
Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease



1
Guangdong Provincial Key Laboratory of Medical Molecular Diagnostics, Institute of Biochemistry and Molecular Biology, Institute of Aging Research, Guangdong Medical University, Dongguan 523808, China
2
Center for Human Tissues and Organs Degeneration, Institute of Biomedicine and Biotechnology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Metabolites 2021, 11(5), 320; https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050320
Submission received: 6 April 2021
/
Revised: 10 May 2021
/
Accepted: 12 May 2021
/
Published: 17 May 2021
(This article belongs to the Special Issue Steroids in Non-alcoholic Fatty Liver Disease)
Abstract
:Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease and may progress to cirrhosis or even hepatocellular carcinoma. A number of steroid hormones are important regulators of lipid homeostasis through fine tuning the expression of genes related to lipid synthesis, export, and metabolism. Dysregulation of such pathways has been implicated in the pathogenesis of NAFLD. The aim of this review is to clarify the potential impact of steroid hormones on NAFLD. We also highlight potential interventions through modulating steroid hormone levels or the activities of their cognate receptors as therapeutic strategies for preventing NAFLD.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) has been increasingly recognized as a worldwide health issue and is estimated to affect 20–30% of the general population [1]. It is a progressive disease ranging from steatosis to nonalcoholic steatohepatitis (NASH), which is a risk for the development of cirrhosis and hepatocellular carcinoma (HCC) [2,3]. Most patients with NAFLD have simple steatosis in the absence of hepatocyte injury. However, approximately 10–30% of patients with NAFLD develop NASH characterized by inflammation and fibrosis [4]. Over the last decade, it has been shown that NAFLD is a multisystem disease, affecting several extrahepatic organs and regulatory pathways [5]. NAFLD is tightly associated with obesity, metabolic syndrome, and type 2 diabetes [6]. All of these complications related to NAFLD pose significant health and economic burdens on patients and society.
The pathogenesis of NAFLD is complex and described by a “multiple-hit” hypothesis, which has been proposed to supersede the outdated “two-hit” hypothesis [7]. The “multiple-hit” hypothesis highlights the importance of genetic and epigenetic factors, nutritional factors, intestinal microbiota, insulin resistance, and hormones secreted from adipose tissue [7,8]. All forms of NAFLD tightly correlate with hepatic and peripheral insulin resistance in epidemiological, experimental, and human studies [9,10,11]. Insulin resistance upregulates de novo lipogenesis, inhibits β-oxidation of free fatty acids (FFAs) and increases the degradation of very-low-density lipoprotein (VLDL) in the liver, leading to an increase in hepatic fat accumulation [12,13,14]. It also promotes lipolysis rates in adipose tissue, leading to more fatty-acid efflux to the liver [15]. The intestinal microbiota has been found to alter bile acid metabolism to affect hepatic lipid handling and alter host immunity contributing to NASH [16]. There is increasing evidence reporting adiponectin as an adipokine with anti-inflammatory and antifibrogenic activity protecting liver parenchyma against steatosis and apoptosis [17].
Lipid accumulation within hepatocytes caused by these risk factors leads to steatosis, defined as the presence of fat comprising 5% of liver weight [18]. Exaggerated lipid accumulation can cause toxicity via diverse mechanisms. For instance, toxic lipid metabolites derived from FFAs impose oxidative stress on hepatocytes [19]. Furthermore, oxidative stress affects the function of the endoplasmic reticulum and mitochondria, as well as contributes to hepatic inflammation and fibrosis [20].
Steroid hormones have been discovered to play important roles in lipid metabolism. Deregulation of steroid hormone level or bioactivity has been implicated in various degree of hepatocellular damages. In Cushing’s syndrome, the high circulating steroid hormone (glucocorticoid) level causes visceral obesity, insulin resistance, and hepatic steatosis [21]. Activation of the renin–angiotensin–aldosterone system modulated by multiple steroid hormones and corresponding receptors causes liver inflammation and fibrosis [22,23]. Moreover, steroid hormone receptor-regulated target genes are involved in both cholesterol and fatty-acid metabolism [24] and, therefore, have been implicated in the pathogenesis of NAFLD.
In this review, we discuss the roles of steroid hormones in the pathogenesis of NAFLD. It is widely known that steroid hormones have crucial clinical and therapeutic implications in many diseases. Such knowledge may provide insight to develop novel therapy strategies for NAFLD.
2. Steroid Hormones and Cognate Receptors
Steroid hormones act as chemical messengers in the body and regulate a wide variety of physiological processes including development, growth, reproduction, and metabolism. They are all derived from cholesterol via a sequential series of enzyme-catalyzed reactions. Further modifications of steroid structure and function can occur in many tissues of the body such as the liver and brain [25]. The main classes of steroid hormones include sex hormones, glucocorticoids, mineralocorticoids, and vitamin D. Sex steroid hormones can be divided into three classes: estrogens, androgens, and progestins. At the cellular level, steroid hormones mediate their physiologic effects through binding to their cognate receptors, most of which are nuclear receptors. Once the hormone is bound to its receptor, dimerization of the receptors occurs [26]. Following the interaction of the steroid hormone–receptor complex with target genes, coregulators are recruited for activation or suppression of specific genes. Mutation or aberrant expression of steroid hormone receptors or coregulators affects the normal function of steroid hormones and leads to the development of diseases [27].
2.1. Steroid Hormones
2.1.1. Estrogens
Estrogens are the main female sex steroid hormones associated with reproductive organs and responsible for the development of female sexual characteristics [28]. Estrogen (E1), estradiol (E2 or 17β-estradiol), and estriol (E3) are the three major forms of physiological estrogens [29].During the menstrual cycle, estrogen levels produced by the gonads fluctuate in response to the changes in follicle-stimulating hormone and pituitary luteinizing hormone. Estrogen and its derivatives have been heavily researched for their clinical applications in postmenopausal women for reducing the risk of coronary arterial disease, osteoporosis, and mortality [30]. Estrogen and its receptors modulate pathological processes including breast and endometrial cancer [31]. They also perform important functions in the secretion of sebum [32], deposition of fat, lipogenesis, and insulin sensitivity [33,34,35].
2.1.2. Androgens
Androgens are the primary male sex steroid hormones that exert significant effects on male sexual and reproductive function [36]. Androgens belong to a group of hormones that include testosterone, dihydrotestosterone (DHT), dehydroepiandrosterone (DHEA), androstenedione, androstenone, and androstenediol. They play essential roles mainly through androgen receptor (AR), which is an important member of the nuclear receptor superfamily [37]. Androgens also seem to influence the development and growth of other organs, such as skeletal muscle, liver, and kidney [38]. Moreover, it has been reported that androgens are beneficial for improving the cognition in the elderly population [39].
2.1.3. Progestogens
Progestogens are also a class of steroid hormones that bind to progesterone receptor (PR) [40]. Progesterone is the major and most important progestogen in the body. It is considered to play essential roles in both the female and the male reproductive system [41]. Specifically, progesterone displays essential roles in the maintenance of pregnancy, regulation of menstrual cycle, and preparation of the mammary glands for lactation and breastfeeding after parturition in women, whereas progesterone regulates testosterone synthesis, spermiogenesis, and sperm capacitation in men. In addition, progestogens were shown to be used in menopausal hormone therapy and transgender hormone therapy for transgender women [42].
2.1.4. Glucocorticoids
Synthesized in the adrenal cortex, glucocorticoids are a class of steroid hormones that bind to glucocorticoid receptor (GR) [43]. Glucocorticoids such as cortisol play roles in mediating stress-related metabolic regulation, as well as immune modulation. It is one of the most potent anti-inflammatory compounds known, and it is used in treating diseases caused by an overactive immune system [44]. An activated glucocorticoid receptor–glucocorticoid complex suppresses inflammation by preventing the translocation of proinflammatory transcription factors from the cytoplasm into the nucleus [45]. Glucocorticoids are also of key significance in fetal development and body fluid homeostasis [46]. Glucocorticoids stimulate protein degradation in muscle, skin, and lymphoid tissue; consequently, the released amino acids can be used for glucose and glycogen synthesis [47]. Glucocorticoids seem to be involved in mitochondrial RNA and protein synthesis, regulating mitochondrial respiration and oxidative phosphorylation [48].
2.1.5. Mineralocorticoids
Mineralocorticoids belong to another class of steroid hormones produced in the adrenal cortex, which are important in the regulation of extracellular volume homeostasis [49,50]. As the primary mineralocorticoid, aldosterone regulates the balance of salt and water. Blood volume and pressure are increased by aldosterone, which mainly promotes active reabsorption of sodium and passive reabsorption of water, as well as the active secretion of potassium into the principal cells of the cortical collecting tubule in the kidney [51]. Aldosterone increases blood pressure, and activation of the mineralocorticoid receptor (MR) can affect cardiac function via induction of inflammation and fibrosis [52]. Moreover, MR is reportedly associated with adipocyte dysfunction and vascular abnormalities, which may lead to obesity and insulin resistance [53,54,55].
2.1.6. Vitamin D
Vitamin D is recognized as a class of steroid hormone. Among all types of vitamin D, vitamin D3 (known as cholecalciferol) and vitamin D2 (ergocalciferol) are the most important forms [56]. Cholecalciferol can be produced in the skin exposed to ultraviolet (UV) rays from sunlight. The ingestion of cholecalciferol and ergocalciferol from the diet is also a source of vitamin D [57]. The active form, calcitriol, which takes effects via vitamin D receptor (VDR), is responsible for increasing the absorption of magnesium, calcium, and phosphate [58]. It is essential in calcium homeostasis and metabolism affecting bone growth and remodeling. Moreover, calcitriol has been reported to attenuate systemic inflammation and plays a protective role in liver structure [59].
2.2. Steroid Hormone Receptors
Steroid receptors are critical for normal liver function, while NAFLD is associated with inappropriate transcriptional regulation by steroid receptors. The classical steroid hormone receptor family includes estrogen receptor (ER), AR, PR, GR, MR [60,61], and VDR. It is well known that most of these receptors are nuclear receptors. Nuclear receptors are transcription factors which sense changing environmental or hormonal signals and regulate gene expression. To support this function, all nuclear receptors share the same basic structure: an amino-terminal transcriptional activation domain containing activating function 1 (AF-1); a DNA-binding domain; a hinge region; a carboxy-terminal ligand-binding domain containing AF-2 [62]. Following the binding of steroid hormones, all these regions contribute to the changes in gene expression occurring through the combined action of coregulators and chromatin modifiers.
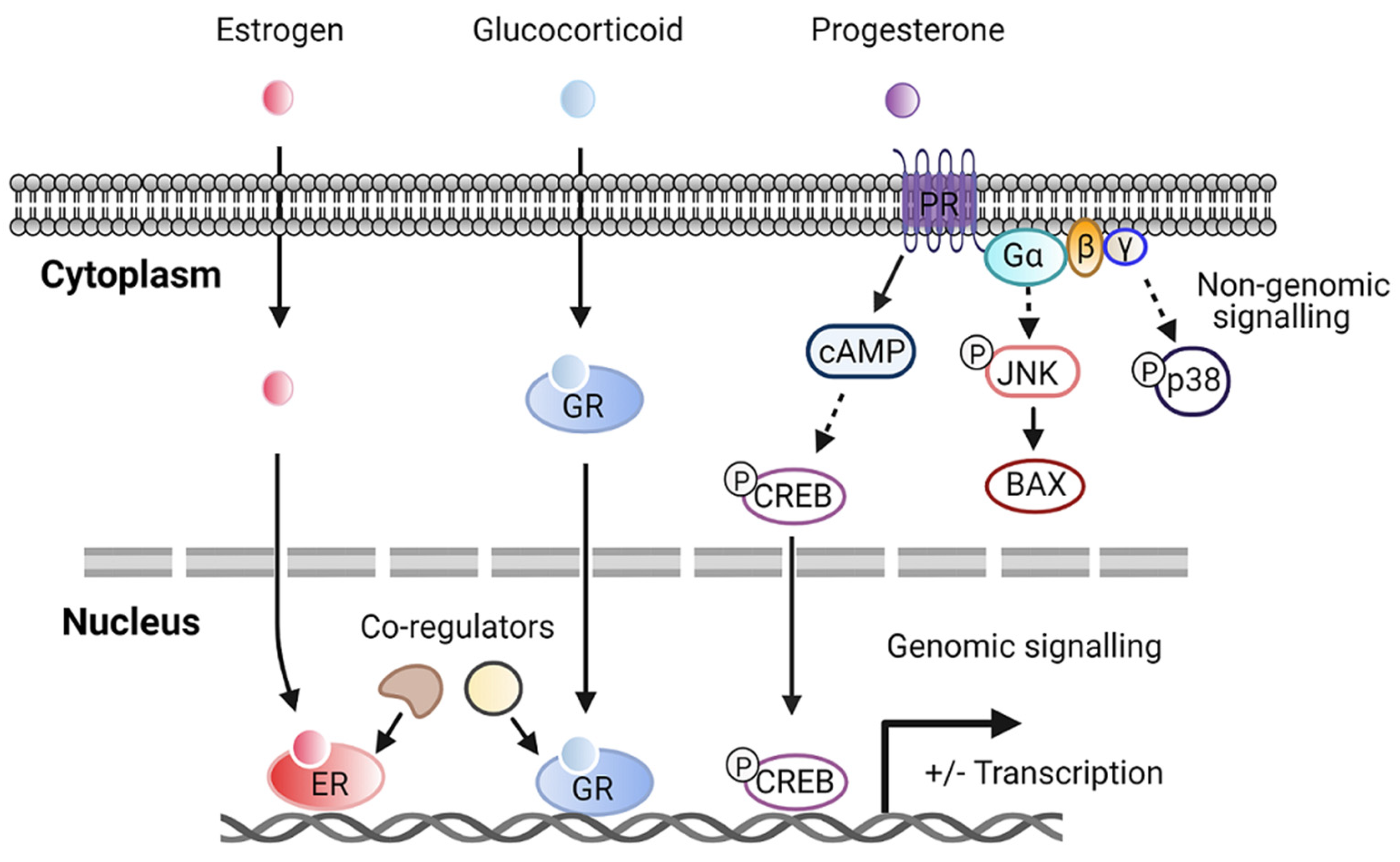
Although steroid hormone receptors are notably nuclear receptors, it has been reported that different steroid receptors in fact exist in extranuclear cellular pools. Steroid hormones may enter cells through the plasma membrane and bind to their receptors localized in the cytoplasm before translocating to the nucleus to regulate the transcription of target genes. In addition, new pathways of steroid hormones signaling through cell surface receptors contribute to more rapid, “non-nuclear”, or non-genomic signaling. Activated steroid receptors in the plasma membrane can stimulate second messenger cascades, interacting with several signaling molecules such as phosphatidylinositol 3-kinase (PI3K)/Akt, Ras/Raf-1, protein kinase A (PKA), and protein kinase C (PKC), leading to cell proliferation [63]. The molecular function of steroid hormones depends on the subcellular distribution of the receptors (Figure 1). Some sex hormone receptors, such as estrogen receptor α and β (ERα, ERβ), are found mainly in the nucleus [64,65]. Glucocorticoid receptors are predominantly localized in the cytoplasm and translocate to the nucleus upon binding to the ligand [66]. Recent evidence has demonstrated that membrane PRs mediate most of the nongenomic signaling of progesterone actions [67]. Progesterone-bound membrane PR potentiates cAMP production and cAMP responsive element-binding protein (CREB)-mediated transcription [68]. Furthermore, the activation of G protein via membrane PR induces the activation of JNK and p38 signaling pathways [67,68]. For novel therapeutic targets under consideration, priority should be given to these steroid receptors in nuclear or plasma membranes.
3. Role of Steroid Hormones in Hepatic Steatosis and Metabolism
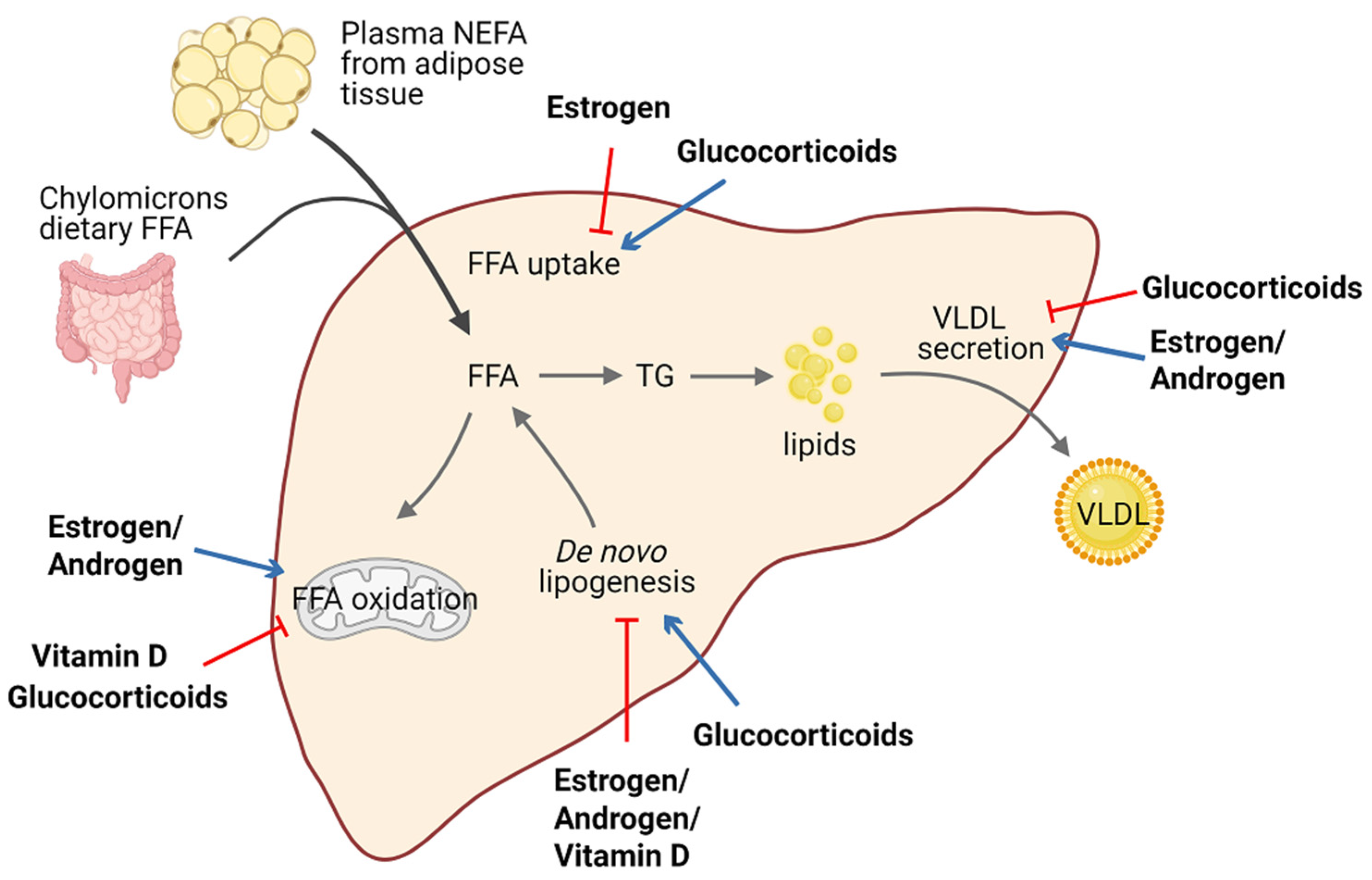
Lipid homeostasis is fine-tuned through four major pathways: uptake of circulating lipids, de novo lipogenesis, fatty-acid oxidation, and export of lipids in VLDL [69]. Hepatic steatosis occurs when there is an imbalance between lipid acquisition and lipid disposal. Increasing data have shown that steroid hormones are involved in hepatic steatosis by modulating these processes (Figure 2). We summarize steroid hormone-mediated effects of hepatic steatosis in animal models in Table 1.
It is well established that estrogen and ERs are involved in the pathogenesis of NAFLD. For example, ERα-deficient mice have disrupted lipid metabolism, such as decreased fatty-acid oxidation and increased de novo lipogenesis, leading to elevated lipid accumulation in the liver compared with normal mice [70,71,72]. Similarly, downstream blocking of ERα contributed to higher visceral fat accumulation and reduced energy expenditure in female mice [34,70,73]. Meanwhile, E2 treatment promoted fatty-acid oxidation in the liver by increasing the expression of the fatty-acid transport protein, carnitine palmitoyltransferase 1 (CPT-1) [34]. In ovariectomized (OVX) female mice, estrogen replacement improves insulin sensitivity and facilitates the VLDL-mediated export of lipids from the liver by increasing hepatic VLDL-TG production [74]. Interestingly, long-term therapy of tamoxifen, a selective estrogen receptor modulator, can increase the risk of NAFLD in breast cancer patients [75].

{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of steroid hormones effects on hepatic steatosis. ↑ increased; ↓ decreased.
| Steroid Hormones | Model(s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - Female ERα-deficient mice fed HFD for 10 weeks [71] - Male hepatic ERα-deficient mice fed HFD for 12 weeks [72] - OVX mice treated with E2 and fed HFD for 6 weeks [74] | - Liver weight, hepatic steatosis, and ALT level ↑ - Hepatic steatosis and insulin resistance ↑ - Hepatic steatosis and insulin- mediated suppression of VLDL secretion ↓ |
| Androgen | - Male hepatic AR-deficient mice fed HFD for 8 weeks [76] | - Body weight, hepatic steatosis, and insulin resistance ↑ |
| Glucocorticoid | - db/db mice treated with GC shRNA for 14 days [77] - SD rats treated with exogenous corticosterone and fed HFD for 16 days [78] | - Hepatic steatosis and genes critical for lipid storage and transport ↓ - Hepatic steatosis, uptake of FA into liver, and ALT level ↑ |
| Mineralocorticoid | - Myeloid MR-deficient ob/ob mice [79] - Aldosterone synthase-deficient mice fed HFD for 12 weeks [80] | - Hepatic steatosis, lipogenesis, and insulin resistance ↓ - HFD-feeding-induced hepatic steatosis ↓ |
| Vitamin D | - C57BL6 mice fed a high-fat/ high-sucrose diet followed by treatment with vitamin D for 15 weeks [81] - SD rats fed HFD followed by treatment with vitamin D for 16 weeks [82] | - Hepatic steatosis and hepatic de novo lipogenesis ↓ - Liver weight, hepatic steatosis, and ALT level ↓ |
A meta-analysis revealed that lower serum testosterone levels are associated with men with NAFLD [83,84]. Testosterone deficiency in men displayed an increased accumulation of visceral adipose tissue and insulin resistance, which are factors favoring the development of hepatic steatosis [85]. Similar results were confirmed in hepatic AR-deficient mice, indicating that AR might play a role in the suppression of NAFLD [76,86]. Indeed, androgen/AR signaling was revealed to suppress fatty-acid synthesis by decreasing the expression of sterol regulatory element-binding protein (SREBP) and to induce insulin sensitivity by modulating phosphoinositide-3 kinase activity [24].
Conversely, glucocorticoids drive the expression of lipogenic genes including fatty-acid synthase (FASN) and acetyl-coA carboxylase 1 (Acaca) [87], stimulate de novo lipogenesis, and block VLDL secretion, thus resulting in hepatic steatosis [88]. Corticosterone, administered to rodent models to increase basal glucocorticoids levels, also induces an increase in Cluster Determinant 36 (CD36) expression that facilitates fatty-acid uptake, favoring the progression of hepatic steatosis [78]. Of note, patients with glucocorticoid excess may develop hepatic steatosis, as well as obesity and insulin resistance, in a significant proportion of cases [89,90]. Moreover, fatty liver development also represents a typical side-effect of long-term systemic glucocorticoids treatment during anti-inflammatory therapy. Additionally, mice deficient in hepatic GR displayed lower hepatic TG content and elevated levels of ketone bodies in the serum, indicating that disruption of GR expression reduces steatosis in db/db animals partly by triggering hepatic fatty-acid oxidation and ketogenesis [77].
Macrophage-specific deficiency of MR protects mice from hepatic steatosis and insulin resistance through the ERα/HGF/Met pathway [79]. In addition, aldosterone deficiency attenuates high-fat feeding-induced hepatic steatosis, although the underlying mechanism remains elusive [80]. On the other hand, elevated aldosterone level seems to enhance hepatic FFAs via modulation of lipogenesis and lipolysis [91].
Vitamin D has been proven to protect against high-fat diet-induced fatty liver by acting on the expression of de novo lipogenesis-related genes, including FASN and Acaca, and fatty-acid oxidation-related genes such as the acetyl-coA oxidase (Acox) [81,82]. In humans, hepatic VDR expression is inversely correlated with steatosis severity [92]. Activation of VDR in hepatic macrophages via its cognate ligand has been uncovered to ameliorate steatosis and insulin resistance in experimental studies [93].
4. Role of Steroid Hormones in Hepatic Inflammation and Fibrosis
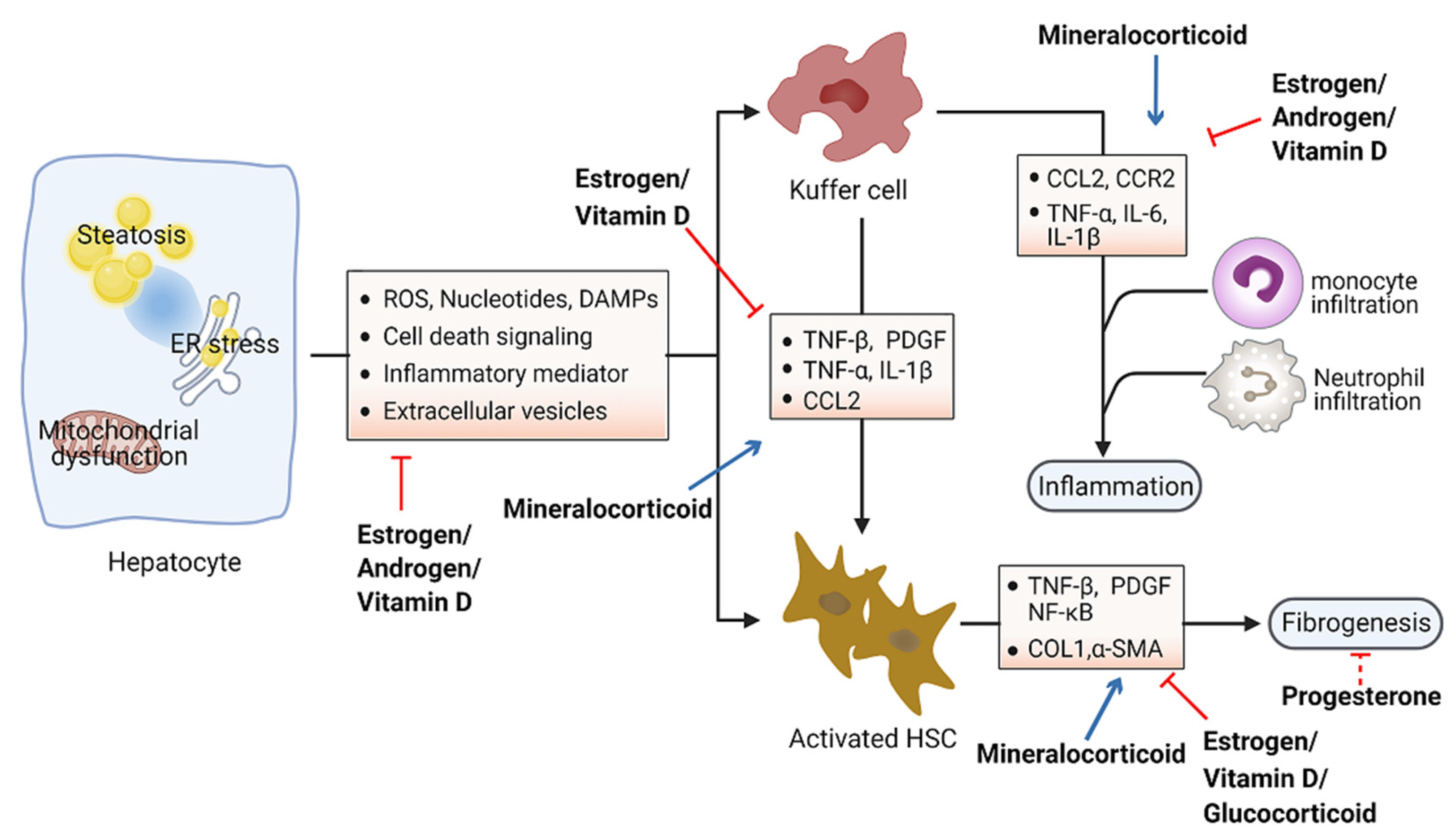
Accumulation of toxic lipids leads to ROS, endoplasmic reticulum (ER) stress, cell death, release of damage-associated molecular patterns (DAMPs), and secretion of inflammatory mediators and extracellular vesicles, which stimulate an inflammatory response in Kupffer cells and a fibrotic response in hepatic stellate cells (HSC) [94,95]. The inflammatory process includes recruitment of macrophages and neutrophils to the site of injured tissue, followed by production of proinflammatory chemokines and cytokines such as interleukin 1 (IL-1) and tumor necrosis factor alpha (TNF-α) [96,97]. Furthermore, initiation of the inflammatory acute-phase response of the liver is induced by these cytokines together with IL-6 [98]. Liver inflammation can lead to fibrosis that may eventually induce cirrhosis [99]. Hepatic fibrosis is characterized by the accumulation of extracellular matrix. As a crucial driver of fibrosis, HSCs can be transdifferentiated into proliferative and fibrogenic myofibroblasts [100]. Profibrogenic cytokines such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and transforming growth factor-β (TGF-β) are involved in the activation of HSCs and hepatic abnormal wound repair response [101,102,103]. Collectively, hepatocellular damage, inflammation, and fibrosis are the main characteristics of NASH [104,105]. Numerous studies have demonstrated the important effects of steroid hormones in the pathological progression of NASH (Figure 3). Here, we summarize steroid hormone-mediated effects on hepatic inflammation and fibrosis in animal models in Table 2.
Steroid hormones primarily display anti-inflammation and antifibrosis properties in the context of metabolic disorders. Estrogens have been found to directly suppress inflammatory processes in the liver. Women after menopause have decreased fatty-acid oxidation and increased lipogenesis in the liver, leading to the excessive accumulation of hepatic fat and culminating with inflammation [106,107]. Moreover, the generation of ROS and proinflammation cytokines was prevented by estrogen signaling in hepatocytes [108,109]. Estradiol treatment reduces hepatic inflammation in the animal model of NASH induced by methionine and a choline-deficient diet [110]. Furthermore, female mice with estrogen deficiency induced by OVX operation were observed with enhanced levels of macrophages infiltration and proinflammatory cytokines in the liver, including TNF-α, IL-6, C–C motif chemokine ligand 2 (CCL2), and C–C motif chemokine receptor 2 (CCR2) [111]. Hepatic fibrosis has also been shown to be associated with estrogen level. The risk of liver fibrosis increased significantly in a zebrafish experiment with ovarian senescence [112]. Similarly, a long duration of estrogen deficiency poses a high risk of hepatic fibrosis among postmenopausal women with NAFLD [113]. However, postmenopausal estrogen therapy may be considered for preventing the occurrence of liver fibrosis [114]. OVX female mice fed with a high-fat and high-cholesterol diet had obvious liver fibrosis with upregulated expression of collagen I α1, which could be improved by estrogen replacement therapy [109]. Additionally, β-LGND2, an ER-β-selective agonist, partially prevented inflammatory cell infiltration and liver fibrosis, providing a therapeutic benefit in NASH [115].
It has been shown that testosterone may increase the level of anti-inflammatory cytokines and reduce the level of proinflammatory cytokines, thereby exerting anti-inflammatory properties in patients with diabetes mellitus or coronary artery disease [116]. In experimental animal models of NAFLD, androgens were shown to be able to prevent NASH progression by acting on proinflammatory cytokines such as TNF-α and IL-6 [117].
Although increasing levels of progesterone have been associated with the development of systemic insulin resistance [118], little is known regarding the role of serum progesterone in NAFLD. Early studies have shown that treatment with progesterone in rabbits exerts a protective effect on hepatocytes against vacuolization and inflammation, accompanied by a low level of fibrosis [119].
Glucocorticoid hormones are considered the most widely used anti-inflammatory drugs. However, the effects of glucocorticoids on hepatic inflammation are still unclear, particularly in the progression of NAFLD. One study suggested that glucocorticoids may suppress the development of liver inflammation, considering that the perivascular infiltration of small mononuclear cells was diminished in the liver of mice following treatment with dexamethasone, a synthetic GR ligand [120]. Meanwhile, glucocorticoids suppress fibrotic gene expression including collagen I α1/2 by activating GR, which differentially regulates liver injury and hepatic fibrosis in HSCs or immune cells [120].
Table 2.
Summary of the effects of steroid hormones on hepatic inflammation and fibrosis. ↑ increased; ↓ decreased.
Table 2.
Summary of the effects of steroid hormones on hepatic inflammation and fibrosis. ↑ increased; ↓ decreased.
| Steroid Hormones | Model (s) Used | Major Phenotypes Examined |
|---|---|---|
| Estrogen | - OVX mice fed HFD and high-fructose water for 12 weeks [111] - OVX mice fed a high-fat and high-cholic-acid diet for 6 weeks [109] - Old female zebrafish fed a high-calorie diet for 24 weeks [112] - Orchidectomized C57/BL6 mice treated with estradiol benzoate-fed MCD for 4 weeks [110] - Male C57BL6 mice treated with β-LGND2 and fed HFD for 10 weeks [115] | - Hepatic inflammation and fibrosis, ALT level and ballooning degeneration ↑ - Liver fibrosis, inflammation, and hepatocyte ballooning degeneration ↑ - Liver fibrosis, IL-6, and TNF-β ↑ - Hepatic inflammation, MyD88, and IL-6 ↓ - Hepatic steatosis and insulin resistance ↓ |
| Androgen | - Orchidectomized male SD rats treated with dihydrotestosterone and fed HFD for 75 days [117] | - Portal inflammation, TNF-α, and IL-6 ↓ |
| Progesterone | - Hepatic fibrosis model of New Zealand male rabbits treated with progesterone for 180 days [119] | - Liver fibrosis, fat metamorphosis, and inflammatory infiltrate ↓ |
| Glucocorticoid | - Immune cell-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [120] - HSC-specific GR-knockout mice treated with CCl4 and dexamethasone for 6 weeks [120] | - Inflammation and monocyte recruitment ↓ - Hepatic fibrosis and fibrotic gene expression ↓ |
| Mineralocorticoid | - C57BL6 mice fed HFFD mixed with eplerenone for 12 weeks [121] - Male C57BL6 mice fed a CDAA diet for 22 weeks with eplerenone [122] - Male SD rats treated with aldosterone for 4 weeks [123] | - Lipid accumulation, lobular inflammation, and collagen deposition ↓ - Hepatic fibrosis, steatosis, and inflammation ↓ - Hepatic fibrosis, oxidative stress, and DNA double-strand breaks ↑ |
| Vitamin D | - Vitamin D-deficient SD rats fed WD for 10 weeks [124] - CDAA diet-induced rat NASH model with phototherapy for 6 or 12 weeks [125] | - Foci of lobular inflammation and ballooning degeneration ↑ - Collagen fibrosis, insulin and leptin resistance, inflammation, and HSC activation ↓ |
Aldosterone was observed to promote liver inflammation. ROS production can be stimulated by aldosterone in several tissues [126]. Blockade of aldosterone interaction with MR by eplerenone impeded macrophage infiltration and suppressed the expression of TNF-α and multiple copies in T-cell lymphoma-1 (MCT-1) in Kupffer cells, consequently ameliorating the development of NASH in mice [121]. Unlike the other steroids, mineralocorticoids foster the development of fibrosis by upregulating serum- and glucocorticoid-inducible kinase 1 (SGK1), which increases NF-κB, thus promoting fibrosis [127]. Additionally, aldosterone treatment could lead to liver fibrosis in male rats independent of blood pressure via promoting the expression of several fibrosis-associated genes (e.g., TGF-β, α-SMA) [123]. Furthermore, the expression of MR in hepatic stellate cells correlates with inflammation and fibrosis development in choline-deficient and amino-acid-defined diet-induced NASH [122]. Specific MR blockade with eplerenone effectively ameliorated histological steatosis and hepatic fibrosis in a mouse model of NASH. These data provide the basis for therapeutic exploitation of MR blockade for treatment of NASH [122].
Hepatic inflammation can also be affected by vitamin D through downregulation of the expression of Toll-like receptors on Kupffer cells [128]. Vitamin D deficiency causes upregulation of inflammation and oxidative stress genes [124]. Furthermore, restoration of vitamin D could effectively improve TNF-α-modulated immunological abnormalities in a diet-induced steatohepatitis rat model [125]. Moreover, serum vitamin D was significantly decreased in NAFLD patients with advanced liver fibrosis, suggesting that vitamin D might be associated with the progression of liver fibrosis [129]. Further study showed that vitamin D plays an antifibrotic role by suppressing the activation of the HSC-mediated TGF-β signaling pathway, inhibiting the accumulation of profibrotic extracellular matrix proteins [130] and the expression of profibrotic genes including collagen and α-smooth muscle actin (α-SMA) [131,132].
5. Conclusions and Perspective
Steroid hormones bound to cognate receptors perform diverse functions in lipid metabolism, modulating hepatic steatosis, as well as inflammation and fibrosis. In particular, the physiological changes can be made via the relevant gene expressions of lipid metabolism being affected by receptors directly or indirectly [63,117]. Specifically, estrogens, androgens, and vitamin D and their corresponding receptors play anti-steatosis, anti-inflammatory, and antifibrosis roles, while glucocorticoids play pro-steatosis, anti-inflammatory, and antifibrosis roles, and aldosterone plays pro-steatosis, proinflammation, and pro-fibrosis roles.
The complexity of NAFLD is presented not only in its association with other clinical conditions which include intricate organ crosstalk but also in its various and enigmatic biochemical mechanisms. Indeed, the increasing fatty acids from lipolysis of fat adipose tissue, the proceeding de novo lipogenesis, and the inhibition of β-oxidation of fatty acid in the mitochondria and inflammation from liver injury cause abnormal hepatic lipid accumulation and, eventually, NAFLD development [13]. Considering the existing differential biological functions of each steroid hormone under physiological conditions, especially related to these metabolism pathways, it is not surprising that steroid hormones have major impacts on the pathogenesis of NAFLD.
A number of studies on the general adult population have reported a higher prevalence of NAFLD in men than women [133,134,135]. Interestingly, the prevalence of NAFLD becomes more common in aged women [134,136,137,138]. Specifically, the prevalence of NAFLD was significantly higher in postmenopausal obese women (60.2%) than in premenopausal obese women (42.9%) [139]. The prevalence of NAFLD in men and postmenopausal women compared with premenopausal women highlights the nature of the sex disparity of NAFLD influenced by sex steroid hormones [140,141]. Indeed, recent studies have confirmed that estrogen or androgen (and their receptors) is able to decrease the incidence of NAFLD in women or men, respectively. Understanding the molecular mechanisms that promote the sex difference in NAFLD development might help pave ways to developing better therapeutic strategies.
Although steady progress has been disclosed in the pathogenesis of NAFLD, significant challenges still exist in the development of a therapeutic agent. This review summarized the action of several steroid hormones on NAFLD, not only providing the molecular mechanisms underlying this action, but also implicating the potential strategies for treating NAFLD in the future, for example, the development of novel steroid hormone analogues and cautious application of HRT based on the effects of sex steroid hormones in the sex disparity of NAFLD. Furthermore, several recent studies have provided evidence for the roles of steroid hormones in NAFLD. Specifically, it has been shown that total testosterone is inversely related to NAFLD in men, and the previously increased use of testosterone replacement may be encouraged if more exploration studies could be warranted [142,143]. Another recent study implied that vitamin D supplementation has no effect in patients with NAFLD since there are no changes in lipid profile or liver enzymes; however, when combined with vitamin E, calcium, or omega-3 fatty-acid supplementation, potential benefits emerged in those patients [144]. Nevertheless, new therapeutic strategies may be required to reduce the impact of NAFLD on modern society.
Funding
This work was funded by the National Natural Science Foundation of China, grant numbers 82072493 and 81770882, Shenzhen Science and Technology Research Funding, grant numbers KQJSCX20180330170052049 and 20170502171625936, and the Guangdong Special Support Program, grant number 2017TQ04R394.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of Nafld and Nash: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Jou, J.; Choi, S.S.; Diehl, A.M. Mechanisms of Disease Progression in Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2008, 28, 370–379. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis among a Largely Middle-Aged Population Utilizing Ultrasound and Liver Biopsy: A Prospective Study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic Complications of Nonalcoholic Fatty Liver Disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef]
- Cusi, K. Role of Obesity and Lipotoxicity in the Development of Nonalcoholic Steatohepatitis: Pathophysiology and Clinical Implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (Nafld). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.T.; Francque, S.; Staels, B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (Nafld) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef]
- Utzschneider, K.M.; Kahn, S.E. Review: The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhang, M.; Liu, Q.; Xu, T.; Huang, T.; Yao, D.; Wong, C.W.; Liu, J.; Guan, M. 18beta-Glycyrrhetinic Acid Acts through Hepatocyte Nuclear Factor 4 Alpha to Modulate Lipid and Carbohydrate Metabolism. Pharmacol. Res. 2020, 157, 104840. [Google Scholar] [CrossRef]
- Guan, M.; Qu, L.; Tan, W.; Chen, L.; Wong, C.W. Hepatocyte Nuclear Factor-4 Alpha Regulates Liver Triglyceride Metabolism in Part through Secreted Phospholipase a(2) Gxiib. Hepatology 2011, 53, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Gusdon, A.M.; Song, K.X.; Qu, S. Nonalcoholic Fatty Liver Disease: Pathogenesis and Therapeutics from a Mitochondria-Centric Perspective. Oxid. Med. Cell. Longev. 2014, 2014, 637027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yang, M.; Wang, N.; Liu, Q.; Wang, B.; Huang, T.; Tong, Y.; Ming, Y.; Wong, C.W.; Liu, J.; et al. Andrographolide Modulates Hnf4alpha Activity Imparting on Hepatic Metabolism. Mol. Cell. Endocrinol. 2020, 513, 110867. [Google Scholar] [CrossRef] [PubMed]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of Non-Alcoholic Fatty Liver Disease. QJM 2010, 103, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of Nash. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef] [Green Version]
- Handa, P.; Maliken, B.D.; Nelson, J.E.; Morgan-Stevenson, V.; Messner, D.J.; Dhillon, B.K.; Klintworth, H.M.; Beauchamp, M.; Yeh, M.M.; Elfers, C.T.; et al. Reduced Adiponectin Signaling Due to Weight Gain Results in Nonalcoholic Steatohepatitis through Impaired Mitochondrial Biogenesis. Hepatology 2014, 60, 133–145. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Gramlich, T.; Liu, Y.C.; Matteoni, C.; Petrelli, M.; Goldblum, J.; Rybicki, L.; McCullough, A.J. Nonalcoholic Fatty Liver Disease: Assessment of Variability in Pathologic Interpretations. Mod. Pathol. 1998, 11, 560–565. [Google Scholar]
- Beste, L.A.; Leipertz, S.L.; Green, P.K.; Dominitz, J.A.; Ross, D.; Ioannou, G.N. Trends in Burden of Cirrhosis and Hepatocellular Carcinoma by Underlying Liver Disease in Us Veterans, 2001-2013. Gastroenterology 2015, 149, 1471–1482.e5. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic Reticulum Stress and Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Tarantino, G.; Finelli, C. Pathogenesis of Hepatic Steatosis: The Link between Hypercortisolism and Non-Alcoholic Fatty Liver Disease. World J. Gastroenterol. 2013, 19, 6735–6743. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Aloni-Grinstein, R.; Osher, E.; Rotter, V. Liver and Steroid Hormones-Can a Touch of P53 Make a Difference? Front. Endocrinol. 2019, 10, 374. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.M.; Fletcher, J.A.; Thyfault, J.P.; Rector, R.S. The Role of Angiotensin Ii in Nonalcoholic Steatohepatitis. Mol. Cell. Endocrinol. 2013, 378, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.A.; Lacin, S.; Abdel-Wahab, R.; Uemura, M.; Hassan, M.; Rashid, A.; Duda, D.G.; Kaseb, A.O. Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma: Is There a Role for the Androgen Receptor Pathway? Onco Targets Ther. 2017, 10, 1403–1412. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.L.; Auchus, R.J. The Molecular Biology, Biochemistry, and Physiology of Human Steroidogenesis and Its Disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.E.; Karpati, E.; Schuszter, K.; Toth, E.A.; Kiss, E.; Kulcsar, M.; Laszlo, G.; Matko, J. A Dynamic Network of Estrogen Receptors in Murine Lymphocytes: Fine-Tuning the Immune Response. J. Leukoc. Biol. 2014, 96, 857–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, M.K.; Paramanik, V. Role of Steroid Hormone Coregulators in Health and Disease. Horm. Res. 2009, 71, 194–200. [Google Scholar] [CrossRef]
- McLachlan, J.A.; Newbold, R.R. Estrogens and Development. Environ Health Perspect 1987, 75, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Shen, Y.; Li, R. Estrogen Synthesis and Signaling Pathways During Aging: From Periphery to Brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Lobo, R.A.; Pickar, J.H.; Stevenson, J.C.; Mack, W.J.; Hodis, H.N. Back to the Future: Hormone Replacement Therapy as Part of a Prevention Strategy for Women at the Onset of Menopause. Atherosclerosis 2016, 254, 282–290. [Google Scholar] [CrossRef]
- Trabert, B.; Wentzensen, N.; Yang, H.P.; Sherman, M.E.; Hollenbeck, A.R.; Park, Y.; Brinton, L.A. Is Estrogen Plus Progestin Menopausal Hormone Therapy Safe with Respect to Endometrial Cancer Risk? Int. J. Cancer 2013, 132, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Pierard-Franchimont, C.; Pierard, G.E. Postmenopausal Aging of the Sebaceous Follicle: A Comparison between Women Receiving Hormone Replacement Therapy or Not. Dermatology 2002, 204, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Camporez, J.P.; Lyu, K.; Goldberg, E.L.; Zhang, D.; Cline, G.W.; Jurczak, M.J.; Dixit, V.D.; Petersen, K.F.; Shulman, G.I. Anti-Inflammatory Effects of Oestrogen Mediate the Sexual Dimorphic Response to Lipid-Induced Insulin Resistance. J. Physiol. 2019, 597, 3885–3903. [Google Scholar] [CrossRef]
- Palmisano, B.T.; Zhu, L.; Stafford, J.M. Role of Estrogens in the Regulation of Liver Lipid Metabolism. Adv. Exp. Med. Biol. 2017, 1043, 227–256. [Google Scholar] [PubMed] [Green Version]
- Shen, M.; Kumar, S.P.; Shi, H. Estradiol Regulates Insulin Signaling and Inflammation in Adipose Tissue. Horm. Mol. Biol. Clin. Investig. 2014, 17, 99–107. [Google Scholar] [CrossRef]
- Wilson, J.D.; Griffin, J.E.; Leshin, M.; George, F.W. Role of Gonadal Hormones in Development of the Sexual Phenotypes. Hum. Genet. 1981, 58, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Quigley, C.A.; De Bellis, A.; Marschke, K.B.; el-Awady, M.K.; Wilson, E.M.; French, F.S. Androgen Receptor Defects: Historical, Clinical, and Molecular Perspectives. Endocr. Rev. 1995, 16, 271–321. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Morley, J.E.; Korenman, S.G. Biological Actions of Androgens. Endocr. Rev. 1987, 8, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Zhao, J.; Wang, H.; Tan, J.; Yang, M.; Li, Y.; Deng, S.; Gao, S.; Li, H.; et al. Distinct Cardiac Energy Metabolism and Oxidative Stress Adaptations between Obese and Non-Obese Type 2 Diabetes Mellitus. Theranostics 2020, 10, 2675–2695. [Google Scholar] [CrossRef]
- Pikler, G.M.; Webster, R.A.; Spelsberg, T.C. Nuclear Binding of Progesterone in Hen Oviduct. Binding to Multiple Sites in Vitro. Biochem. J. 1976, 156, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Oettel, M.; Mukhopadhyay, A.K. Progesterone: The Forgotten Hormone in Men? Aging Male 2004, 7, 236–257. [Google Scholar] [CrossRef] [PubMed]
- Manyonda, I.; Talaulikar, V.S.; Pirhadi, R.; Onwude, J. Progestogens Are the Problem in Hormone Replacement Therapy: Time to Reappraise Their Use. Post Reprod. Health 2020, 26, 26–31. [Google Scholar] [CrossRef]
- Adcock, I.M.; Mumby, S. Glucocorticoids. Handb. Exp. Pharmacol. 2017, 237, 171–196. [Google Scholar]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids--New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazirandeh, A.; Xue, Y.; Prestegaard, T.; Jondal, M.; Okret, S. Effects of Altered Glucocorticoid Sensitivity in the T Cell Lineage on Thymocyte and T Cell Homeostasis. FASEB J. 2002, 16, 727–729. [Google Scholar] [CrossRef]
- Scheller, K.; Sekeris, C.E. The Effects of Steroid Hormones on the Transcription of Genes Encoding Enzymes of Oxidative Phosphorylation. Exp. Physiol. 2003, 88, 129–140. [Google Scholar] [CrossRef] [Green Version]
- Allan, E.H.; Chisholm, A.B.; Titheradge, M.A. The Stimulation of Hepatic Oxidative Phosphorylation Following Dexamethasone Treatment of Rats. Biochim. Biophys. Acta 1983, 725, 71–76. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. Aldosterone: Effects on the Kidney and Cardiovascular System. Nat. Rev. Nephrol. 2010, 6, 261–273. [Google Scholar] [CrossRef]
- Pearce, D.; Bhargava, A.; Cole, T.J. Aldosterone: Its Receptor, Target Genes, and Actions. Vitam. Horm. 2003, 66, 29–76. [Google Scholar]
- Weinberger, M.H. Mineralocorticoids and Blood Pressure. Curr. Opin. Nephrol. Hypertens. 1994, 3, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Contribution of Aldosterone to Cardiovascular and Renal Inflammation and Fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.B.; McGraw, A.P.; Jaffe, I.Z.; Sowers, J.R. Mineralocorticoid Receptor-Mediated Vascular Insulin Resistance: An Early Contributor to Diabetes-Related Vascular Disease? Diabetes 2013, 62, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Feraco, A.; Armani, A.; Mammi, C.; Fabbri, A.; Rosano, G.M.; Caprio, M. Role of Mineralocorticoid Receptor and Renin-Angiotensin-Aldosterone System in Adipocyte Dysfunction and Obesity. J. Steroid Biochem. Mol. Biol. 2013, 137, 99–106. [Google Scholar] [CrossRef]
- Garg, R.; Adler, G.K. Role of Mineralocorticoid Receptor in Insulin Resistance. Curr. Opin. Endocrinol. Diabetes Obes. 2012, 19, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Calvo, M.S.; Whiting, S.J.; Barton, C.N. Vitamin D Intake: A Global Perspective of Current Status. J. Nutr. 2005, 135, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.W. From Vitamin D to Hormone D: Fundamentals of the Vitamin D Endocrine System Essential for Good Health. Am. J. Clin. Nutr. 2008, 88, 491S–499S. [Google Scholar] [CrossRef] [Green Version]
- Alkharfy, K.M.; Al-Daghri, N.M.; Yakout, S.M.; Ahmed, M. Calcitriol Attenuates Weight-Related Systemic Inflammation and Ultrastructural Changes in the Liver in a Rodent Model. Basic Clin. Pharmacol. Toxicol. 2013, 112, 42–49. [Google Scholar] [CrossRef]
- Kumar, R.; Litwack, G. Structural and Functional Relationships of the Steroid Hormone Receptors’ N-Terminal Transactivation Domain. Steroids 2009, 74, 877–883. [Google Scholar] [CrossRef] [Green Version]
- Weigel, N.L. Steroid Hormone Receptors and Their Regulation by Phosphorylation. Biochem. J. 1996, 319 (Pt. 3), 657–667. [Google Scholar] [CrossRef] [Green Version]
- Robinson-Rechavi, M.; Escriva Garcia, H.; Laudet, V. The Nuclear Receptor Superfamily. J. Cell Sci. 2003, 116, 585–586. [Google Scholar] [CrossRef] [Green Version]
- Liao, R.S.; Ma, S.; Miao, L.; Li, R.; Yin, Y.; Raj, G.V. Androgen Receptor-Mediated Non-Genomic Regulation of Prostate Cancer Cell Proliferation. Transl. Androl. Urol. 2013, 2, 187–196. [Google Scholar]
- Pupo, M.; Maggiolini, M.; Musti, A.M. Gper Mediates Non-Genomic Effects of Estrogen. Methods Mol. Biol. 2016, 1366, 471–488. [Google Scholar] [PubMed]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular Mechanism of Estrogen-Estrogen Receptor Signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Scheller, K.; Seibel, P.; Sekeris, C.E. Glucocorticoid and Thyroid Hormone Receptors in Mitochondria of Animal Cells. Int. Rev. Cytol. 2003, 222, 1–61. [Google Scholar] [PubMed]
- Valadez-Cosmes, P.; Vazquez-Martinez, E.R.; Cerbon, M.; Camacho-Arroyo, I. Membrane Progesterone Receptors in Reproduction and Cancer. Mol. Cell. Endocrinol. 2016, 434, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Dressing, G.E.; Goldberg, J.E.; Charles, N.J.; Schwertfeger, K.L.; Lange, C.A. Membrane Progesterone Receptor Expression in Mammalian Tissues: A Review of Regulation and Physiological Implications. Steroids 2011, 76, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular Mechanisms of Hepatic Lipid Accumulation in Non-Alcoholic Fatty Liver Disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.L.; Madak-Erdogan, Z. Estrogens and Female Liver Health. Steroids 2018, 133, 38–43. [Google Scholar] [CrossRef]
- Hart-Unger, S.; Arao, Y.; Hamilton, K.J.; Lierz, S.L.; Malarkey, D.E.; Hewitt, S.C.; Freemark, M.; Korach, K.S. Hormone Signaling and Fatty Liver in Females: Analysis of Estrogen Receptor Alpha Mutant Mice. Int. J. Obes. 2017, 41, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Martinez, M.N.; Emfinger, C.H.; Palmisano, B.T.; Stafford, J.M. Estrogen Signaling Prevents Diet-Induced Hepatic Insulin Resistance in Male Mice with Obesity. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1188–E1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Liu, Q.; Huang, T.; Tan, W.; Qu, L.; Chen, T.; Pan, H.; Chen, L.; Liu, J.; Wong, C.W.; et al. Dysfunction of Estrogen-Related Receptor Alpha-Dependent Hepatic Vldl Secretion Contributes to Sex Disparity in Nafld/Nash Development. Theranostics 2020, 10, 10874–10891. [Google Scholar] [CrossRef]
- Della Torre, S. Non-Alcoholic Fatty Liver Disease as a Canonical Example of Metabolic Inflammatory-Based Liver Disease Showing a Sex-Specific Prevalence: Relevance of Estrogen Signaling. Front. Endocrinol. 2020, 11, 572490. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Pan, H.J.; Lee, C.H. Prevention of Tamoxifen-Related Nonalcoholic Fatty Liver Disease in Breast Cancer Patients. Clin. Breast Cancer 2018, 18, e677–e685. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Yu, I.C.; Wang, R.S.; Chen, Y.T.; Liu, N.C.; Altuwaijri, S.; Hsu, C.L.; Ma, W.L.; Jokinen, J.; Sparks, J.D.; et al. Increased Hepatic Steatosis and Insulin Resistance in Mice Lacking Hepatic Androgen Receptor. Hepatology 2008, 47, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Lemke, U.; Krones-Herzig, A.; Berriel Diaz, M.; Narvekar, P.; Ziegler, A.; Vegiopoulos, A.; Cato, A.C.; Bohl, S.; Klingmuller, U.; Screaton, R.A.; et al. The Glucocorticoid Receptor Controls Hepatic Dyslipidemia through Hes1. Cell Metab. 2008, 8, 212–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, A.M.; Beaudry, J.L.; Szigiato, A.A.; Trumble, S.J.; Snook, L.A.; Bonen, A.; Giacca, A.; Riddell, M.C. Consumption of a High-Fat Diet Rapidly Exacerbates the Development of Fatty Liver Disease That Occurs with Chronically Elevated Glucocorticoids. Am. J. Physiol Gastrointest. Liver Physiol. 2012, 302, G850–G863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Y.; Li, C.; Yao, G.F.; Du, L.J.; Liu, Y.; Zheng, X.J.; Yan, S.; Sun, J.Y.; Liu, Y.; Liu, M.Z.; et al. Deletion of Macrophage Mineralocorticoid Receptor Protects Hepatic Steatosis and Insulin Resistance through Eralpha/Hgf/Met Pathway. Diabetes 2017, 66, 1535–1547. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Dematteo, A.; Wang, Z.; Zhu, L.; Wang, A.; Kim, H.S.; Pozzi, A.; Stafford, J.M.; Luther, J.M. Aldosterone Deficiency Prevents High-Fat-Feeding-Induced Hyperglycaemia and Adipocyte Dysfunction in Mice. Diabetologia 2013, 56, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Marziou, A.; Philouze, C.; Couturier, C.; Astier, J.; Obert, P.; Landrier, J.F.; Riva, C. Vitamin D Supplementation Improves Adipose Tissue Inflammation and Reduces Hepatic Steatosis in Obese C57bl/6j Mice. Nutrients 2020, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.G.; Liu, Y.X.; Wang, H.; Wang, B.P.; Qu, H.Q.; Wang, B.L.; Zhu, M. Active Form of Vitamin D Ameliorates Non-Alcoholic Fatty Liver Disease by Alleviating Oxidative Stress in a High-Fat Diet Rat Model. Endocr. J. 2017, 64, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaruvongvanich, V.; Sanguankeo, A.; Riangwiwat, T.; Upala, S. Testosterone, Sex Hormone-Binding Globulin and Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Ann. Hepatol. 2017, 16, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, H.; Park, J.H.; Cho, B.; Kim, D.; Oh, S.W.; Lee, C.M.; Choi, H.C. A Low Level of Serum Total Testosterone Is Independently Associated with Nonalcoholic Fatty Liver Disease. BMC Gastroenterol. 2012, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, Y.; Eguchi, T.; Mizuta, T.; Ide, Y.; Yasutake, T.; Iwakiri, R.; Hisatomi, A.; Ozaki, I.; Yamamoto, K.; Kitajima, Y.; et al. Visceral Fat Accumulation and Insulin Resistance Are Important Factors in Nonalcoholic Fatty Liver Disease. J. Gastroenterol. 2006, 41, 462–469. [Google Scholar] [CrossRef]
- Ma, W.L.; Lai, H.C.; Yeh, S.; Cai, X.; Chang, C. Androgen Receptor Roles in Hepatocellular Carcinoma, Fatty Liver, Cirrhosis and Hepatitis. Endocr. Relat. Cancer 2014, 21, R165–R182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.C.; Gray, N.E.; Kuo, T.; Harris, C.A. Regulation of Triglyceride Metabolism by Glucocorticoid Receptor. Cell Biosci. 2012, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, H.N.; Gautik, K.M.; Bremer, J.; Spydevold, O. Induction of the Three Peroxisomal Beta-Oxidation Enzymes Is Synergistically Regulated by Dexamethasone and Fatty Acids, and Counteracted by Insulin in Morris 7800c1 Hepatoma Cells in Culture. Eur. J. Biochem. 1992, 208, 705–711. [Google Scholar] [CrossRef]
- Andrews, R.C.; Walker, B.R. Glucocorticoids and Insulin Resistance: Old Hormones, New Targets. Clin. Sci. 1999, 96, 513–523. [Google Scholar] [CrossRef]
- Rockall, A.G.; Sohaib, S.A.; Evans, D.; Kaltsas, G.; Isidori, A.M.; Monson, J.P.; Besser, G.M.; Grossman, A.B.; Reznek, R.H. Hepatic Steatosis in Cushing’s Syndrome: A Radiological Assessment Using Computed Tomography. Eur. J. Endocrinol. 2003, 149, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belden, Z.; Deiuliis, J.A.; Dobre, M.; Rajagopalan, S. The Role of the Mineralocorticoid Receptor in Inflammation: Focus on Kidney and Vasculature. Am. J. Nephrol. 2017, 46, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Carotti, S.; Labbadia, G.; Gentilucci, U.V.; Muda, A.O.; Angelico, F.; Silecchia, G.; Leonetti, F.; Fraioli, A.; Picardi, A.; et al. Liver Vitamin D Receptor, Cyp2r1, and Cyp27a1 Expression: Relationship with Liver Histology and Vitamin D3 Levels in Patients with Nonalcoholic Steatohepatitis or Hepatitis C Virus. Hepatology 2012, 56, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D and Metabolic Dysfunction-Associated Fatty Liver Disease (Mafld): An Update. Nutrients 2020, 12, 3302. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Pathogenesis of Nonalcoholic Steatohepatitis and Hormone-Based Therapeutic Approaches. Front. Endocrinol. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broom, L.J.; Kogut, M.H. Inflammation: Friend or Foe for Animal Production? Poult. Sci. 2018, 97, 510–514. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Kamei, Y.; Xu, L.; Heinzel, T.; Torchia, J.; Kurokawa, R.; Gloss, B.; Lin, S.C.; Heyman, R.A.; Rose, D.W.; Glass, C.K.; et al. A Cbp Integrator Complex Mediates Transcriptional Activation and Ap-1 Inhibition by Nuclear Receptors. Cell 1996, 85, 403–414. [Google Scholar] [CrossRef] [Green Version]
- Koyama, Y.; Brenner, D.A. Liver Inflammation and Fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Parola, M.; Pinzani, M. Liver Fibrosis: Pathophysiology, Pathogenetic Targets and Clinical Issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of Hepatic Fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic Fatty Liver Disease (Nafld) Activity Score and the Histopathologic Diagnosis in Nafld: Distinct Clinicopathologic Meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florentino, G.S.; Cotrim, H.P.; Vilar, C.P.; Florentino, A.V.; Guimaraes, G.M.; Barreto, V.S. Nonalcoholic Fatty Liver Disease in Menopausal Women. Arq. Gastroenterol. 2013, 50, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Abdelmalek, M.F. Nonalcoholic Fatty Liver Disease in Women. Womens Health 2009, 5, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Galmes-Pascual, B.M.; Martinez-Cignoni, M.R.; Moran-Costoya, A.; Bauza-Thorbrugge, M.; Sbert-Roig, M.; Valle, A.; Proenza, A.M.; Llado, I.; Gianotti, M. 17beta-Estradiol Ameliorates Lipotoxicity-Induced Hepatic Mitochondrial Oxidative Stress and Insulin Resistance. Free Radic. Biol. Med. 2020, 150, 148–160. [Google Scholar] [CrossRef]
- Kamada, Y.; Kiso, S.; Yoshida, Y.; Chatani, N.; Kizu, T.; Hamano, M.; Tsubakio, M.; Takemura, T.; Ezaki, H.; Hayashi, N.; et al. Estrogen Deficiency Worsens Steatohepatitis in Mice Fed High-Fat and High-Cholesterol Diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1031–G1043. [Google Scholar] [CrossRef] [Green Version]
- Xin, G.; Qin, S.; Wang, S.; Wang, X.; Zhang, Y.; Wang, J. Sex Hormone Affects the Severity of Non-Alcoholic Steatohepatitis through the Myd88-Dependent Il-6 Signaling Pathway. Exp. Biol. Med. 2015, 240, 1279–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, T.; Kato, M.; Yamasaki, A.; Kuwano, A.; Suzuki, H.; Kohjima, M.; Ogawa, Y. Effects of High Fructose Intake on Liver Injury Progression in High Fat Diet Induced Fatty Liver Disease in Ovariectomized Female Mice. Food Chem. Toxicol. 2018, 118, 190–197. [Google Scholar] [CrossRef]
- Turola, E.; Petta, S.; Vanni, E.; Milosa, F.; Valenti, L.; Critelli, R.; Miele, L.; Maccio, L.; Calvaruso, V.; Fracanzani, A.L.; et al. Ovarian Senescence Increases Liver Fibrosis in Humans and Zebrafish with Steatosis. Dis. Model. Mech. 2015, 8, 1037–1046. [Google Scholar] [CrossRef] [Green Version]
- Klair, J.S.; Yang, J.D.; Abdelmalek, M.F.; Guy, C.D.; Gill, R.M.; Yates, K.; Unalp-Arida, A.; Lavine, J.E.; Clark, J.M.; Diehl, A.M.; et al. A Longer Duration of Estrogen Deficiency Increases Fibrosis Risk among Postmenopausal Women with Nonalcoholic Fatty Liver Disease. Hepatology 2016, 64, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, G.; Shimizu, I.; Cui, X.; Itonaga, M.; Tamaki, K.; Fukuno, H.; Inoue, H.; Honda, H.; Ito, S. Antioxidant and Antiapoptotic Activities of Idoxifene and Estradiol in Hepatic Fibrosis in Rats. Life Sci. 2004, 74, 897–907. [Google Scholar] [CrossRef]
- Ponnusamy, S.; Tran, Q.T.; Thiyagarajan, T.; Miller, D.D.; Bridges, D.; Narayanan, R. An Estrogen Receptor Beta-Selective Agonist Inhibits Non-Alcoholic Steatohepatitis in Preclinical Models by Regulating Bile Acid and Xenobiotic Receptors. Exp. Biol. Med. 2017, 242, 606–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamad, N.V.; Wong, S.K.; Wan Hasan, W.N.; Jolly, J.J.; Nur-Farhana, M.F.; Ima-Nirwana, S.; Chin, K.Y. The Relationship between Circulating Testosterone and Inflammatory Cytokines in Men. Aging Male 2019, 22, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential Effects of Estrogen/Androgen on the Prevention of Nonalcoholic Fatty Liver Disease in the Male Rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, A.; Ishigaki, Y. Gender-Difference in Diabetes Mellitus. Nihon Rinsho 2015, 73, 606–610. [Google Scholar]
- Lanari, A.; Garegnani, T.C.; Heinrichs, G.; Castresana, M.P. [Hepatic Fibrosis and Progesterone]. Acta Gastroenterol. Latinoam. 1988, 18, 161–171. [Google Scholar]
- Kim, K.H.; Lee, J.M.; Zhou, Y.; Harpavat, S.; Moore, D.D. Glucocorticoids Have Opposing Effects on Liver Fibrosis in Hepatic Stellate and Immune Cells. Mol. Endocrinol. 2016, 30, 905–916. [Google Scholar] [CrossRef]
- Wada, T.; Miyashita, Y.; Sasaki, M.; Aruga, Y.; Nakamura, Y.; Ishii, Y.; Sasahara, M.; Kanasaki, K.; Kitada, M.; Koya, D.; et al. Eplerenone Ameliorates the Phenotypes of Metabolic Syndrome with Nash in Liver-Specific Srebp-1c Tg Mice Fed High-Fat and High-Fructose Diet. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1415–E1425. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, M.; Solis, N.; Quintero, P.; Barrera, F.; Cabrera, D.; Rojas-de Santiago, P.; Arab, J.P.; Padilla, O.; Roa, J.C.; Moshage, H.; et al. Beneficial Effects of Mineralocorticoid Receptor Blockade in Experimental Non-Alcoholic Steatohepatitis. Liver Int. 2015, 35, 2129–2138. [Google Scholar] [CrossRef] [Green Version]
- Queisser, N.; Happ, K.; Link, S.; Jahn, D.; Zimnol, A.; Geier, A.; Schupp, N. Aldosterone Induces Fibrosis, Oxidative Stress and DNA Damage in Livers of Male Rats Independent of Blood Pressure Changes. Toxicol. Appl. Pharmacol. 2014, 280, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.T.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D Deficiency in Obese Rats Exacerbates Nonalcoholic Fatty Liver Disease and Increases Hepatic Resistin and Toll-Like Receptor Activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Cheng, Y.F.; Lai, C.Y.; Hsu, L.W.; Chang, Y.C.; Deng, J.Y.; Huang, Y.Z.; Honda, H.; Chen, K.D.; Wang, C.C.; et al. Impact of Artificial Sunlight Therapy on the Progress of Non-Alcoholic Fatty Liver Disease in Rats. J. Hepatol. 2011, 55, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Nolly, M.B.; Caldiz, C.I.; Yeves, A.M.; Villa-Abrille, M.C.; Morgan, P.E.; Amado Mondaca, N.; Portiansky, E.L.; Chiappe de Cingolani, G.E.; Cingolani, H.E.; Ennis, I.L. The Signaling Pathway for Aldosterone-Induced Mitochondrial Production of Superoxide Anion in the Myocardium. J. Mol. Cell. Cardiol. 2014, 67, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Lang, F. Mineralocorticoid and Sgk1-Sensitive Inflammation and Tissue Fibrosis. Nephron Physiol. 2014, 128, 35–39. [Google Scholar] [CrossRef]
- Eliades, M.; Spyrou, E. Vitamin D: A New Player in Non-Alcoholic Fatty Liver Disease? World J. Gastroenterol. 2015, 21, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.B.; Chen, Y.H.; Zhang, C.; Shi, C.E.; Hu, K.F.; Zhou, J.; Xu, D.X.; Chen, X. Low Vitamin D Status Is Associated with Advanced Liver Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Endocrine 2017, 55, 582–590. [Google Scholar] [CrossRef]
- Breitkopf, K.; Godoy, P.; Ciuclan, L.; Singer, M.V.; Dooley, S. Tgf-Beta/Smad Signaling in the Injured Liver. Z. Gastroenterol. 2006, 44, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Cimini, F.A.; Cavallo, M.G. Vitamin D Supplementation and Non-Alcoholic Fatty Liver Disease: Present and Future. Nutrients 2017, 9, 1015. [Google Scholar] [CrossRef] [Green Version]
- Beilfuss, A.; Sowa, J.P.; Sydor, S.; Beste, M.; Bechmann, L.P.; Schlattjan, M.; Syn, W.K.; Wedemeyer, I.; Mathe, Z.; Jochum, C.; et al. Vitamin D Counteracts Fibrogenic Tgf-Beta Signalling in Human Hepatic Stellate Cells Both Receptor-Dependently and Independently. Gut 2015, 64, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Ohbora, A.; Takeda, N.; Fukui, M.; Kato, T. Aging Is a Risk Factor of Nonalcoholic Fatty Liver Disease in Premenopausal Women. World J. Gastroenterol. 2012, 18, 237–243. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, M.; Hu, Z.; Shrestha, U.K. Prevalence of Nonalcoholic Fatty Liver Disease and Its Metabolic Risk Factors in Women of Different Ages and Body Mass Index. Menopause 2015, 22, 667–673. [Google Scholar] [CrossRef]
- Caballeria, L.; Pera, G.; Auladell, M.A.; Toran, P.; Munoz, L.; Miranda, D.; Aluma, A.; Casas, J.D.; Sanchez, C.; Gil, D.; et al. Prevalence and Factors Associated with the Presence of Nonalcoholic Fatty Liver Disease in an Adult Population in Spain. Eur. J. Gastroenterol. Hepatol. 2010, 22, 24–32. [Google Scholar] [CrossRef]
- Camhi, S.M.; Bray, G.A.; Bouchard, C.; Greenway, F.L.; Johnson, W.D.; Newton, R.L.; Ravussin, E.; Ryan, D.H.; Smith, S.R.; Katzmarzyk, P.T. The Relationship of Waist Circumference and Bmi to Visceral, Subcutaneous, and Total Body Fat: Sex and Race Differences. Obesity 2011, 19, 402–408. [Google Scholar] [CrossRef]
- Younossi, Z.M. Non-Alcoholic Fatty Liver Disease—A Global Public Health Perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Xiong, X.; Zhang, H.; Lu, J.; Zheng, S.; Yang, J.; Xia, X.; et al. Hepatic Estrogen Receptor Alpha Improves Hepatosteatosis through Upregulation of Small Heterodimer Partner. J. Hepatol. 2015, 63, 183–190. [Google Scholar] [CrossRef]
- Pan, J.J.; Fallon, M.B. Gender and Racial Differences in Nonalcoholic Fatty Liver Disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Summart, U.; Thinkhamrop, B.; Chamadol, N.; Khuntikeo, N.; Songthamwat, M.; Kim, C.S. Gender Differences in the Prevalence of Nonalcoholic Fatty Liver Disease in the Northeast of Thailand: A Population-Based Cross-Sectional Study. F1000Research 2017, 6, 1630. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.K.; Boulet, S.L.; Mehta, A.; Hotaling, J.; Eisenberg, M.L.; Honig, S.C.; Warner, L.; Kissin, D.M.; Nangia, A.K.; Ross, L.S. Trends in Testosterone Replacement Therapy Use from 2003 to 2013 among Reproductive-Age Men in the United States. J. Urol. 2017, 197, 1121–1126. [Google Scholar] [CrossRef]
- Phan, H.; Richard, A.; Lazo, M.; Nelson, W.G.; Denmeade, S.R.; Groopman, J.; Kanarek, N.; Platz, E.A.; Rohrmann, S. The Association of Sex Steroid Hormone Concentrations with Non-Alcoholic Fatty Liver Disease and Liver Enzymes in Us Men. Liver Int. 2021, 41, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, S.; Meng, Y.; Yu, Q.; Wang, Q.; Xu, H.; Yuan, H.; Li, X.; Chen, L. Effects of Vitamin D Supplementation in Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. Metab. 2020, 18, e97205. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representative steroid hormone genomic and nongenomic pathways. Abbreviations: estrogen receptor (ER); glucocorticoid receptor (GR); progesterone receptor (PR); cyclic adenosine monophosphate (cAMP); cAMP responsive element-binding protein (CREB); JUN N-terminal kinase (JNK); BCL2-associated X (BAX).
Figure 1.
Representative steroid hormone genomic and nongenomic pathways. Abbreviations: estrogen receptor (ER); glucocorticoid receptor (GR); progesterone receptor (PR); cyclic adenosine monophosphate (cAMP); cAMP responsive element-binding protein (CREB); JUN N-terminal kinase (JNK); BCL2-associated X (BAX).

Figure 2.
Schematic view of the roles of steroid hormones in hepatic lipid homeostasis. Steroid hormones have the ability to regulate lipid homeostasis via alteration of multiple major metabolic fates of hepatic FFAs. Blue arrows indicate stimulatory activity and red arrows indicate inhibitory activity. Abbreviations: free fatty acid (FFA); non-estesterified fatty acid (NEFA); triglyceride (TG); very-low-density lipoprotein (VLDL).
Figure 2.
Schematic view of the roles of steroid hormones in hepatic lipid homeostasis. Steroid hormones have the ability to regulate lipid homeostasis via alteration of multiple major metabolic fates of hepatic FFAs. Blue arrows indicate stimulatory activity and red arrows indicate inhibitory activity. Abbreviations: free fatty acid (FFA); non-estesterified fatty acid (NEFA); triglyceride (TG); very-low-density lipoprotein (VLDL).

Figure 3.
Schematic view of the roles of steroid hormones in NASH development. Steroid hormones are involved in guarding against hepatocellular damage, inflammation, and fibrosis in the pathological progression of NASH. Blue arrows indicate promoting activity and red arrows indicate inhibitory activity. Abbreviations: reactive oxygen species (ROS); damage-associated molecular patterns (DAMPs); tumor necrosis factor (TNF); platelet-derived growth factor (PDGF); interleukin (IL); C–C motif chemokine ligand 2 (CCL2); C–C motif chemokine receptor 2 (CCR2); nuclear factor kappa B (NK-kB); collagen I (COL1); α-smooth muscle actin (α-SMA).
Figure 3.
Schematic view of the roles of steroid hormones in NASH development. Steroid hormones are involved in guarding against hepatocellular damage, inflammation, and fibrosis in the pathological progression of NASH. Blue arrows indicate promoting activity and red arrows indicate inhibitory activity. Abbreviations: reactive oxygen species (ROS); damage-associated molecular patterns (DAMPs); tumor necrosis factor (TNF); platelet-derived growth factor (PDGF); interleukin (IL); C–C motif chemokine ligand 2 (CCL2); C–C motif chemokine receptor 2 (CCR2); nuclear factor kappa B (NK-kB); collagen I (COL1); α-smooth muscle actin (α-SMA).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yang, M.; Ma, F.; Guan, M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 320. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050320
AMA Style
Yang M, Ma F, Guan M. Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease. Metabolites. 2021; 11(5):320. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050320
Chicago/Turabian StyleYang, Meng, Feng Ma, and Min Guan. 2021. "Role of Steroid Hormones in the Pathogenesis of Nonalcoholic Fatty Liver Disease" Metabolites 11, no. 5: 320. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11050320
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.