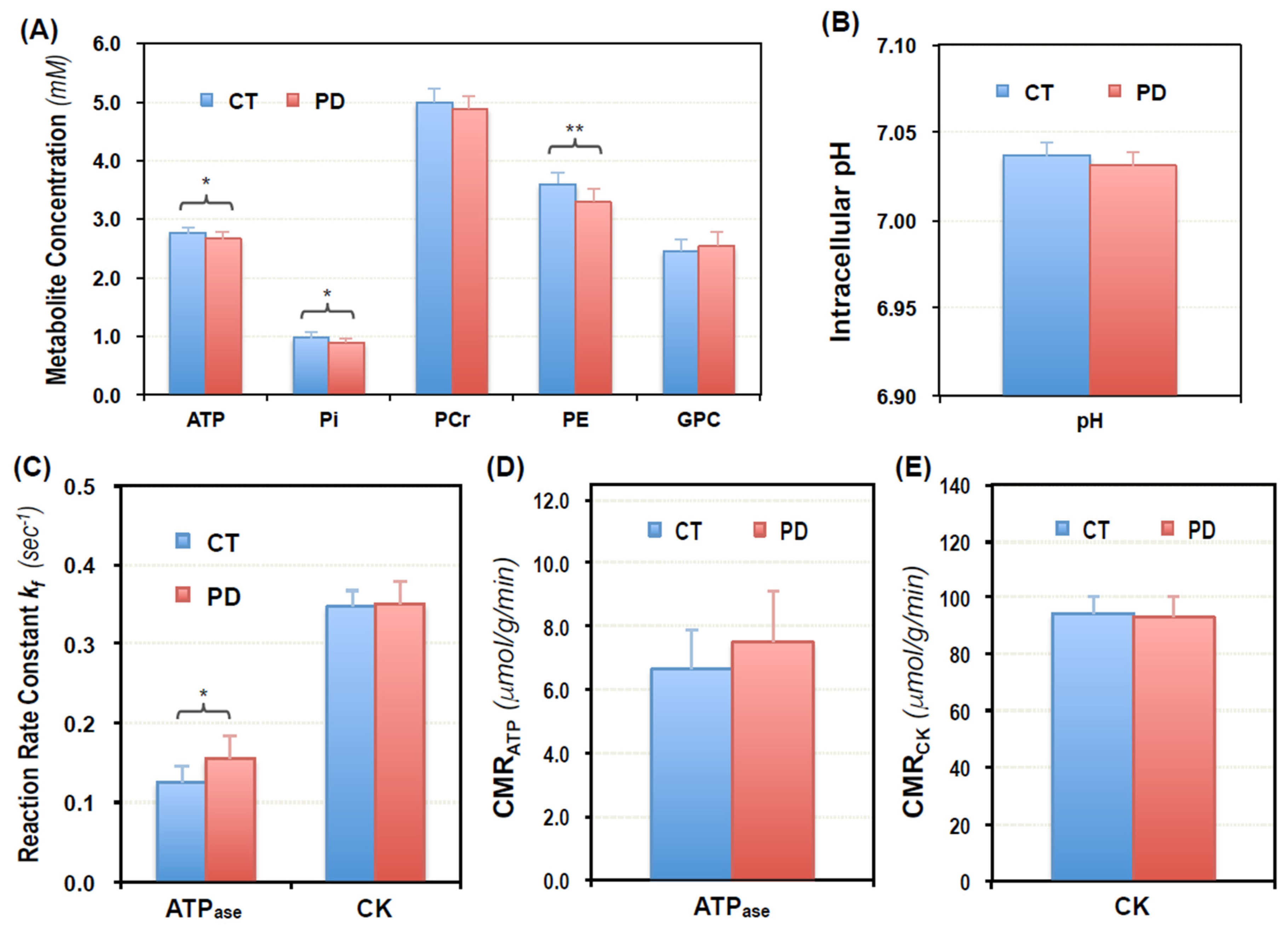
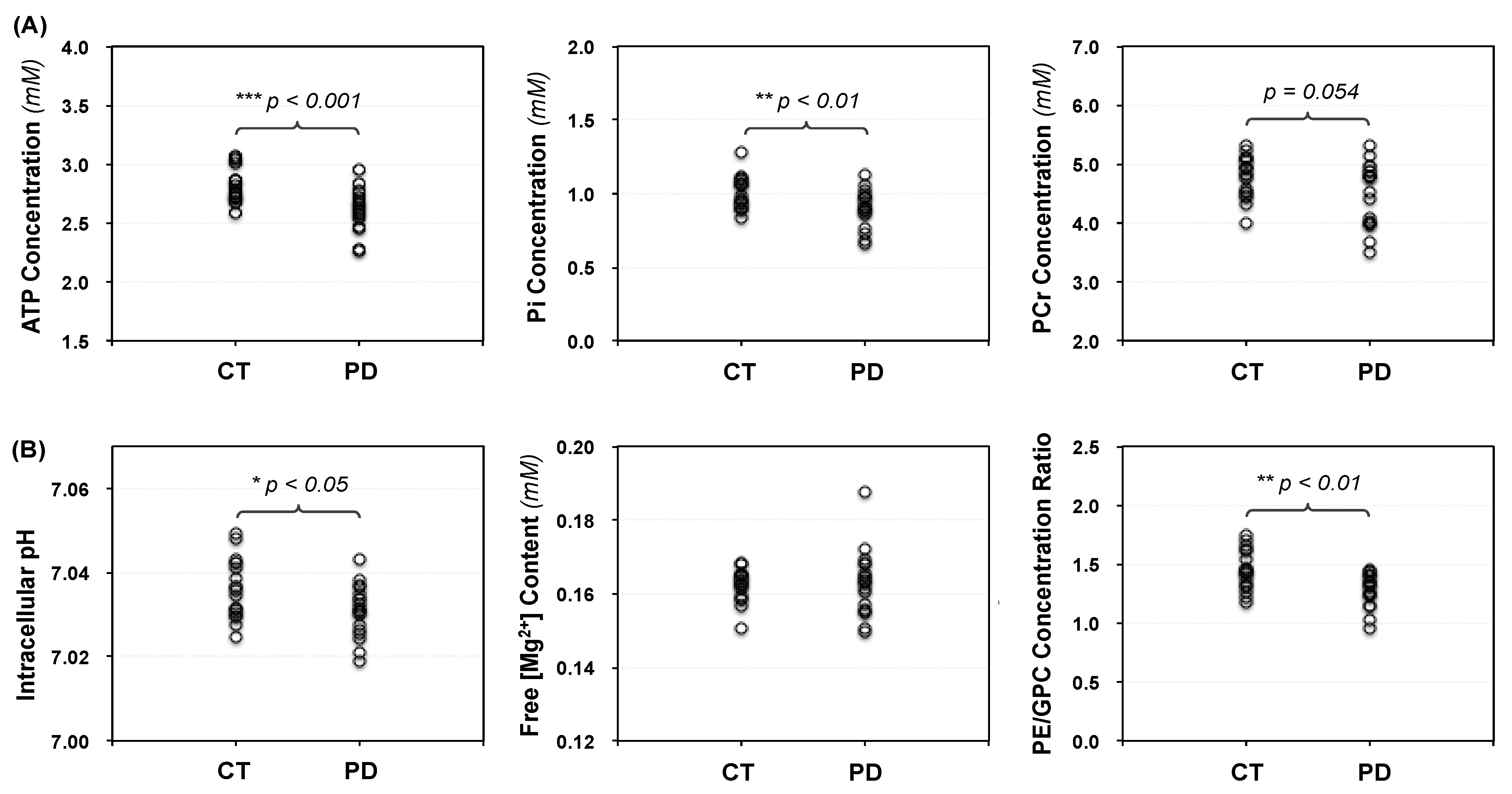
2.2. Cerebral Phosphorous Metabolite Profiles of PD Patients and Controls
2.3. Abnormal Bioenergetics in the Brain of PD Patients
2.4. Pilot Test in Assessing Bioenergetic Effects of UDCA Treatment in PD Brains
3. Discussion
4. Materials and Methods
4.1. Participants and Study Design
4.2. UDCA Therapy and Dosing
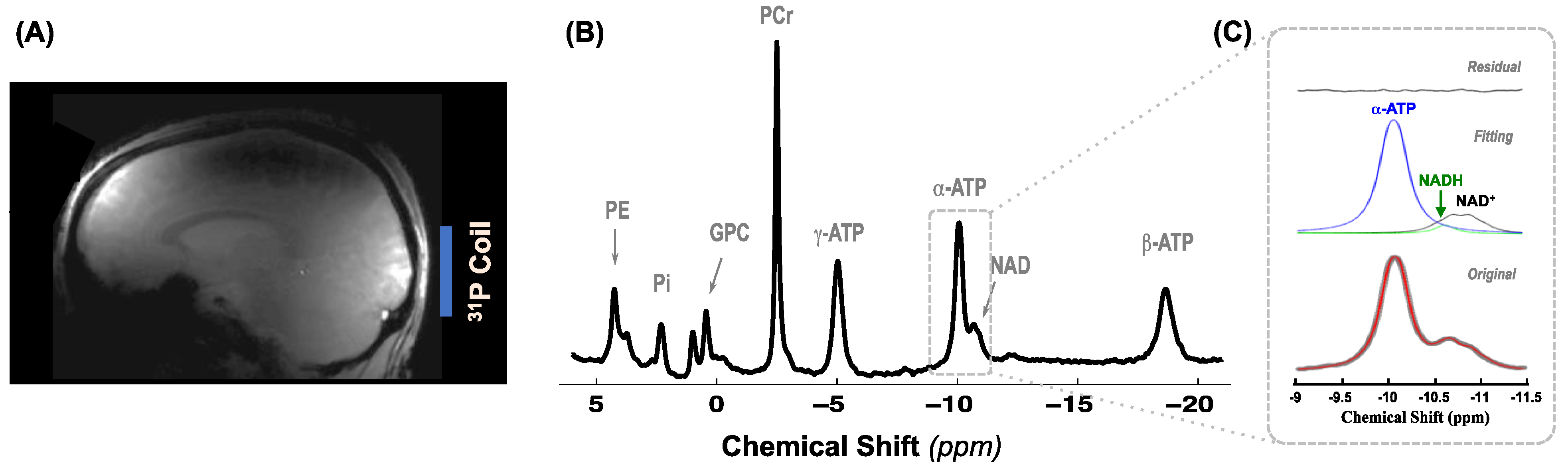
4.3. MR Data Acquisition
4.4. MR Data Analysis and Quantification
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barinaga, M. What makes brain neurons run. Science 1997, 276, 196–198. [Google Scholar] [CrossRef]
- Erecińska, M.; Silver, I.A. ATP and Brain Function. Br. J. Pharmacol. 1989, 9, 2–19. [Google Scholar] [CrossRef] [Green Version]
- Siesjo, B.K. Brain Energy Metabolism; Wiley: Hoboken, NJ, USA, 1978. [Google Scholar]
- Pathak, D.; Berthet, A.; Nakamura, K. Energy failure: Does it contribute to neurodegeneration? Ann. Neurol. 2013, 74, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nat. Cell Biol. 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnane, S.C.; Trushina, E.; Morland, C.; Prigione, A.; Casadesus, G.; Andrews, Z.B.; Beal, M.F.; Bergersen, L.H.; Brinton, R.D.; De La Monte, S.; et al. Brain energy rescue: An emerging therapeutic concept for neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 2020, 19, 609–633. [Google Scholar] [CrossRef] [PubMed]
- Borghammer, P.; Chakravarty, M.; Jonsdottir, K.Y.; Sato, N.; Matsuda, H.; Ito, K.; Arahata, Y.; Kato, T.; Gjedde, A. Cortical hypometabolism and hypoperfusion in Parkinson’s disease is extensive: Probably even at early disease stages. Brain Struct. Funct. 2010, 214, 303–317. [Google Scholar] [CrossRef]
- Brooks, D.J.; Pavese, N. Imaging biomarkers in Parkinson’s disease. Prog. Neurobiol. 2011, 95, 614–628. [Google Scholar] [CrossRef]
- Niccolini, F.; Su, P.; Politis, M. Dopamine receptor mapping with PET imaging in Parkinson’s disease. J. Neurol. 2014, 261, 2251–2263. [Google Scholar] [CrossRef]
- Varley, J.; Brooks, D.J.; Edison, P. Imaging neuroinflammation in Alzheimer’s and other dementias: Recent advances and future directions. Alzheimers Dement. 2014, 1, S9. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L. Advances in Imaging Brain Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 485–515. [Google Scholar] [CrossRef]
- Kemp, G. Non-Invasive Methods for Studying Brain Energy Metabolism: What They Show and What It Means. Dev. Neurosci. 2000, 22, 418–428. [Google Scholar] [CrossRef]
- Ross, B.; Bluml, S. Magnetic resonance spectroscopy of the human brain. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2001, 265, 54–84. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-H.; Chen, W. In vivo X-Nuclear MRS Imaging Methods for Quantitative Assessment of Neuroenergetic Biomarkers in Studying Brain Function and Aging. Front. Aging Neurosci. 2018, 10, 394. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Rodado, V.; Brender, J.R.; Cherukuri, M.K.; Gilbert, M.R.; Larion, M. Magnetic resonance spectroscopy for the study of cns malignancies. Prog. Nucl. Magn. Reson. Spectrosc. 2021, 122, 23–41. [Google Scholar] [CrossRef]
- Blüml, S.; Moreno, A.; Hwang, J.-H.; Ross, B.D. 1-13C glucose magnetic resonance spectroscopy of pediatric and adult brain disorders. NMR Biomed. 2001, 14, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.Q.; Bluml, S.; Dubowitz, D.J.; Moats, R.; Kopyov, O.; Jacques, D.; Ross, B.D. Quantitative proton-decoupled 31P MRS and 1H MRS in the evaluation of Huntington’s and Parkinson’s diseases. Neurology 1998, 50, 1033–1040. [Google Scholar] [CrossRef]
- Öz, G.; Alger, J.R.; Barker, P.B.; Bartha, R.; Bizzi, A.; Boesch, C.; Bolan, P.J.; Brindle, K.M.; Cudalbu, C.; Dinçer, A.; et al. Clinical Proton MR Spectroscopy in Central Nervous System Disorders. Radiology 2014, 270, 658–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettegrew, J.W.; Klunk, W.E.; Panchalingam, K.; McClure, R.J.; Stanley, J.A. Magnetic resonance spectroscopic changes in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1997, 826, 282–306. [Google Scholar] [CrossRef]
- Rango, M.; Bonifati, C.; Bresolin, N. Parkinson’s disease and brain mitochondrial dysfunction: A functional phosphorus magnetic resonance spectroscopy study. J. Cereb. Blood Flow Metab. 2006, 26, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Mortiboys, H.; Macdonald, R.; Payne, T.; Sassani, M.; Jenkins, T.; Bandmann, O. Translational approaches to restoring mitochondrial function in Parkinson’s disease. FEBS Lett. 2018, 592, 776–792. [Google Scholar] [CrossRef] [Green Version]
- Barbiroli, B.; Martinelli, P.; Patuelli, A.; Lodi, R.; Iotti, S.; Cortelli, P.; Montagna, P. Phosphorus magnetic resonance spectroscopy in multiple system atrophy and Parkinson’s disease. Mov. Disord. 1999, 14, 430–435. [Google Scholar] [CrossRef]
- Buchli, R.; Duc, C.O.; Martin, E.; Boesiger, P. Assessment of absolute metabolite concentrations in human tissue by 31P MRS in vivo. Part I: Cerebrum, cerebellum, cerebral gray and white matter. Magn. Reson. Med. 1994, 32, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Eleff, S.M.; Barker, P.B.; Blackband, S.J.; Chatham, J.C.; Lutz, N.W.; Johns, D.R.; Bryan, R.N.; Hurko, O. Phosphorus magnetic resonance spectroscopy of patients with mitochondrial cytopathies demonstrates decreased levels of brain phosphocreatine. Ann. Neurol. 1990, 27, 626–630. [Google Scholar] [CrossRef]
- Hattingen, E.; Magerkurth, J.; Pilatus, U.; Mozer, A.; Seifried, C.; Steinmetz, H.; Zanella, F.; Hilker, R. Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 2009, 132, 3285–3297. [Google Scholar] [CrossRef] [Green Version]
- Henchcliffe, C.; Shungu, D.C.; Mao, X.; Huang, C.; Nirenberg, M.J.; Jenkins, B.G.; Beal, M.F. Multinuclear magnetic resonance spectroscopy for in vivo assessment of mitochondrial dysfunction in Parkin-son’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 206–220. [Google Scholar] [CrossRef]
- Hu, M.T.M.; Taylor-Robinson, S.D.; Chaudhuri, K.R.; Bell, J.D.; Labbe, C.; Cunningham, V.J.; Koepp, M.J.; Hammers, A.; Morris, R.G.; Brooks, D.J.; et al. Cortical dysfunction in non-demented Parkinson’s disease patients: A combined (31)P-MRS and (18)FDG-PET study. Brain 2000, 123, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rango, M.; Bozzali, M.; Prelle, A.; Scarlato, G.; Bresolin, N. Brain Activation in Normal Subjects and in Patients Affected by Mitochondrial Disease without Clinical Central Nervous System Involvement: A Phosphorus Magnetic Resonance Spectroscopy Study. Br. J. Pharmacol. 2001, 21, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijpma, A.; van der Graaf, M.; Meulenbroek, O.; Rikkert, M.G.O.; Heerschap, A. Altered brain high-energy phosphate metabolism in mild Alzheimer’s disease: A 3-dimensional 31P MR spectroscopic imaging study. Neuroimage Clin. 2018, 18, 254–261. [Google Scholar] [CrossRef]
- Weiduschat, N.; Mao, X.; Beal, M.F.; Nirenberg, M.J.; Shungu, D.C.; Henchcliffe, C. Usefulness of proton and phosphorus MR spectroscopic imaging for early diagnosis of Parkinson’s disease. J. Neuroimaging 2015, 25, 105–110. [Google Scholar] [CrossRef]
- Buchli, R.; Martin, E.; Boesiger, P. Comparison of calibration strategies for the in vivo determination of absolute metabolite con-centrations in the human brain by 31P MRS. NMR Biomed. 1994, 7, 225–230. [Google Scholar] [CrossRef]
- Lei, H.; Zhu, X.-H.; Zhang, X.-L.; Ugurbil, K.; Chen, W. In vivo 31P magnetic resonance spectroscopy of human brain at 7 T: An initial experience. Magn. Reson. Med. 2003, 49, 199–205. [Google Scholar] [CrossRef]
- Lu, M.; Chen, E.; Zhu, X.H. Field dependence study of in vivo brain 31P MRS up to 16.4 T. NMR Biomed. 2014, 27, 1135–1141. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Zhu, X.-H.; Qiao, H.; Zhang, X.; Chen, W. Efficient in vivo 31P magnetization transfer approach for noninvasively determining multiple kinetic parameters and metabolic fluxes of ATP metabolism in the human brain. Magn. Reson. Med. 2006, 57, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Ugurbil, K.; Chen, W. Measurement of unidirectional Pi to ATP flux in human visual cortex at 7 T by using in vivo 31P magnetic resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 14409–14414. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Zhu, X.-H.; Chen, W. In vivo 31P MRS assessment of intracellular NAD metabolites and NAD+/NADH redox state in human brain at 4 T. NMR Biomed. 2016, 29, 1010–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Zhu, X.-H.; Zhang, Y.; Chen, W. Intracellular redox state revealed by in vivo 31P MRS measurement of NAD+ and NADH contents in brains. Magn. Reson. Med. 2014, 71, 1959–1972. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-H.; Lu, M.; Lee, B.-Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-H.; Lee, B.-Y.; Chen, W. Functional energetic responses and individual variance of the human brain revealed by quantitative imaging of adenosine triphosphate production rates. Br. J. Pharmacol. 2018, 38, 959–972. [Google Scholar] [CrossRef] [PubMed]
- Keene, C.D.; Rodrigues, C.M.; Eich, T.; Chhabra, M.S.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10671–10676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, C.M.; Steer, C.J. The therapeutic effects of ursodeoxycholic acid as an anti-apoptotic agent. Expert Opin. Investig. Drugs 2001, 10, 1243–1253. [Google Scholar] [CrossRef]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Health Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [Green Version]
- Alger, J.R.; Shulman, R.G. NMR studies of enzymatic rates in vitro and in vivo by magnetization transfer. Q. Rev. Biophys. 1984, 17, 83–124. [Google Scholar] [CrossRef]
- Yin, F.; Boveris, A.; Cadenas, E. Mitochondrial Energy Metabolism and Redox Signaling in Brain Aging and Neurodegeneration. Antioxid. Redox Signal. 2014, 20, 353–371. [Google Scholar] [CrossRef] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nat. Cell Biol. 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnen, N.I.; Koeppe, R.A.; Minoshima, S.; Giordani, B.; Albin, R.L.; Frey, K.A.; Kuhl, D.E. Cerebral Glucose Metabolic Features of Parkinson Disease and Incident Dementia: Longitudinal Study. J. Nucl. Med. 2011, 52, 848–855. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-Y.; Cohen, B.M.; Chen, X.; Lukas, S.E.; Shinn, A.K.; Yuksel, A.C.; Li, T.; Du, F.; Öngür, D. Redox Dysregulation in Schizophrenia Revealed by in vivo NAD+/NADH Measurement. Schizophr. Bull. 2016, 43, 197–204. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef]
- Cantó, C.; Auwerx, J. NAD+ as a Signaling Molecule Modulating Metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Cantό, C.; Wanders, R.J.; Auwerx, J. The Secret Life of NAD+: An Old Metabolite Controlling New Metabolic Signaling Pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Chen, H.; He, X.; Nie, H.; Hong, Y.; Sheng, C.; Wang, Q.; Xia, W.; Ying, W. NAD+ Metabolism and NAD+-Dependent Enzymes: Promising Therapeutic Targets for Neurological Diseases. Curr. Drug Targets 2012, 13, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Auwerx, J. NAD⁺ metabolism: A therapeutic target for age-related metabolic disease. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöndorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V.; et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson’s Disease. Cell Rep. 2018, 23, 2976–2988. [Google Scholar] [CrossRef] [PubMed]
- Puri, B. Indexation of cerebral cell membrane phospholipid catabolism by the non-invasively determined cerebral 31-phosphorus neurospectroscopic phosphodiester peak. Med. Hypotheses 2012, 78, 312–314. [Google Scholar] [CrossRef]
- Puri, B.; Treasaden, I. A human in vivo study of the extent to which 31-phosphorus neurospectroscopy phosphomonoesters index cerebral cell membrane phospholipid anabolism. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 307–308. [Google Scholar] [CrossRef]
- Forester, B.P.; Berlow, Y.A.; Harper, D.G.; Jensen, J.E.; Lange, N.; Froimowitz, M.P.; Ravichandran, C.; Iosifescu, D.V.; Lukas, S.E.; Renshaw, P.F.; et al. Age-related changes in brain energetics and phospholipid metabolism. NMR Biomed. 2009, 23, 242–250. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Mak, E.; Su, L.; Williams, G.B.; Firbank, M.J.; Lawson, R.A.; Yarnall, A.J.; Duncan, G.W.; Owen, A.M.; Khoo, T.K.; Brooks, D.J.; et al. Baseline and longitudinal grey matter changes in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain 2015, 138, 2974–2986. [Google Scholar] [CrossRef]
- Wilson, H.; Niccolini, F.; Pellicano, C.; Politis, M. Cortical thinning across Parkinson’s disease stages and clinical correlates. J. Neurol. Sci. 2019, 398, 31–38. [Google Scholar] [CrossRef]
- Piechnik, S.; Evans, J.; Bary, L.; Wise, R.; Jezzard, P. Functional changes in CSF volume estimated using measurement of waterT2relaxation. Magn. Reson. Med. 2009, 61, 579–586. [Google Scholar] [CrossRef]
- Van der Veen, J.W.; Shen, J. Regional difference in GABA levels between medial prefrontal and occipital cortices. J. Magn. Reason. Imaging 2013, 38, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Du, F.; Zhu, X.-H.; Zhang, Y.; Friedman, M.; Zhang, N.; Ugurbil, K.; Chen, W. Tightly coupled brain activity and cerebral ATP metabolic rate. Proc. Natl. Acad. Sci. USA 2008, 105, 6409–6414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacelli, C.; Giguère, N.; Bourque, M.-J.; Lévesque, M.; Slack, R.S.; Trudeau, L.É. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Caldas, M.; Carvalho, A.N.; Rodrigues, E.; Henderson, C.J.; Wolf, C.R.; Rodrigues, C.M.P.; Gama, M.J. Tauroursodeoxycholic Acid Prevents MPTP-Induced Dopaminergic Cell Death in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2012, 46, 475–486. [Google Scholar] [CrossRef]
- Duan, W.M.; Rodrigures, C.M.; Zhao, L.R.; Steer, C.J.; Low, W.C. Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson’s disease. Cell Transpl. 2002, 11, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, S.M.; Barnes, K.; Clemmens, H.; Al-Rafiah, A.R.; Al-Ofi, E.A.; Leech, V.; Bandmann, O.; Shaw, P.J.; Blackburn, D.J.; Ferraiuolo, L.; et al. Ursodeoxycholic Acid Improves Mitochondrial Function and Redistributes Drp1 in Fibroblasts from Patients with Either Sporadic or Familial Alzheimer’s Disease. J. Mol. Biol. 2018, 430, 3942–3953. [Google Scholar] [CrossRef]
- Mortiboys, H.J.A.; Bandmann, O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkin-son’s disease. Brain 2013, 136, 3038–3050. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.M.; Fan, G.; Ma, X.; Kren, B.T.; Steer, C.J. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane per-turbation. J. Clin. Investig. 1998, 101, 2790–2799. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.M.; Ma, X.; Linehan-Stieers, C.; Fan, G.; Kren, B.T.; Steer, C.J. Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 1999, 6, 842–854. [Google Scholar] [CrossRef] [Green Version]
- Parry, G.J.; Rodrigues, C.M.; Aranha, M.M.; Hilbert, S.J.; Davey, C.; Kelkar, P.; Low, W.C.; Steer, C.J. Safety, Tolerability, and Cerebrospinal Fluid Penetration of Ursodeoxycholic Acid in Patients with Amyotrophic Lateral Sclerosis. Clin. Neuropharmacol. 2010, 33, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Sathe, A.G.; Tuite, P.; Chen, C.; Ma, Y.; Chen, W.; Cloyd, J.; Low, W.C.; Steer, C.J.; Lee, B.-Y.; Coles, L.D.; et al. Pharmacokinetics, Safety and Tolerability of Orally Administered Ursodeoxycholic Acid in Patients with Parkin-son’s Disease—A Pilot Study. J. Clin. Pharmacol. 2020, 60, 744–750. [Google Scholar] [CrossRef]
- Chen, C.; Gonzalez, F.J.; Idle, J.R. LC-MS-Based Metabolomics in Drug Metabolism. Drug Metab. Rev. 2007, 39, 581–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Graaf, R.; Luob, Y.; Garwood, M.; Nicolay, K. B1-Insensitive, Single-Shot Localization and Water Suppression. J. Magn. Reson. Ser. B 1996, 113, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Van den Boogaart, A.; Van Hecke, P.; Van Huffel, S.; Graveron-Demilly, D.; Van Ormondt, D.; De Beer, R. MRUI: A graphical user interface for accurate routine MRS data analysis. In Proceedings of the 13th Annual Meeting of ESMRMB, Prague, Czech Republic, 27 April–3 May 1996. [Google Scholar]
- Vanhammea, L.; Boogaart, A.V.D.; Van Huffel, S. Improved Method for Accurate and Efficient Quantification of MRS Data with Use of Prior Knowledge. J. Magn. Reson. 1997, 129, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Iotti, S.; Malucelli, E. Free magnesium concentration in the human brain. In Magnesium in the Central Nervous System; Vink, R., Nechifor, M., Eds.; University of Adelaide Press: Adelaide, AU, USA, 2011; pp. 3–12. [Google Scholar]
- Xiong, Q.; Du, F.; Zhu, X.; Zhang, P.; Suntharalingam, P.; Ippolito, J.; Kadmar, F.D.; Chen, W.; Zhang, J. ATP production rate via creatine kinase or ATP synthase in vivo: A novel superfast magnetization saturation transfer method. Circ. Res. 2011, 108, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-H.; Qiao, H.; Du, F.; Xiong, Q.; Liu, X.; Zhang, X.; Ugurbil, K.; Chen, W. Quantitative imaging of energy expenditure in human brain. NeuroImage 2012, 60, 2107–2117. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cohort I | Cohort II | Subset of Cohort II-PD | ||||
|---|---|---|---|---|---|---|
| PD | CT | PD | CT | Pre-UDCA | Post-UDCA | |
| Subject Number | 8 | 8 | 11 | 11 | 3 | 3 |
| Gender | 4M/4F | 4M/4F | 5M/6F | 5M/6F | 2M/1F | 2M/1F |
| Age (years) | 62 ± 7 | 60 ± 8 | 64 ± 8 | 61 ± 8 | 67 ± 11 | |
| UDPRS Score | 24 ± 15 | - | 36 ± 11 | - | 40 ± 10 | 37 ± 12 |
| MoCA Score | 27 ± 3 | 29 ± 2 | 28 ± 2 | 29 ± 1 | 27 ± 2 | 28 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, X.-H.; Lee, B.-Y.; Tuite, P.; Coles, L.; Sathe, A.G.; Chen, C.; Cloyd, J.; Low, W.C.; Steer, C.J.; Chen, W. Quantitative Assessment of Occipital Metabolic and Energetic Changes in Parkinson’s Patients, Using In Vivo 31P MRS-Based Metabolic Imaging at 7T. Metabolites 2021, 11, 145. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11030145
Zhu X-H, Lee B-Y, Tuite P, Coles L, Sathe AG, Chen C, Cloyd J, Low WC, Steer CJ, Chen W. Quantitative Assessment of Occipital Metabolic and Energetic Changes in Parkinson’s Patients, Using In Vivo 31P MRS-Based Metabolic Imaging at 7T. Metabolites. 2021; 11(3):145. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11030145
Chicago/Turabian StyleZhu, Xiao-Hong, Byeong-Yeul Lee, Paul Tuite, Lisa Coles, Abhishek G. Sathe, Chi Chen, Jim Cloyd, Walter C. Low, Clifford J. Steer, and Wei Chen. 2021. "Quantitative Assessment of Occipital Metabolic and Energetic Changes in Parkinson’s Patients, Using In Vivo 31P MRS-Based Metabolic Imaging at 7T" Metabolites 11, no. 3: 145. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11030145