
Rapid Identification of Common Secondary Metabolites of Medicinal Herbs Using High-Performance Liquid Chromatography with Evaporative Light Scattering Detector in Extracts

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. HPLC-ELSD Optimization
2.2. Method Validation
2.3. LC-ESI-MS/MS Analysis
2.4. Comparison with Reported Methods
2.5. Analysis of Plant Extracts
3. Materials and Methods
3.1. Chemicals and Standard Solutions
3.2. Plant Extract Preparation
3.3. Stock and Standard Solutions
3.4. HPLC Analysis
3.5. HPLC-ESI-MS/MS Analysis
3.6. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Masubuchi, M. Dereplication of microbial extracts and related analytical technologies. J. Antibiot. 2014, 67, 353–360. [Google Scholar] [CrossRef]
- Kosalec, I.; Cvek, J.; Tomić, S. Contaminants of medicinal herbs and herbal products. Arh. Za Hig. Rada I Toksikol. 2009, 60, 485–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Deng, J.; Gao, Y.; Zhu, L.; He, R.; Xu, Y. The screening toolbox of bioactive substances from natural products: A review. Fitoterapia 2011, 82, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Singh, V.P.; Singh, R.K.; Joshi, C.S.; Sharma, V. 4—Isolation and characterization of bioactive compounds from natural resources: Metabolomics and molecular approaches. In Evolutionary Diversity as a Source for Anticancer Molecules; Srivastava, A.K., Kannaujiya, V.K., Singh, R.K., Singh, D., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 77–101. [Google Scholar]
- Locatelli, M.; Macchione, N.; Ferrante, C.; Chiavaroli, A.; Recinella, L.; Carradori, S.; Zengin, G.; Cesa, S.; Leporini, L.; Leone, S.; et al. Graminex Pollen: Phenolic Pattern, Colorimetric Analysis and Protective Effects in Immortalized Prostate Cells (PC3) and Rat Prostate Challenged with LPS. Molecules 2018, 23, 1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products As Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gligor, O.; Mocan, A.; Moldovan, C.; Locatelli, M.; Crișan, G.; Ferreira, I.C. Enzyme-assisted extractions of polyphenols—A comprehensive review. Trends Food Sci. Technol. 2019, 88, 302–315. [Google Scholar] [CrossRef]
- Wolfender, J.-L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, F.-J.; Wang, Y.-M.; Li, H.-J. Dereplication-guided isolation of novel hepatoprotective triterpenoid saponins from Celosiae Semen by high-performance liquid chromatography coupled with electrospray ionization tandem quadrupole–time-of-flight mass spectrometry. J. Pharm. Biomed. Anal. 2017, 132, 148–155. [Google Scholar] [CrossRef]
- Silva, M.F.S.; Silva, L.M.A.; Quintela, A.L.; dos Santos, A.G.; Silva, F.A.N.; de Oliveira, F.d.C.E.; Alves Filho, E.G.; de Brito, E.S.; Canuto, K.M.; Pessoa, C.; et al. UPLC-HRMS and NMR applied in the evaluation of solid-phase extraction methods as a rational strategy of dereplication of Phyllanthus spp. aiming at the discovery of cytotoxic metabolites. J. Chromatogr. B 2019, 1120, 51–61. [Google Scholar] [CrossRef]
- Gallon, M.E.; Jaiyesimi, O.A.; Gobbo-Neto, L. LC-UV-HRMS dereplication of secondary metabolites from Brazilian Vernonieae (Asteraceae) species supported through in-house database. Biochem. Syst. Ecol. 2018, 78, 5–16. [Google Scholar] [CrossRef]
- Samulski, G.B.; Gontijo, D.C.; Moreira, N.C.; Brandão, G.C.; Braga de Oliveira, A. Dereplication of Palicourea sessilis ethanol extracts by UPLC-DAD-ESI-MS/MS discloses the presence of hydroxycinnamic acid amides and the absence of monoterpene indole alkaloids. Biochem. Syst. Ecol. 2020, 92, 104114. [Google Scholar] [CrossRef]
- Zengin, G.; Locatelli, M.; Stefanucci, A.; Macedonio, G.; Novellino, E.; Mirzaie, S.; Dvorácskó, S.; Carradori, S.; Brunetti, L.; Orlando, G.; et al. Chemical characterization, antioxidant properties, anti-inflammatory activity, and enzyme inhibition of Ipomoea batatas L. leaf extracts. Int. J. Food Prop. 2017, 20, 1907–1919. [Google Scholar] [CrossRef] [Green Version]
- Locatelli, C.; Melucci, D.; Locatelli, M. Toxic Metals in Herbal Medicines. A Review. Curr. Bioact. Compd. 2014, 10, 181–188. [Google Scholar] [CrossRef]
- Melucci, D.; Locatelli, M.; De Laurentiis, F.; Zengin, G.; Locatelli, C. Herbal Medicines: Application of a Sequential Voltammetric Procedure to the Determination of Mercury, Copper, Lead, Cadmium and Zinc at Trace Level. Lett. Drug Des. Discov. 2018, 15, 270–280. [Google Scholar] [CrossRef]
- Martelanc, M.; Vovk, I.; Simonovska, B. Separation and identification of some common isomeric plant triterpenoids by thin-layer chromatography and high-performance liquid chromatography. J. Chromatogr. A 2009, 1216, 6662–6670. [Google Scholar] [CrossRef]
- Perez-Vizcaino, F.; Duarte, J. Flavonols and cardiovascular disease. Mol. Asp. Med. 2010, 31, 478–494. [Google Scholar] [CrossRef]
- Blunder, M.; Orthaber, A.; Bauer, R.; Bucar, F.; Kunert, O. Efficient identification of flavones, flavanones and their glycosides in routine analysis via off-line combination of sensitive NMR and HPLC experiments. Food Chem. 2017, 218, 600–609. [Google Scholar] [CrossRef]
- Devi, R.S.; Basha, S. Screening of Flavonoids in Selected High Valued Medicinal Plants (HVM) of Tirumala, India. Int. J. Curr. Microbiol. App. Sci 2017, 6, 2844–2850. [Google Scholar]
- Nagula, R.L.; Wairkar, S. Recent advances in topical delivery of flavonoids: A review. J. Control. Release 2019, 296, 190–201. [Google Scholar] [CrossRef]
- Diuzheva, A.; Locatelli, M.; Tartaglia, A.; Goga, M.; Ferrone, V.; Carlucci, G.; Andruch, V. Application of liquid-phase microextraction to the analysis of plant and herbal samples. Phytochem. Anal. 2020, 31, 687–699. [Google Scholar] [CrossRef]
- Busta, L.; Serra, O.; Kim, O.T.; Molinas, M.; Peré-Fossoul, I.; Figueras, M.; Jetter, R. Oxidosqualene cyclases involved in the biosynthesis of triterpenoids in Quercus suber cork. Sci. Rep. 2020, 10, 8011. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chu, Y.-L.; Sridhar, K.; Tsai, P.-J. Analysis and determination of phytosterols and triterpenes in different inbred lines of Djulis (Chenopodium formosanum Koidz.) hull: A potential source of novel bioactive ingredients. Food Chem. 2019, 297, 124948. [Google Scholar] [CrossRef]
- Mathe, C.; Culioli, G.; Archier, P.; Vieillescazes, C. High-performance liquid chromatographic analysis of triterpenoids in commercial frankincense. Chromatographia 2004, 60, 493–499. [Google Scholar] [CrossRef]
- Vu, T.P.D.; Khong, T.Q.; Nguyen, T.M.N.; Kim, Y.H.; Kang, J.S. Phytochemical profile of Syzygium formosum (Wall.) Masam leaves using HPLC–PDA–MS/MS and a simple HPLC–ELSD method for quality control. J. Pharm. Biomed. Anal. 2019, 168, 1–12. [Google Scholar]
- Tyszczuk-Rotko, K.; Domańska, K.; Sadok, I.; Wójciak-Kosior, M.; Sowa, I. Voltammetric procedure for the determination of oleanolic and ursolic acids in plant extracts. Anal. Methods 2015, 7, 9435–9441. [Google Scholar] [CrossRef]
- Yang, G.; Fen, W.; Xiao, W.; Sun, H. Study on determination of pentacyclic triterpenoids in Chaenomeles by HPLC-ELSD. J. Chromatogr. Sci. 2009, 47, 718–722. [Google Scholar] [CrossRef] [Green Version]
- Costa, P.P.; Mendes, T.D.; Salum, T.F.; Pacheco, T.F.; Braga, S.C.; de Almeida, J.R.M.; Gonçalves, S.B.; Damaso, M.C.; Rodrigues, C.M. Development and validation of HILIC-UHPLC-ELSD methods for determination of sugar alcohols stereoisomers and its application for bioconversion processes of crude glycerin. J. Chromatogr. A 2019, 1589, 56–64. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Zhang, C.; Ren, R.; Liu, R. Simultaneous determination of seven major triterpenoids in Pyrola decorata H. Andres by LC-MS method. Die Pharm. Int. J. Pharm. Sci. 2012, 67, 822–826. [Google Scholar]
- Zhang, H.-q.; Liu, P.; Duan, J.-a.; Dong, L.; Shang, E.-x.; Qian, D.-w.; Xiao, P.; Zhao, M.; Li, W.-w. Hierarchical extraction and simultaneous determination of flavones and triterpenes in different parts of Trichosanthes kirilowii Maxim. by ultra-high-performance liquid chromatography coupled with tandem mass spectrometry. J. Pharm. Biomed. Anal. 2019, 167, 114–122. [Google Scholar] [CrossRef]
- Falev, D.I.; Kosyakov, D.S.; Ul’yanovskii, N.V.; Ovchinnikov, D.V. Rapid simultaneous determination of pentacyclic triterpenoids by mixed-mode liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2020, 1609, 460458. [Google Scholar] [CrossRef] [PubMed]
- Moreno-González, R.; Juan, M.E.; Planas, J.M. Profiling of pentacyclic triterpenes and polyphenols by LC-MS in Arbequina and Empeltre table olives. LWT 2020, 126, 109310. [Google Scholar] [CrossRef]
- Khan, F.; Dastagir, N.; Lateef, M.; Yousuf, M.; Mirani, Z.A.; Mesaik, A.; Faizi, S.; Kazmi, S.U. Immunomodulatory activities of extracts of Caesalpinia pulcherrima. J. Herbs Spices Med. Plants 2018, 24, 245–256. [Google Scholar] [CrossRef]
- Faizi, S.; Fayyaz, S.; Bano, S.; Yawar Iqbal, E.; Lubna; Siddiqi, H.; Naz, A. Isolation of Nematicidal Compounds from Tagetes patula L. Yellow Flowers: Structure–Activity Relationship Studies against Cyst Nematode Heterodera zeae Infective Stage Larvae. J. Agric. Food Chem. 2011, 59, 9080–9093. [Google Scholar] [CrossRef] [PubMed]
- Faizi, S.; Zikr-Ur-Rehman, S.; Versiani, M.A. Shamiminol: A New Aromatic Glycoside from the Stem Bark of Bombax ceiba. Nat. Prod. Commun. 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Saleem, R.; Ahmed, M.; Ahmed, S.I.; Azeem, M.; Khan, R.A.; Rasool, N.; Saleem, H.; Noor, F.; Faizi, S. Hypotensive activity and toxicology of constituents from root bark of Polyalthia longifolia var. pendula. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2005, 19, 881–884. [Google Scholar]
- Siddiqui, F.; Naqvi, S.; Abidi, L.; Faizi, S.; Avesi, L.; Mirza, T.; Farooq, A.D. Opuntia dillenii cladode: Opuntiol and opuntioside attenuated cytokines and eicosanoids mediated inflammation. J. Ethnopharmacol. 2016, 182, 221–234. [Google Scholar] [CrossRef]
- Zikr-ur-Rehman, S.; Versiani, M.A.; Saleem, R.; Sadaf, F.; Azmat, A.; Ahmad, M.; Dar, A.; Faizi, S. Hypotensive and Antioxidant Constituents of Stem Wood of Bombax ceiba. Chem. Nat. Compd. 2019, 55, 939–941. [Google Scholar] [CrossRef]
- Saleem, R.; Rani, R.; Ahmed, M.; Sadaf, F.; Ahmad, S.I.; ul Zafar, N.; Khan, S.S.; Siddiqui, B.S.; Lubna; Ansari, F.; et al. Effect of cream containing Melia azedarach flowers on skin diseases in children. Phytomedicine 2008, 15, 231–236. [Google Scholar] [CrossRef]



{kind=link}
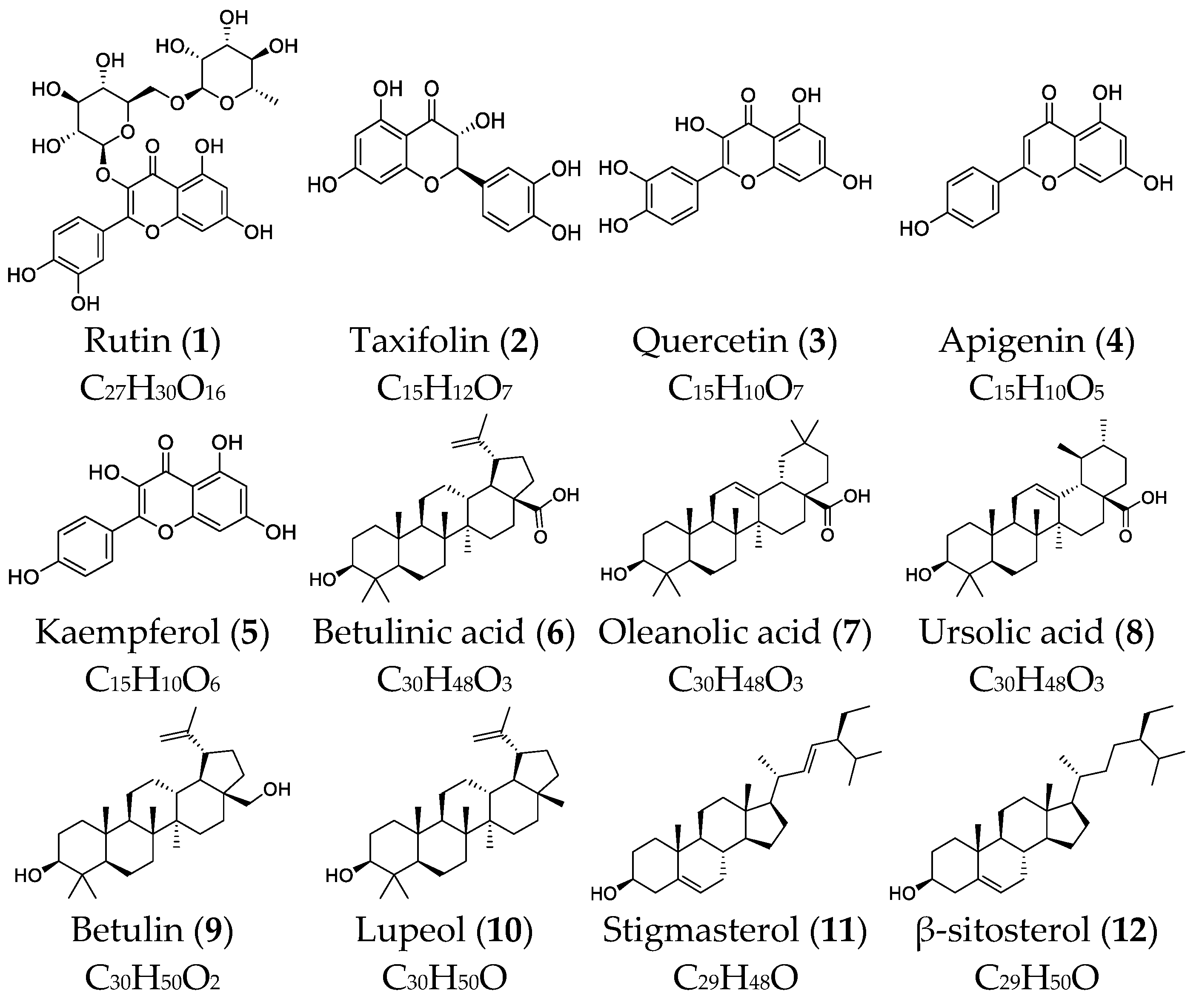{kind=link}
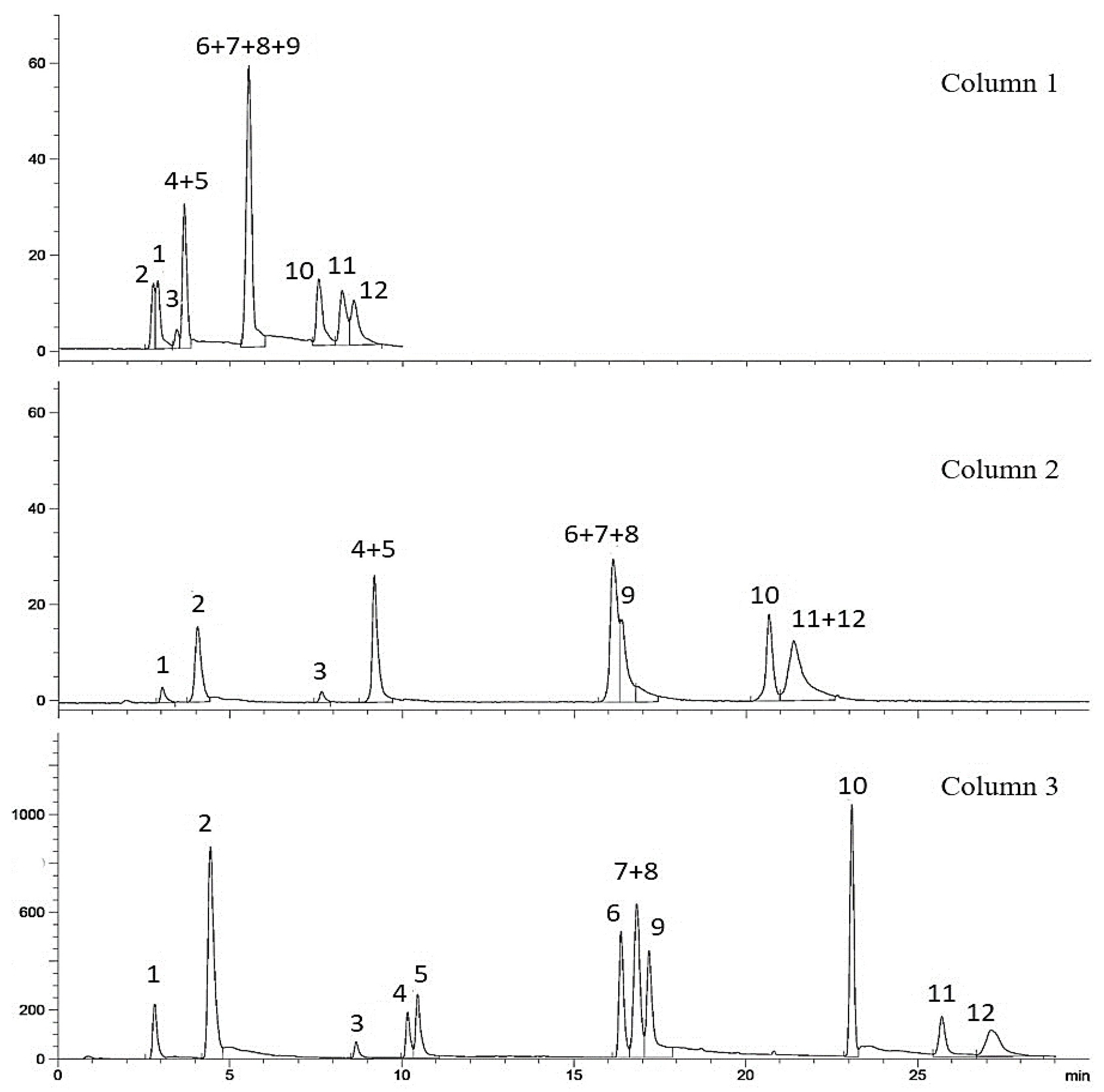{kind=link}
| S. No. | Manufacturer | Column | Dimensions | Flow Rate | Temperature (°C) | Gradient Used | Well Resolved Peaks |
|---|---|---|---|---|---|---|---|
| (mL/min), Time (min) | (Gradient No.) | ||||||
| 1 | Agilent | Poroshell 120 EC-C18 | 3 × 50 mm, 2.7 µm | 1 | 30 | (4) 10% B, 0–1 min; 10–98% B, 1–5 min; 98% B, 5–7.5 min; 98–10% B, 7.5–9 min; 10% B, 9–10 min. | 4 |
| 2 | Agilent | ZORBAX Eclipse | 4.6 × 75 mm, 3.5 µm | 0.5 | 30 | (4) 20% B, 0–2 min; 20–70% B, 2–15 min; 70–80% B, 15–20 min; 80–85% B, 20–21 min; 85–98% B, 21–27 min; 98–20% B, 27–28 min; 20% B, 28–30 min. | 6 |
| XDB-Phenyl | |||||||
| 3 | Macherey-Nagel | EC, NUCLEODUR C18 Gravity | 100 × 3 mm, 1.8 µm | 0.6, 0–17 | 30 | (7) 20% B, 0–2 min; 20–45% B, 2–10 min; 45–85% B, 10–11 min; 85–88% B, 11–17 min; 88–100% B, 17–18 min; 100% B, 18–25 min; 100–20% B, 25–27 min; 20%B, 27–29 min. | 11 |
| 1.3, 17–25 | |||||||
| 0.6, 25–29 |
| Compound | Rt a (Min) | Width (Min) | Rs b |
|---|---|---|---|
| Rutin | 2.33 | 0.13 | 9.11 |
| Taxifolin | 3.73 | 0.17 | 9.11 |
| Quercetin | 8.10 | 0.13 | 11.47 |
| Apigenin | 9.62 | 0.13 | 1.63 |
| Kaempferol | 9.91 | 0.16 | 0.17 |
| Betulinic acid | 16.03 | 0.15 | 2.62 |
| Ursolic acid + Oleanolic acid | 16.46 | 0.17 | 1.37 |
| Betulin | 16.74 | 0.23 | 1.37 |
| Lupeol | 22.55 | 0.11 | 1.68 |
| Stigmasterol | 24.50 | 0.15 | 6.10 |
| β-sitosterol | 25.44 | 0.16 | 6.10 |
| ELSD Parameters | Optimization Parameters Condition | |||
|---|---|---|---|---|
| a | b | c | d | |
| Nebulizer gas pressure (psi) | 50 ± 3 | 50 ± 3 | 45 ± 3, 55 ± 3 | 55 ± 3 |
| Photomultiplier gain | 1, 3, 5, 7, 9, 10, 12 | 10 | 10 | 10 |
| Drift tube temperature (°C) | 80 | 60, 70, 90 | 60 | 60 |
| S. No | Compound Name | Linear Calibration Range | Regression Equation | R2 | LOD | LOQ |
|---|---|---|---|---|---|---|
| (µg/mL) | (µg/mL) | (µg/mL) | ||||
| 1 | Rutin | 100–1000 | y = 1.837x − 0.1568 | 0.997 | 29.4 | 89.1 |
| 2 | Taxifolin | 100–1000 | y = 1.8246x − 0.153 | 0.996 | 31.3 | 94.9 |
| 3 | Quercetin | 100–1000 | y = 3.5603x − 0.4755 | 0.999 | 19.1 | 57.9 |
| 4 | Apigenin | 200–1000 | y = 2.6904x − 0.3257 | 0.997 | 38.3 | 116.1 |
| 5 | Kaempferol | 50–1000 | y = 4.909x − 0.2025 | 0.999 | 15.8 | 47.9 |
| 6 | Betulinic acid | 50–1000 | y = 3.8292x − 0.4187 | 0.999 | 7.8 | 23.5 |
| 7 | Oleanolic acid + Ursolic acid | 100–1000 | y = 9.6962x − 0.84 | 0.996 | 29.9 | 90.5 |
| 8 | Betulin | 100–1000 | y = 5.5929x − 0.6313 | 0.999 | 15.3 | 46.3 |
| 9 | Lupeol | 100–1000 | y = 3.6515x − 0.4517 | 0.999 | 21.5 | 65.0 |
| 10 | Stigmasterol | 100–1000 | y = 2.2809x − 0.2659 | 0.999 | 25.6 | 77.5 |
| HPLC Column | Mode of Elution | Mobile Phase | Compounds | Run Time | Technique | Ref. | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | ||||||
| Hypersil BDS C 18 (250 × 3 mm) 3 μm | Isocratic | 6.5% H2O in acetonitrile | - | - | - | - | - | - | - | - | - | + | + | + | 35 min | HPLC-UV | [13,17] |
| Acquity BEH C18 (100 mm × 2.1 mm, 1.7 μm) | Gradient | Ultra-pure water and methanol | + | - | + | + | + | - | - | - | - | - | - | - | 13 min | HPLC-MS | [25,31] |
| Mixed-Mode WAX-1 (2.1 × 150 mm) 3 μm | Isocratic | Formate buffer solution in acetonitrile | - | - | - | - | - | + | + | + | + | + | - | - | 7 min | MMLC-MS | [26,32] |
| Zorbax Eclipse PAH (150 mm × 4.60 mm, 3.50 μm) | Gradient 1 | Ultra-pure water with acetic acid 0.05% and methanol with acetic acid 0.05% | - | - | - | - | - | - | + | + | - | - | - | - | 24 min | LC-APCI-MS | [27,33] |
| Zorbax Eclipse XDB-C18 (150 mm × 4.60 mm, 5.00 μm) | Gradient 2 | Ultra-pure water with 0.025% acetic acid and solvent B, acetonitrile with 5% acetone | + | - | + | + | - | - | - | - | - | - | - | - | 8.5 min | LC-ESI-MS/MS | |
| EC, NUCLEODUR C18 Gravity (100 mm × 3 mm), 1.8 µm | Gradient | Ultra-pure water with 0.1% formic acid and ACN with 0.1% formic acid | + | + | + | + | + | + | ± | ± | + | + | + | + | 29 min | HPLC-ELSD | Current study |
| S. No | Extract Code | Plant Source (Solvent) | Rutin | Taxifolin | Quercetin | Apigenin | Kaempferol | Betulinic Acid | Oleanolic Acid + Ursolic Acid | Betulin | Lupeol | Stigmasterol |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Shf-1 | Caesalpinia pulcherrima, flowers (ethyl acetate) | 485.97 | + | 234.56 | + | 284.01 | - | - | 227.33 | + | + |
| 2 | Shf-2 | Caesalpinia pulcherrima fresh pods (ethyl acetate) | 212.45 | 137.24 | 315.07 | + | 304.01 | + | - | 112.76 | + | + |
| 3 | Shf-3 | Citrus lemon seeds cover (ethyl acetate) | 146.71 | - | + | + | 568.31 | - | - | - | + | + |
| 4 | Shf-4 | Opuntia dellenii cladodes (ethyl acetate) | + | + | - | 435.70 | 374.07 | - | - | 212.24 | - | - |
| 5 | Shf-5 | Bauhinia variegata pod cover, (ethyl acetate) | 482.39 | + | 237.84 | 468.80 | + | - | + | 265.92 | - | + |
| 6 | Shf-6 | Polyalthia longifolia var. pendula (P) root bark, (methanol) | + | - | 579.51 | + | 53.06 | - | - | + | + | 413.35 |
| 7 | Shf-7 | Bombax ceiba, wood (methanol) | - | + | + | 475.00 | + | + | + | 564.81 | - | + |
| 8 | Shf-8 | Phlox drummondii aerial part (Methanol:H2O) | - | + | + | + | + | - | - | - | + | - |
| 9 | Shf-9 | Olea europea leaves, (ethyl acetate) | 197.17 | 185.71 | 94.50 | + | 483.12 | - | - | 581.83 | + | + |
| 10 | Shf-10 | Caesalpinia pulcherrima flowers, (pet ether) | - | - | + | + | + | + | + | + | 110.01 | + |
| 11 | Shf-11 | Polyalthia longifolia var. pendula (P) dried leaves, (methanol) | + | 399.64 | + | 185.58 | 207.84 | - | 506.22 | + | + | 362.03 |
| 12 | Shf-12 | Tagetes patula flowers, capitulam (Pet ether) | + | - | - | 399.88 | - | - | - | 466.82 | + | - |
| 13 | Shf-13 | Bombax ceiba stem bark (pet ether) | - | - | - | - | + | - | 156.67 | 490.67 | + | - |
| 14 | Shf-14 | Melia azedarach flowers (Methanol) | 223.89 | + | + | + | 496.56 | - | + | 378.49 | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, K.; Ali, A.; Khan, M.N.; Rahman, S.; Faizi, S.; Ali, M.S.; Khalifa, S.A.M.; El-Seedi, H.R.; Musharraf, S.G. Rapid Identification of Common Secondary Metabolites of Medicinal Herbs Using High-Performance Liquid Chromatography with Evaporative Light Scattering Detector in Extracts. Metabolites 2021, 11, 489. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11080489
Ali K, Ali A, Khan MN, Rahman S, Faizi S, Ali MS, Khalifa SAM, El-Seedi HR, Musharraf SG. Rapid Identification of Common Secondary Metabolites of Medicinal Herbs Using High-Performance Liquid Chromatography with Evaporative Light Scattering Detector in Extracts. Metabolites. 2021; 11(8):489. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11080489
Chicago/Turabian StyleAli, Kiran, Arslan Ali, Muhammad Noman Khan, Saeedur Rahman, Shaheen Faizi, Muhammad Shaiq Ali, Shaden A. M. Khalifa, Hesham R. El-Seedi, and Syed Ghulam Musharraf. 2021. "Rapid Identification of Common Secondary Metabolites of Medicinal Herbs Using High-Performance Liquid Chromatography with Evaporative Light Scattering Detector in Extracts" Metabolites 11, no. 8: 489. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo11080489