
Towards Standards for Human Fecal Sample Preparation in Targeted and Untargeted LC-HRMS Studies

 , , and
, , and
Abstract
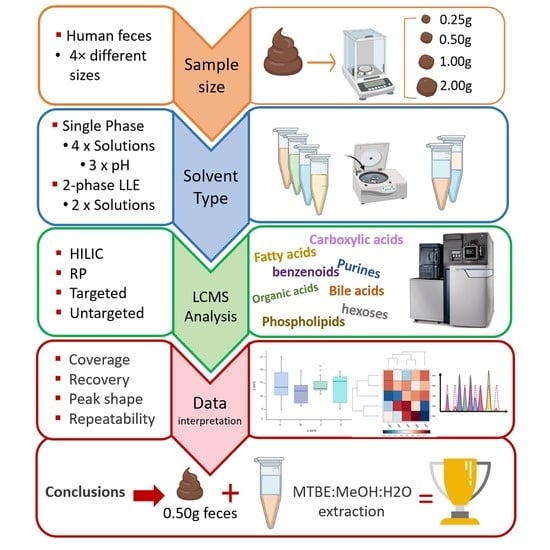
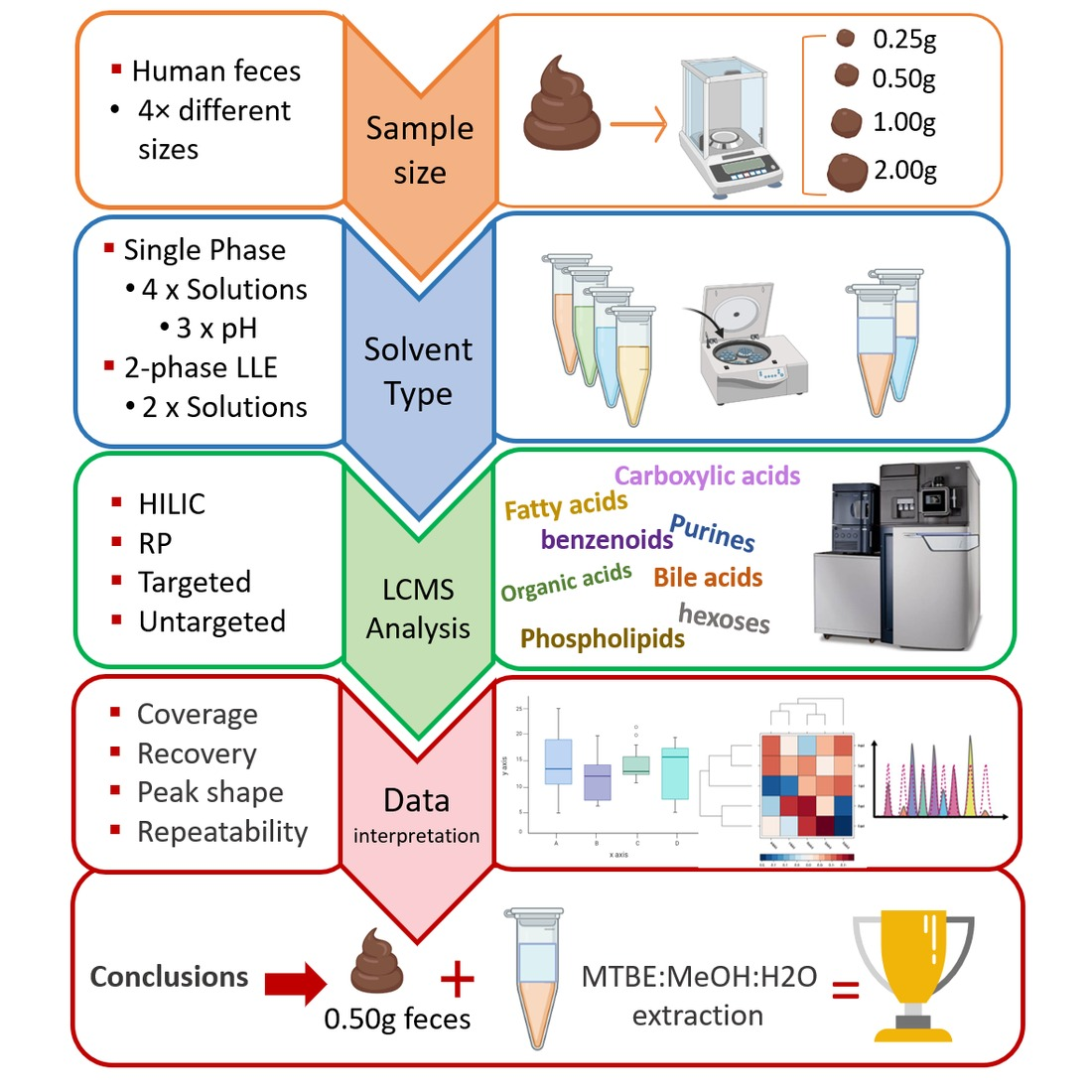
:
1. Introduction
2. Results
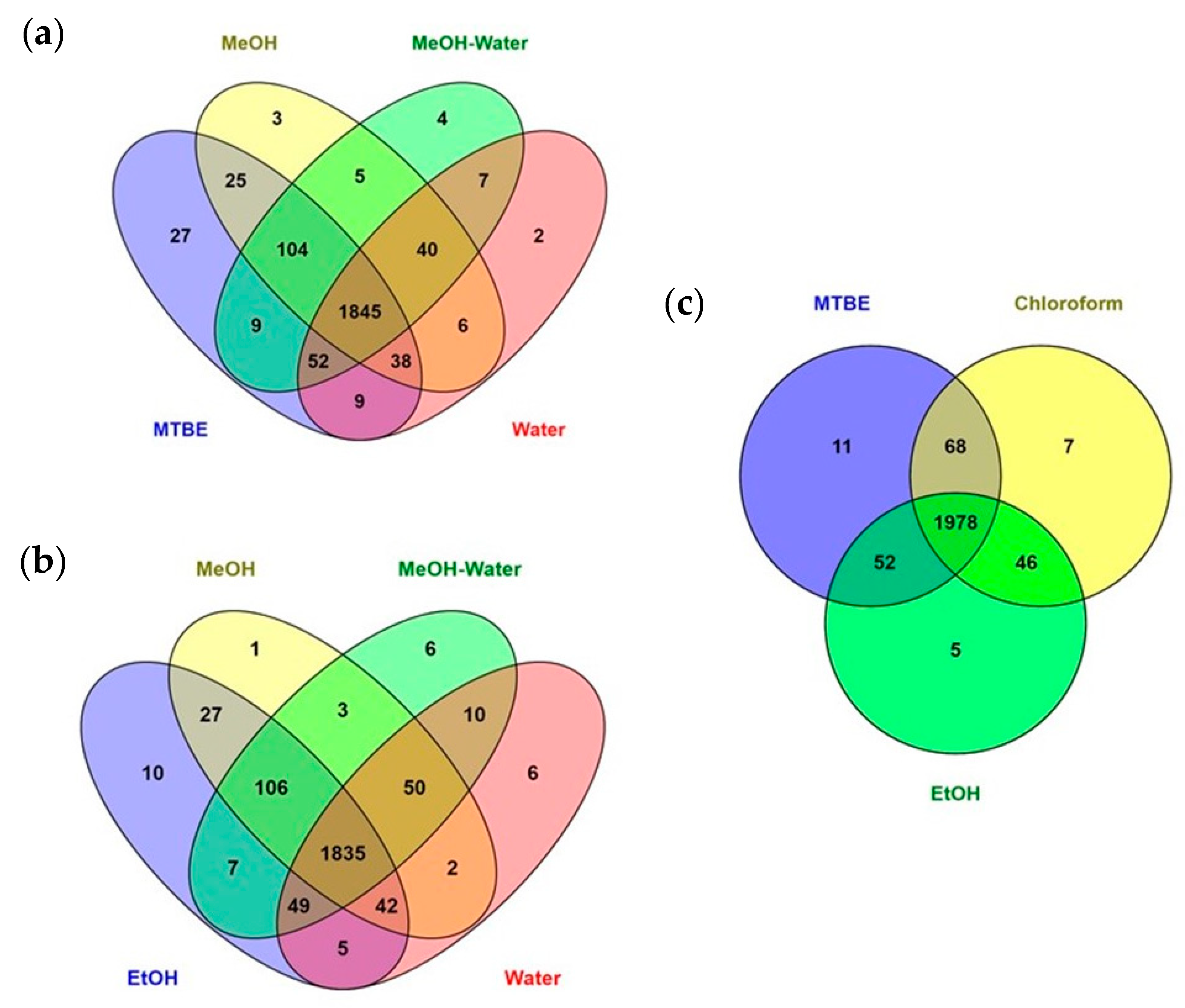
2.1. Sample Preparation Methods and Workflow
2.2. Quantity of Starting Material
2.3. Assessment of Extraction Method, Solvent and pH for Optimal Extraction Efficiency
2.3.1. pH Effect of Extraction Solvent
2.3.2. Effect of Solvent Selection
2.3.3. LLE Solvent Effect
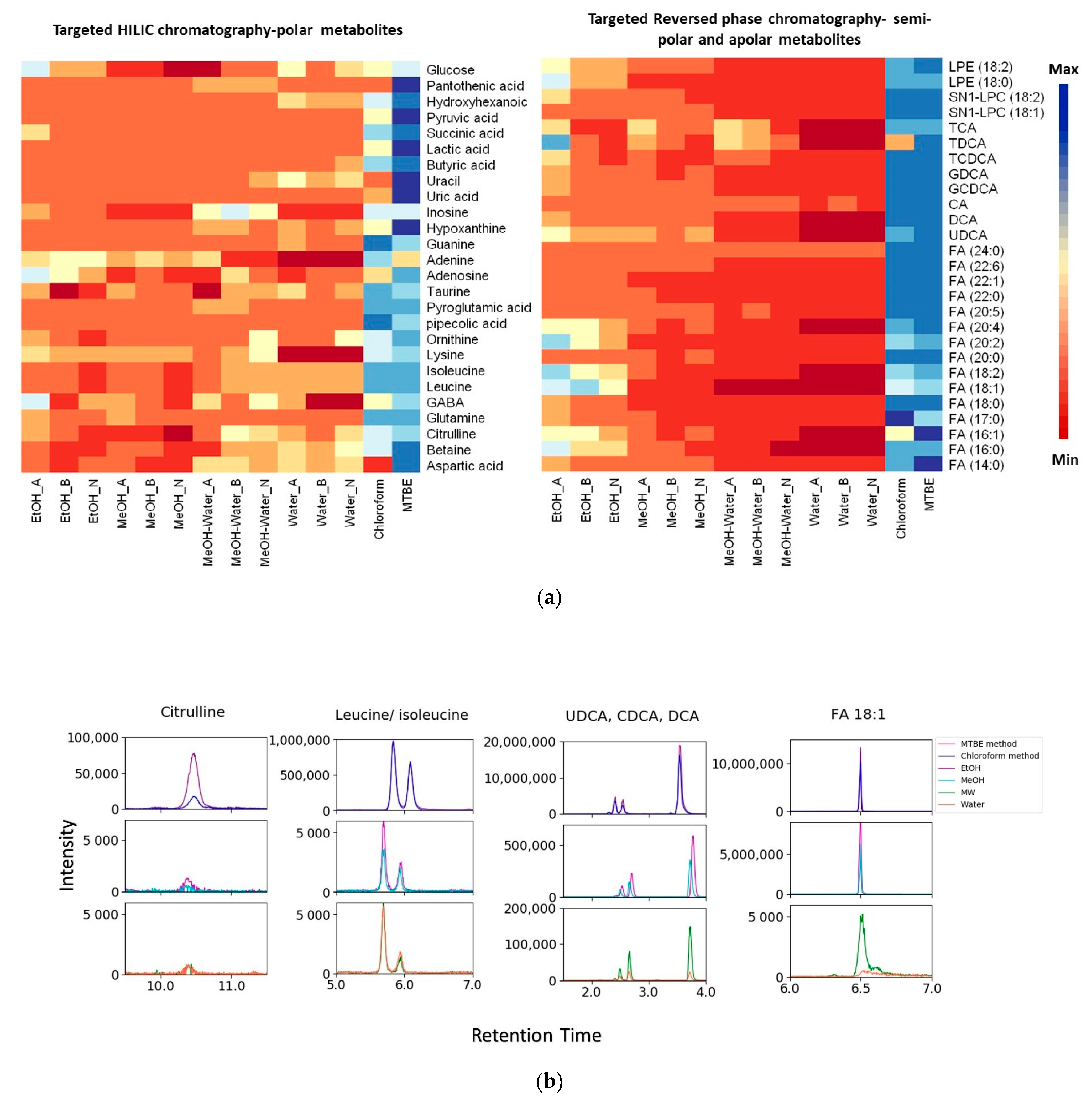
2.3.4. Metabolic Coverage
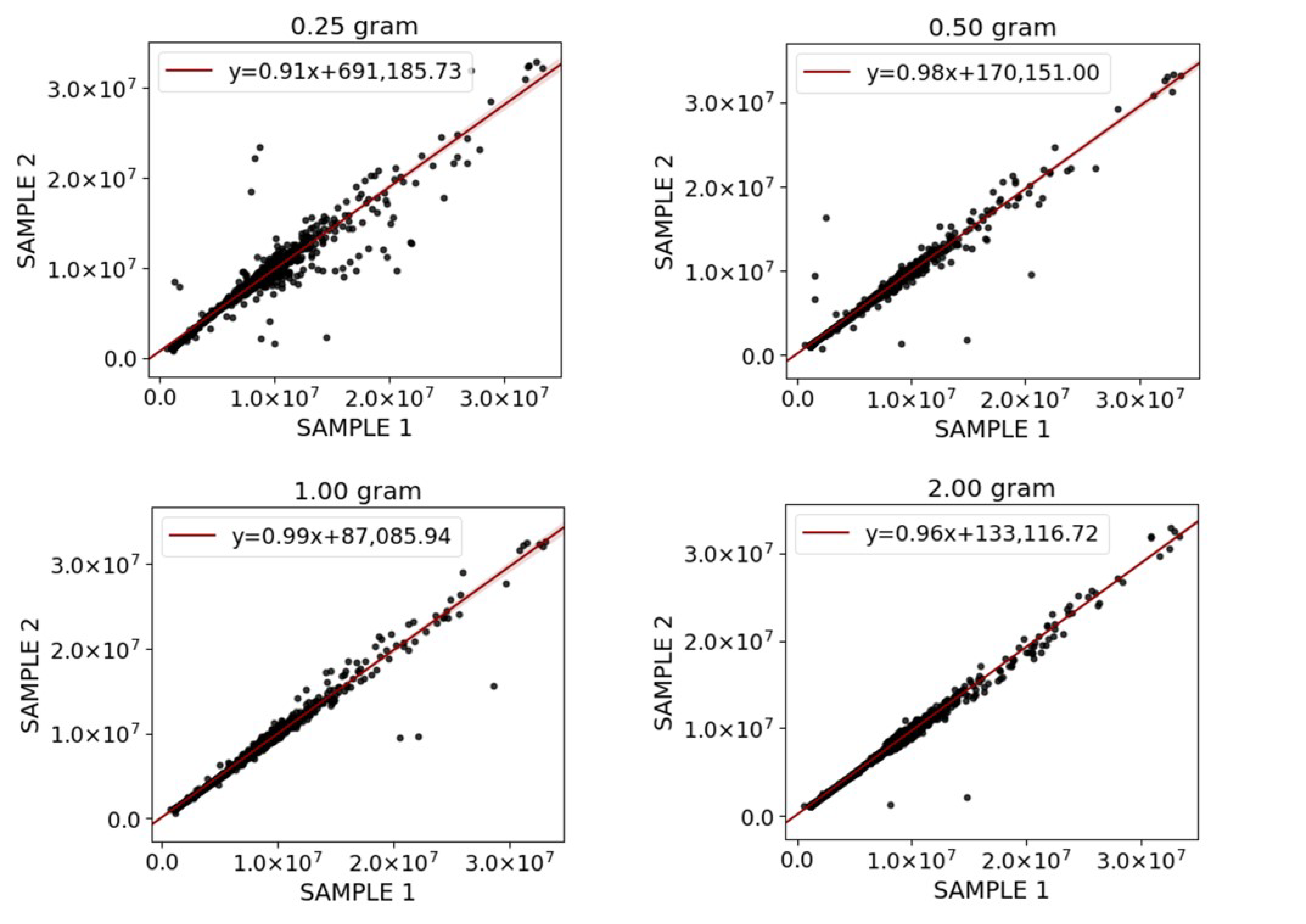
2.4. Extraction Repeatability and Recovery for Selected Methods
3. Discussion
3.1. Quantity of Starting Material
3.2. Assessment of Extraction Method, Solvent and pH for Optimal Extraction Efficiency
3.3. Extraction Coverage
3.4. Extraction Repeatability and Recovery for Selected Methods
3.5. Standardization
4. Materials and Methods
4.1. Chemical and Reagents
4.2. LCMS Analysis
4.3. Quantity of Starting Material
4.4. Assessment of Extraction Method, Solvent and pH for Optimal Extraction Efficiency
4.5. Assessment of Extraction Repeatibility and Recovery
4.6. Data Pre-Processing and Software
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Olive, A.J.; Sassetti, C.M. Metabolic crosstalk between host and pathogen: Sensing, adapting and competing. Nat. Rev. Microbiol. 2016, 14, 221–234. [Google Scholar] [CrossRef]
- Hosseinkhani, F.; Heinken, A.; Thiele, I.; Lindenburg, P.W.; Harms, A.C.; Hankemeier, T. The contribution of gut bacterial metabolites in the human immune signaling pathway of non-communicable diseases. Gut Microbes 2021, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zierer, J.; Jackson, M.A.; Kastenmüller, G.; Mangino, M.; Long, T.; Telenti, A.; Mohney, R.P.; Small, K.S.; Bell, J.T.; Steves, C.J.; et al. The fecal metabolome as a functional readout of the gut microbiome. Nat. Genet. 2018, 50, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool Microbiome and Metabolome Differences between Colorectal Cancer Patients and Healthy Adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Jiang, H.; Gu, S.; Feng, N.; Zhang, N.; Lv, L.; Liu, F. Fecal Metabolites Were Altered, Identified as Biomarkers and Correlated With Disease Activity in Patients With Systemic Lupus Erythematosus in a GC-MS-Based Metabolomics Study. Front. Immunol. 2020, 11, 2138. [Google Scholar] [CrossRef]
- Nagpal, R.; Wang, S.; Ahmadi, S.; Hayes, J.; Gagliano, J.; Subashchandrabose, S.; Kitzman, D.W.; Becton, T.; Read, R.; Yadav, H. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddavide, R.; Rotolo, O.; Caruso, M.G.; Stasi, E.; Notarnicola, M.; Miraglia, C.; Nouvenne, A.; Meschi, T.; De’ Angelis, G.L.; Di Mario, F.; et al. The role of diet in the prevention and treatment of inflammatory bowel diseases. Acta Biomed. 2018, 89, 60–75. [Google Scholar] [CrossRef]
- Karu, N.; Deng, L.; Slae, M.; Chi, A.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. Analytica Chimica Acta A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24. [Google Scholar] [CrossRef]
- Fiori, J.; Turroni, S.; Candela, M.; Gotti, R. Assessment of gut microbiota fecal metabolites by chromatographic targeted approaches. J. Pharm. Biomed. Anal. 2020, 177, 112867. [Google Scholar] [CrossRef]
- Blekhman, R.; Tang, K.; Archie, E.A.; Barreiro, L.B.; Johnson, Z.P.; Wilson, M.E.; Kohn, J.; Yuan, M.L.; Gesquiere, L.; Grieneisen, L.E.; et al. Common methods for fecal sample storage in field studies yield consistent signatures of individual identity in microbiome sequencing data. Sci. Rep. 2016, 6, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Bundgaard-Nielsen, C.; Hagstrøm, S.; Sørensen, S. Interpersonal Variations in Gut Microbiota Profiles Supersedes the Effects of Differing Fecal Storage Conditions. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Carroll, I.M.; Ringel-Kulka, T.; Siddle, J.P.; Klaenhammer, T.R.; Ringel, Y. Characterization of the Fecal Microbiota Using High-Throughput Sequencing Reveals a Stable Microbial Community during Storage. PLoS ONE 2012, 7, e46953. [Google Scholar] [CrossRef]
- Liang, Y.; Dong, T.; Chen, M.; He, L.; Wang, T.; Liu, X.; Chang, H.; Mao, J.-H.; Hang, B.; Snijders, A.M.; et al. Systematic Analysis of Impact of Sampling Regions and Storage Methods on Fecal Gut Microbiome and Metabolome Profiles. mSphere 2020, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnov, K.S.; Maier, T.V.; Walker, A.; Heinzmann, S.S.; Forcisi, S.; Martinez, I.; Walter, J.; Schmitt-Kopplin, P. Challenges of metabolomics in human gut microbiota research. Int. J. Med. Microbiol. 2016, 306, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Q.; Zheng, J.; Yuan, B.; Feng, Y. Trends in Analytical Chemistry Mass spectrometry-based fecal metabolome analysis. Trends Anal. Chem. 2019, 112, 161–174. [Google Scholar] [CrossRef]
- Wilson, A.; Teft, W.A.; Morse, B.L.; Choi, Y.H.; Woolsey, S.; DeGorter, M.K.; Hegele, R.A.; Tirona, R.G.; Kim, R.B. Trimethylamine-N-oxide: A Novel Biomarker for the Identification of Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 3620–3630. [Google Scholar] [CrossRef]
- Bobeldijk, I.; Hekman, M.; de Vries-van der Weij, J.; Coulier, L.; Ramaker, R.; Kleemann, R.; Kooistra, T.; Rubingh, C.; Freidig, A.; Verheij, E. Quantitative profiling of bile acids in biofluids and tissues based on accurate mass high resolution LC-FT-MS: Compound class targeting in a metabolomics workflow. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 871, 306–313. [Google Scholar] [CrossRef]
- Buijink, M.R.; van Weeghel, M.; Gülersönmez, M.C.; Harms, A.C.; Rohling, J.H.T.; Meijer, J.H.; Hankemeier, T.; Michel, S. The influence of neuronal electrical activity on the mammalian central clock metabolome. Metabolomics 2018, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Connors, J.; Dawe, N.; Van Limbergen, J. The role of succinate in the regulation of intestinal inflammation. Nutrients 2019, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, S.; Sen, P.; Alves, M.A.; Ribeiro, H.C.; Raunioniemi, P.; Hyötyläinen, T.; Orešič, M. Linking Gut Microbiome and Lipid Metabolism: Moving beyond Associations. Metabolites 2021, 11, 55. [Google Scholar] [CrossRef]
- Jain, A.; Li, X.H.; Chen, W.N. An untargeted fecal and urine metabolomics analysis of the interplay between the gut microbiome, diet and human metabolism in Indian and Chinese adults. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Rosa, V.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Pujos-Guillot, E.; Martin, J.F.; Galan, P.; Juste, C.; Jia, W.; Sebedio, J.L. Metabolite analysis of human fecal water by gas chromatography/mass spectrometry with ethyl chloroformate derivatization. Anal. Biochem. 2009, 393, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Garner, C.E.; Smith, S.; Lacy Costello, B.; White, P.; Spencer, R.; Probert, C.S.J.; Ratcliffem, N.M. Volatile organic compounds from feces and their potential for diagnosis of gastrointestinal disease. FASEB J. 2007, 21, 1675–1688. [Google Scholar] [CrossRef] [Green Version]
- Gratton, J.; Phetcharaburanin, J.; Mullish, B.H.; Williams, H.R.T.; Thursz, M.; Nicholson, J.K.; Holmes, E.; Marchesi, J.R.; Li, J.V. Optimized Sample Handling Strategy for Metabolic Profiling of Human Feces. Anal. Chem. 2016, 88, 4661–4668. [Google Scholar] [CrossRef]
- Cajka, T.; Fiehn, O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal. Chem. 2016, 88, 524–545. [Google Scholar] [CrossRef]
- Deda, O.; Chatziioannou, A.C.; Fasoula, S.; Palachanis, D.; Raikos, Ν.; Theodoridis, G.A.; Gika, H.G. Sample preparation optimization in fecal metabolic profiling. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1047, 115–123. [Google Scholar] [CrossRef]
- López-Bascón, M.A.; Calderón-Santiago, M.; Argüello, H.; Morera, L.; Garrido, J.J.; Priego-Capote, F. Comprehensive analysis of pig feces metabolome by chromatographic techniques coupled to mass spectrometry in high resolution mode: Influence of sample preparation on the identification coverage. Talanta 2019, 199, 303–309. [Google Scholar] [CrossRef]
- Turroni, S.; Fiori, J.; Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Barone, M.; Biagi, E.; Fanelli, F.; Mezzullo, M.; Crittenden, A.N.; et al. Fecal metabolome of the Hadza hunter-gatherers: A host-microbiome integrative view. Sci. Rep. 2016, 1–9. [Google Scholar] [CrossRef]
- Sostare, J.; Di Guida, R.; Kirwan, J.; Chalal, K.; Palmer, E.; Dunn, W.B.; Viant, M.R. Analytica Chimica Acta Comparison of modi fi ed Matyash method to conventional solvent systems for polar metabolite and lipid extractions. Anal. Chim. Acta 2018, 1037, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Patterson, R.E. Comparison of blood plasma sample preparation methods for combined LC-MS lipidomics and metabolomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 176, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.; Jehmlich, N.; Alfaro, T.; Dohnalkova, A.; Capek, P.; Bell, S.L.; Hofmockel, K.S. Incomplete cell disruption of resistant microbes. Sci. Rep. 2019, 9, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Chou, J.; Hou, S.; Liu, X.; Yu, J.; Zhao, X.; Li, Y.; Liu, L.; Sun, C. Analytica Chimica Acta Evaluation of two-step liquid-liquid extraction protocol for untargeted metabolic pro fi ling of serum samples to achieve broader metabolome coverage by UPLC-Q-TOF-MS. Anal. Chim. Acta 2018, 1035, 96–107. [Google Scholar] [CrossRef]
- Whiley, L.; Godzien, J.; Ruperez, F.J.; Legido-quigley, C.; Barbas, C. In-Vial Dual Extraction for Direct LC-MS Analysis of Plasma for Comprehensive and Highly Reproducible Metabolic Fingerprinting. Anal. Chem. 2012, 84, 5992–5999. [Google Scholar] [CrossRef]
- Sitnikov, D.G.; Monnin, C.S.; Vuckovic, D. Systematic Assessment of Seven Solvent and Solid-Phase Extraction Methods for Metabolomics Analysis of Human Plasma by LC-MS. Nat. Publ. Gr. 2016, 1–11. [Google Scholar] [CrossRef]
- Chambers, M.C.; MacLean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, C.; Tautenhahn, R.; Böttcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | LLE-MTBE | LLE-Chloroform | EtOH |
|---|---|---|---|
| D4-DCA | 20 | 14 | 21 |
| D4-CA | 3 | 42 | 21 |
| FA 20(4)-d8 | 6.5 | 10 | 10 |
| FA 22 (6)-d5 | 6.3 | 15 | 3.5 |
| D5-TUDCA | 4.5 | 5.6 | 10.8 |
| D4-GDCA | 10 | 28 | 10 |
| FA18(2) d4 | 5 | 19 | 11.5 |
| LPE (17:1) | 28.5 | 16 | 41 |
| D3-Leucine | 5 | 18 | 8 |
| D4-Succinate | 8 | 11 | 2 |
| U13-C5-valine | 4 | 1.5 | 3 |
| D6-Ornithine | 7.5 | 7 | 24 |
| U 13C6- Lysine | 6.5 | 14 | 17 |
| D3-9-15N-aspartate | 12 | 28 | 37.5 |
| D2-Glycine | 5 | 8 | 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosseinkhani, F.; Dubbelman, A.-C.; Karu, N.; Harms, A.C.; Hankemeier, T. Towards Standards for Human Fecal Sample Preparation in Targeted and Untargeted LC-HRMS Studies. Metabolites 2021, 11, 364. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11060364
Hosseinkhani F, Dubbelman A-C, Karu N, Harms AC, Hankemeier T. Towards Standards for Human Fecal Sample Preparation in Targeted and Untargeted LC-HRMS Studies. Metabolites. 2021; 11(6):364. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11060364
Chicago/Turabian StyleHosseinkhani, Farideh, Anne-Charlotte Dubbelman, Naama Karu, Amy C. Harms, and Thomas Hankemeier. 2021. "Towards Standards for Human Fecal Sample Preparation in Targeted and Untargeted LC-HRMS Studies" Metabolites 11, no. 6: 364. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11060364