
Correcting for Naturally Occurring Mass Isotopologue Abundances in Stable-Isotope Tracing Experiments with PolyMID

{kind=link}
{kind=link}
{kind=link}
Abstract
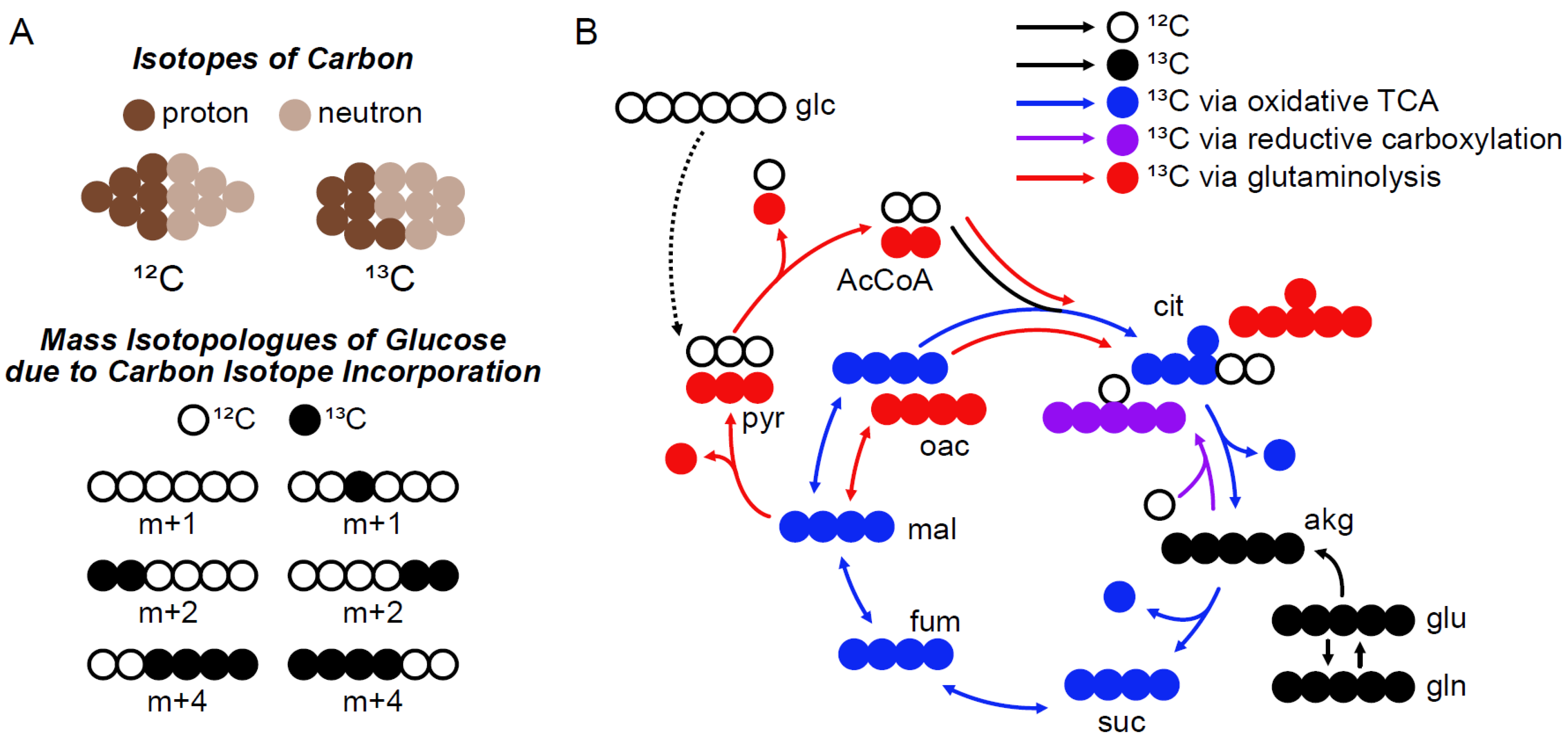
:1. Introduction
2. Results
2.1. General Correction Algorithm
2.2. The Correction Matrix
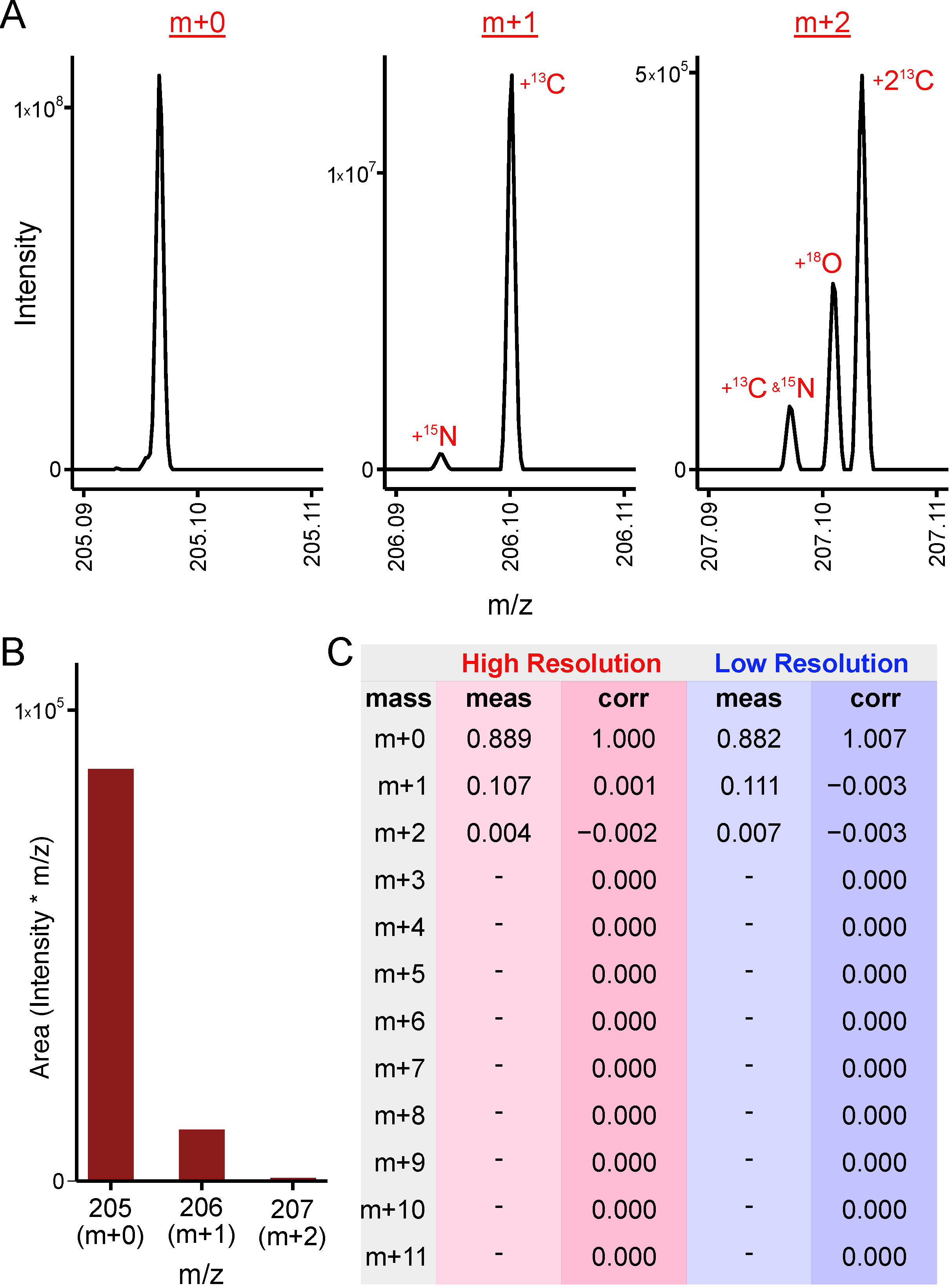
2.3. High Resolution Data
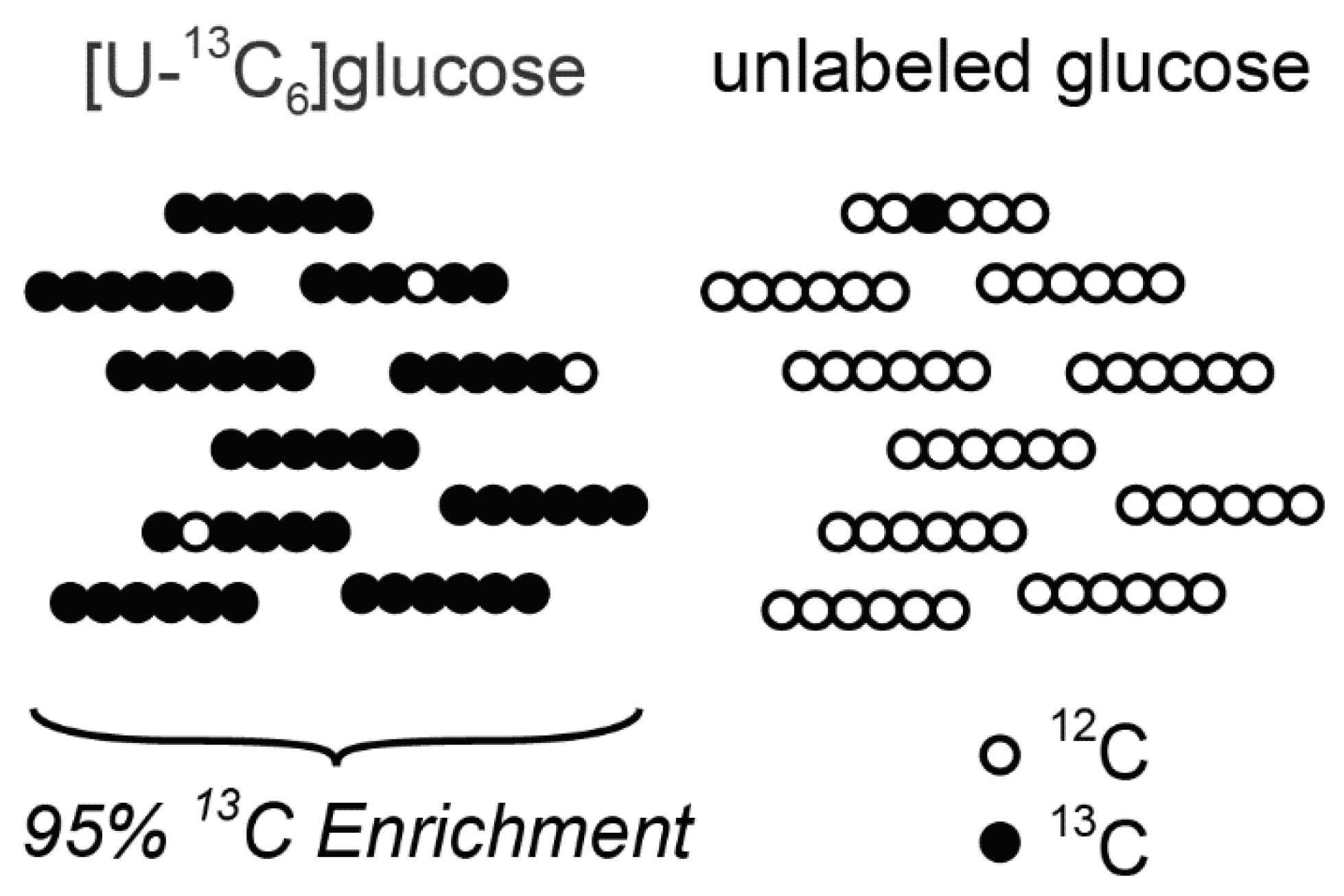
2.4. Tracer Purity and Tracer Atom Enrichment
3. Methods
3.1. PolyMID Availability and Installation
3.2. Running PolyMID-Correct
3.2.1. Inputs from a Text File
3.2.2. Inputs from the Command Line
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosman, K.J.R.; Taylor, P.D.P. Isotopic Compositions of the Elements 1997. Pure Appl. Chem. 1998, 70, 217–235. [Google Scholar] [CrossRef]
- Su, X.; Lu, W.; Rabinowitz, J.D. Metabolite Spectral Accuracy on Orbitraps. Anal. Chem. 2017, 89, 5940–5948. [Google Scholar] [CrossRef] [PubMed]
- Millard, P.; Delépine, B.; Guionnet, M.; Heuillet, M.; Bellvert, F.; Létisse, F. IsoCor: Isotope correction for high-resolution MS labeling experiments. Bioinformatics 2019, 35, 4484–4487. [Google Scholar] [CrossRef]
- Heinrich, P.; Kohler, C.; Ellmann, L.; Kuerner, P.; Spang, R.; Oefner, P.J.; Dettmer, K. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci. Rep. 2018, 8, 17910. [Google Scholar] [CrossRef]
- Carreer, W.J.; Flight, R.M.; Moseley, H.N. A Computational Framework for High-Throughput Isotopic Natural Abundance Correction of Omics-Level Ultra-High Resolution FT-MS Datasets. Metabolites 2013, 3, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strang, G. Introduction to Linear Algebra, 5th ed.; Wellesley-Cambridge Press: Wellesley, MA, USA, 2016. [Google Scholar]
- Fernandez, C.A.; Rosiers, C.D.; Previs, S.F.; David, F.; Brunengraber, H. Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom. 1996, 31, 255–262. [Google Scholar] [CrossRef]
- Lee, W.N.; Byerley, L.O.; Bergner, E.A.; Edmond, J. Mass isotopomer analysis: Theoretical and practical considerations. Biol. Mass Spectrom. 1991, 20, 451–458. [Google Scholar] [CrossRef]
- van Winden, W.A.; Wittmann, C.; Heinzle, E.; Heijnen, J.J. Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol. Bioeng. 2002, 80, 477–479. [Google Scholar] [CrossRef] [PubMed]
- Yergey, J. A General Approach to Calculating Isotopic Distributions for Mass Spectrometry. Int. J. Mass Spectrom. Ion Phys. 1983, 52, 337–349. [Google Scholar] [CrossRef]
- Vacanti, N.M.; Divakaruni, A.S.; Green, C.R.; Parker, S.J.; Henry, R.R.; Ciaraldi, T.P.; Murphy, A.N.; Metallo, C.M. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol. Cell 2014, 56, 425–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Heiden, M.G.V.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.P.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 481, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 2016, 12, 15–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, M.; Green, C.R.; Roberts, L.S.; Lee, Y.M.; McCarville, J.L.; Sanchez-Gurmaches, J.; Meurs, N.; Gengatharan, J.M.; Hover, J.D.; Phillips, S.A.; et al. Enzyme promiscuity drives branched-chain fatty acid synthesis in adipose tissues. Nat. Chem. Biol. 2018, 14, 1021–1031. [Google Scholar] [CrossRef]
- Zhang, H.; Badur, M.G.; Divakaruni, A.S.; Parker, S.J.; Jäger, C.; Hiller, K.; Murphy, A.N.; Metallo, C.M. Distinct Metabolic States Can Support Self-Renewal and Lipogenesis in Human Pluripotent Stem Cells under Different Culture Conditions. Cell Rep. 2016, 16, 1536–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacanti, N.M.; Metallo, C.M. Exploring metabolic pathways that contribute to the stem cell phenotype. Biochim. Biophys. Acta 2013, 1830, 2361–2369. [Google Scholar] [CrossRef]
- Ghosh, A.; Ando, D.; Gin, J.; Runguphan, W.; Denby, C.; Wang, G.; Baidoo, E.E.K.; Shymansky, C.; Keasling, J.D.; Martín, H.G. (13)C Metabolic Flux Analysis for Systematic Metabolic Engineering of S. cerevisiae for Overproduction of Fatty Acids. Front. Bioeng. Biotechnol. 2016, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Berry, D.; Loy, A. Stable-Isotope Probing of Human and Animal Microbiome Function. Trends Microbiol. 2018, 26, 999–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, H.J.; Socciarelli, F.; Vacanti, N.M.; Haugen, M.H.; Zhu, Y.; Siavelis, I.; Fernandez-Woodbridge, A.; Aure, M.R.; Sennblad, B.; Vesterlund, M.; et al. Breast cancer quantitative proteome and proteogenomic landscape. Nat. Commun. 2019, 10, 1600. [Google Scholar] [CrossRef] [PubMed]
- Sacco, F.; Seelig, A.; Humphrey, S.J.; Krahmer, N.; Volta, F.; Reggio, A.; Marchetti, P.; Gerdes, J.; Mann, M. Phosphoproteomics Reveals the GSK3-PDX1 Axis as a Key Pathogenic Signaling Node in Diabetic Islets. Cell Metab. 2019, 29, 1422–1432.e3. [Google Scholar] [CrossRef]
- Humphrey, S.J.; James, D.E.; Mann, M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Panizza, E.; Branca, R.M.M.; Oliviusson, P.; Orre, L.M.; Lehtiö, J. Isoelectric point-based fractionation by HiRIEF coupled to LC-MS allows for in-depth quantitative analysis of the phosphoproteome. Sci. Rep. 2017, 7, 4513. [Google Scholar] [CrossRef]
- Panizza, E.; Zhang, L.; Fontana, J.M.; Hamada, K.; Svensson, D.; Akkuratov, E.E.; Scott, L.; Mikoshiba, K.; Brismar, H.; Lehtiö, J.; et al. Ouabain-regulated phosphoproteome reveals molecular mechanisms for Na(+), K(+)-ATPase control of cell adhesion, proliferation, and survival. FASEB J. 2019, 33, 10193–10206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vacanti, N.M. The Fundamentals of Constructing and Interpreting Heat Maps. Methods Mol. Biol. 2019, 1862, 279–291. [Google Scholar] [PubMed]
- Jeong, H.; Vacanti, N.M. Systemic vitamin intake impacting tissue proteomes. Nutr. Metab. Lond. 2020, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Metallo, C.M.; Vander Heiden, M.G. Understanding metabolic regulation and its influence on cell physiology. Mol. Cell 2013, 49, 388–398. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, H.; Yu, Y.; Johansson, H.J.; Schroeder, F.C.; Lehtiö, J.; Vacanti, N.M. Correcting for Naturally Occurring Mass Isotopologue Abundances in Stable-Isotope Tracing Experiments with PolyMID. Metabolites 2021, 11, 310. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050310
Jeong H, Yu Y, Johansson HJ, Schroeder FC, Lehtiö J, Vacanti NM. Correcting for Naturally Occurring Mass Isotopologue Abundances in Stable-Isotope Tracing Experiments with PolyMID. Metabolites. 2021; 11(5):310. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050310
Chicago/Turabian StyleJeong, Heesoo, Yan Yu, Henrik J. Johansson, Frank C. Schroeder, Janne Lehtiö, and Nathaniel M. Vacanti. 2021. "Correcting for Naturally Occurring Mass Isotopologue Abundances in Stable-Isotope Tracing Experiments with PolyMID" Metabolites 11, no. 5: 310. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050310