
Glutamate Efflux across the Blood–Brain Barrier: New Perspectives on the Relationship between Depression and the Glutamatergic System

 , ,
, , 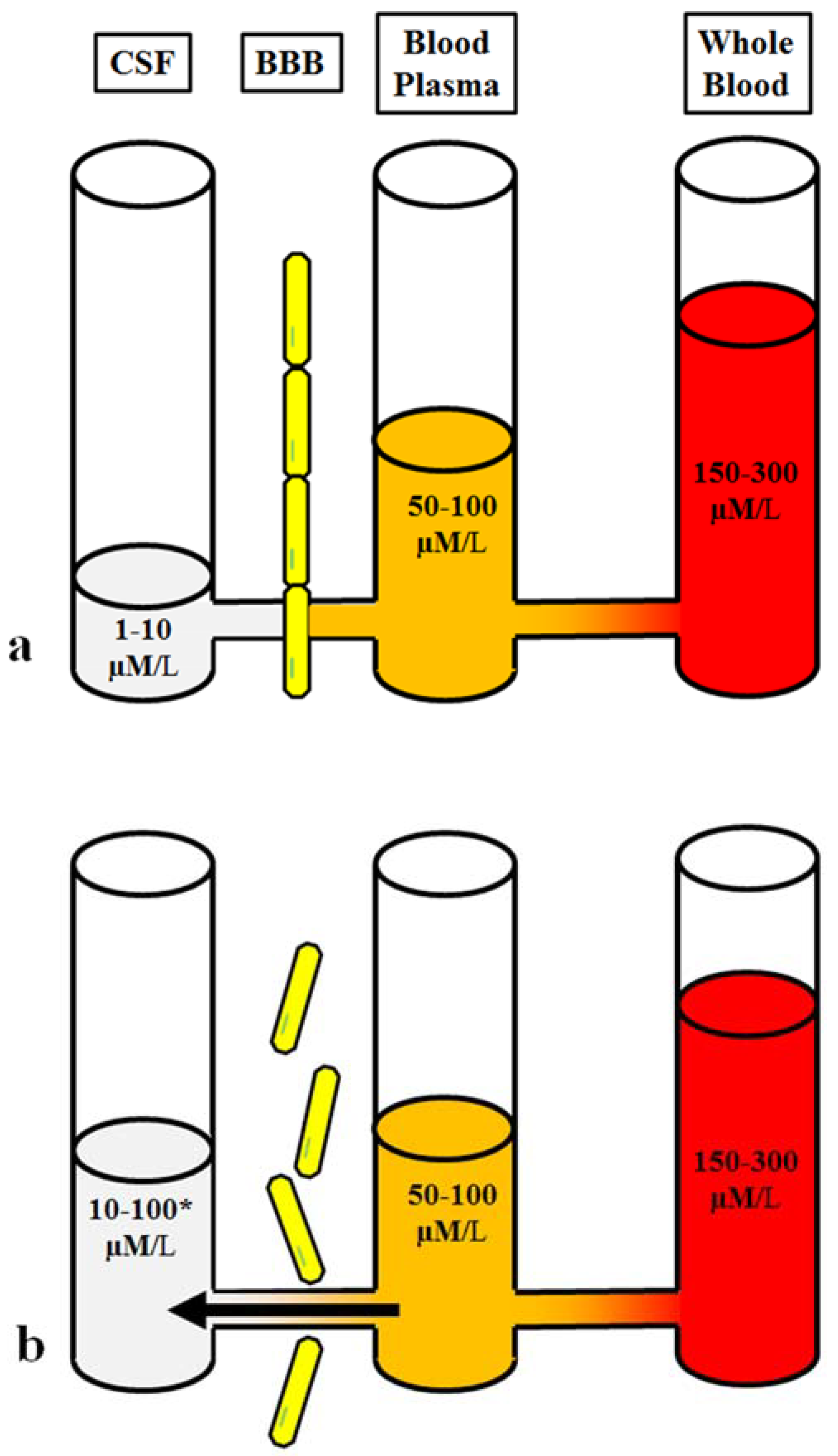{kind=link}
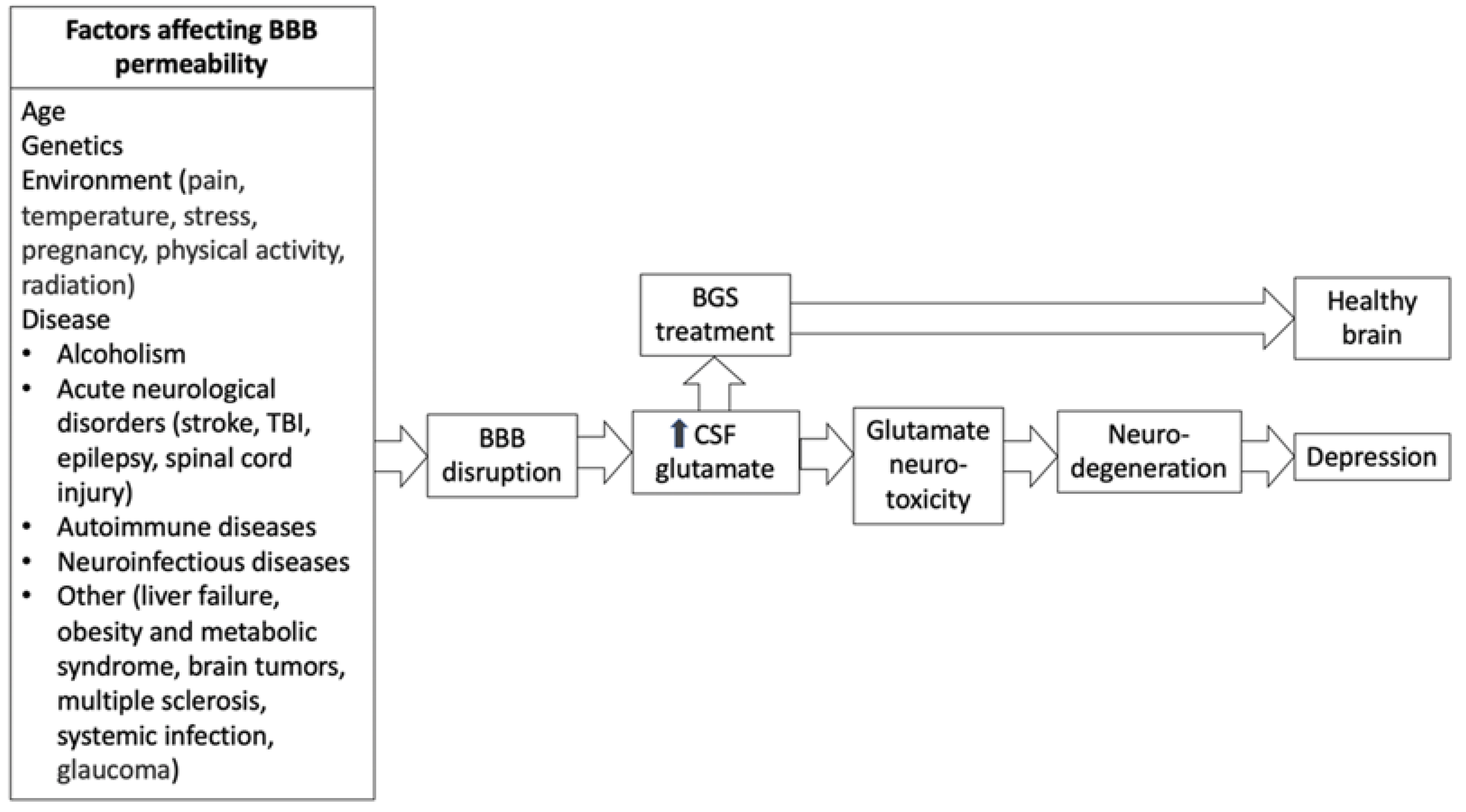{kind=link}
Abstract
:1. Introduction
2. Glutamate
3. Glutamatergic Hypothesis of Depression
4. Depression and the BBB
5. The Imbalance of Blood-Glutamate Concentration Gradient Following Brain Injury
6. Blood as the Source of Elevated Brain Glutamate
7. Neurological Conditions Associated with BBB Permeability and Depression
7.1. Stroke
7.2. TBI
7.3. Other
8. Manipulation of Brain-Blood Glutamate Equilibrium
9. Contrary Findings of Impaired BBB Permeability and Brain-Blood Glutamate Equilibrium
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Musazzi, L.; Treccani, G.; Popoli, M. Glutamate hypothesis of depression and its consequences for antidepressant treatments. Expert Rev. Neurother. 2012, 12, 1169–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladding, C.M.; Raymond, L.A. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol. Cell. Neurosci. 2011, 48, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, C.G.; Sanacora, G.; Duman, R.S.; Krystal, J.H. Ketamine and rapid-acting antidepressants: A window into a new neurobiology for mood disorder therapeutics. Annu. Rev. Med. 2015, 66, 509–523. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Bading, H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003, 26, 81–89. [Google Scholar] [CrossRef]
- Kavalali, E.T.; Monteggia, L.M. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am. J. Psychiatry 2012, 169, 1150–1156. [Google Scholar] [CrossRef]
- Wang, S.-M.; Han, C.; Bahk, W.-M.; Lee, S.-J.; Patkar, A.A.; Masand, P.S.; Pae, C.-U. Addressing the side effects of contemporary antidepressant drugs: A comprehensive review. Chonnam Med. J. 2018, 54, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Sanacora, G.; Treccani, G.; Popoli, M. Towards a glutamate hypothesis of depression: An emerging frontier of neuropsychopharmacology for mood disorders. Neuropharmacology 2012, 62, 63–77. [Google Scholar] [CrossRef] [Green Version]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef]
- DuBourdieu, D. Glutamine supplementation: Hope, hype, or stay tuned? In Nutraceuticals; Elsevier: Amsterdam, The Netherlands, 2021; pp. 1027–1036. [Google Scholar]
- Pralong, E.; Magistretti, P.; Stoop, R. Cellular perspectives on the glutamate–monoamine interactions in limbic lobe structures and their relevance for some psychiatric disorders. Prog. Neurobiol. 2002, 67, 173–202. [Google Scholar] [CrossRef]
- Trullas, R.; Skolnick, P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur. J. Pharmacol. 1990, 185, 1–10. [Google Scholar] [CrossRef]
- Berman, R.M.; Cappiello, A.; Anand, A.; Oren, D.A.; Heninger, G.R.; Charney, D.S.; Krystal, J.H. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000, 47, 351–354. [Google Scholar] [CrossRef]
- Lener, M.S.; Niciu, M.J.; Ballard, E.D.; Park, M.; Park, L.T.; Nugent, A.C.; Zarate, C.A., Jr. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol. Psychiatry 2017, 81, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Schmid-Burgk, W.; Claus, D.; Kornhuber, H. Increased serum glutamate in depressed patients. Arch. Für Psychiatr. Und Nervenkrankh. 1982, 232, 299–304. [Google Scholar] [CrossRef]
- Maes, M.; Verkerk, R.; Vandoolaeghe, E.; Lin, A.; Scharpe, S. Serum levels of excitatory amino acids, serine, glycine, histidine, threonine, taurine, alanine and arginine in treatment-resistant depression: Modulation by treatment with antidepressants and prediction of clinical responsivity. Acta Psychiatr. Scand. 1998, 97, 302–308. [Google Scholar] [CrossRef]
- Küçükibrahimoğlu, E.; Saygın, M.Z.; Çalışkan, M.; Kaplan, O.K.; Ünsal, C.; Gören, M.Z. The change in plasma GABA, glutamine and glutamate levels in fluoxetine-or S-citalopram-treated female patients with major depression. Eur. J. Clin. Pharmacol. 2009, 65, 571–577. [Google Scholar] [CrossRef]
- Mitani, H.; Shirayama, Y.; Yamada, T.; Maeda, K.; Ashby, C.R., Jr.; Kawahara, R. Correlation between plasma levels of glutamate, alanine and serine with severity of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2006, 30, 1155–1158. [Google Scholar] [CrossRef]
- Levine, J.; Panchalingam, K.; Rapoport, A.; Gershon, S.; McClure, R.J.; Pettegrew, J.W. Increased cerebrospinal fluid glutamine levels in depressed patients. Biol. Psychiatry 2000, 47, 586–593. [Google Scholar] [CrossRef]
- Moriguchi, S.; Takamiya, A.; Noda, Y.; Horita, N.; Wada, M.; Tsugawa, S.; Plitman, E.; Sano, Y.; Tarumi, R.; ElSalhy, M. Glutamatergic neurometabolite levels in major depressive disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol. Psychiatry 2019, 24, 952–964. [Google Scholar] [CrossRef] [Green Version]
- Deschwanden, A.; Karolewicz, B.; Feyissa, A.M.; Treyer, V.; Ametamey, S.M.; Johayem, A.; Burger, C.; Auberson, Y.P.; Sovago, J.; Stockmeier, C.A. Reduced metabotropic glutamate receptor 5 density in major depression determined by [11C] ABP688 PET and postmortem study. Am. J. Psychiatry 2011, 168, 727–734. [Google Scholar] [CrossRef] [Green Version]
- Nudmamud-Thanoi, S.; Reynolds, G.P. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neurosci. Lett. 2004, 372, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Beneyto, M.; Kristiansen, L.V.; Oni-Orisan, A.; McCullumsmith, R.E.; Meador-Woodruff, J.H. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology 2007, 32, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Feyissa, A.M.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 70–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyissa, A.M.; Woolverton, W.L.; Miguel-Hidalgo, J.J.; Wang, Z.; Kyle, P.B.; Hasler, G.; Stockmeier, C.A.; Iyo, A.H.; Karolewicz, B. Elevated level of metabotropic glutamate receptor 2/3 in the prefrontal cortex in major depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2010, 34, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Karolewicz, B.; Feyissa, A.M.; Chandran, A.; Legutko, B.; Ordway, G.A.; Rajkowska, G.; Stockmeier, C.A. Glutamate receptors expression in postmortem brain from depressed subjects. Biol. Psychiatry 2009, 65, 177S. [Google Scholar]
- Luykx, J.; Laban, K.; Van Den Heuvel, M.; Boks, M.; Mandl, R.; Kahn, R.; Bakker, S. Region and state specific glutamate downregulation in major depressive disorder: A meta-analysis of 1H-MRS findings. Neurosci. Biobehav. Rev. 2012, 36, 198–205. [Google Scholar] [CrossRef]
- Onaolapo, A.Y.; Onaolapo, O.J. Glutamate and depression: Reflecting a deepening knowledge of the gut and brain effects of a ubiquitous molecule. World J. Psychiatry 2021, 11, 297. [Google Scholar] [CrossRef]
- Godlewska, B.R.; Masaki, C.; Sharpley, A.L.; Cowen, P.J.; Emir, U.E. Brain glutamate in medication-free depressed patients: A proton MRS study at 7 Tesla. Psychol. Med. 2018, 48, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Frank, D.; Kuts, R.; Tsenter, P.; Gruenbaum, B.F.; Grinshpun, Y.; Zvenigorodsky, V.; Shelef, I.; Natanel, D.; Brotfain, E.; Zlotnik, A. The effect of pyruvate on the development and progression of post-stroke depression: A new therapeutic approach. Neuropharmacology 2019, 155, 173–184. [Google Scholar] [CrossRef]
- Frank, D.; Gruenbaum, B.F.; Shelef, I.; Severynovska, O.; Gal, R.; Dubilet, M.; Zlotnik, A.; Kofman, O.; Boyko, M. Blood glutamate scavenging with pyruvate as a novel preventative and therapeutic approach for depressive-like behavior following traumatic brain injury in a rat model. Front. Neurosci. 2022, 21, 832478. [Google Scholar] [CrossRef]
- Gruenbaum, B.F.; Kutz, R.; Zlotnik, A.; Boyko, M. Blood glutamate scavenging as a novel glutamate-based therapeutic approach for post-stroke depression. Ther. Adv. Psychopharmacol. 2020, 10, 2045125320903951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, W.; Zhou, Y.-G. Homeostasis of the intraparenchymal-blood glutamate concentration gradient: Maintenance, imbalance, and regulation. Front. Mol. Neurosci. 2017, 10, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sifat, A.E.; Vaidya, B.; Abbruscato, T.J. Blood-brain barrier protection as a therapeutic strategy for acute ischemic stroke. AAPS J. 2017, 19, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Tajes, M.; Ramos-Fernández, E.; Weng-Jiang, X.; Bosch-Morato, M.; Guivernau, B.; Eraso-Pichot, A.; Salvador, B.; Fernandez-Busquets, X.; Roquer, J.; Munoz, F.J. The blood-brain barrier: Structure, function and therapeutic approaches to cross it. Mol. Membr. Biol. 2014, 31, 152–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, R.A.; Simpson, I.A.; Mokashi, A.; Viña, J.R. Pyroglutamate stimulates Na+-dependent glutamate transport across the blood–brain barrier. FEBS Lett. 2006, 580, 4382–4386. [Google Scholar] [CrossRef] [Green Version]
- Klin, Y.; Zlotnik, A.; Boyko, M.; Ohayon, S.; Shapira, Y.; Teichberg, V.I. Distribution of radiolabeled l-glutamate and d-aspartate from blood into peripheral tissues in naive rats: Significance for brain neuroprotection. Biochem. Biophys. Res. Commun. 2010, 399, 694–698. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases—what is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Rickards, H. Depression in neurological disorders: An update. Curr. Opin. Psychiatry 2006, 19, 294–298. [Google Scholar] [CrossRef]
- Hellmann-Regen, J.; Piber, D.; Hinkelmann, K.; Gold, S.M.; Heesen, C.; Spitzer, C.; Endres, M.; Otte, C. Depressive syndromes in neurological disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263 (Suppl. S2), S123–S136. [Google Scholar] [CrossRef]
- Yang, S.-J.; Kim, E.-A.; Chang, M.-J.; Kim, J.; Na, J.-M.; Choi, S.Y.; Cho, S.-W. N-Adamantyl-4-methylthiazol-2-amine attenuates glutamate-induced oxidative stress and inflammation in the brain. Neurotox. Res. 2017, 32, 107–120. [Google Scholar] [CrossRef]
- Kress, B.T.; Iliff, J.J.; Xia, M.; Wang, M.; Wei, H.S.; Zeppenfeld, D.; Xie, L.; Kang, H.; Xu, Q.; Liew, J.A. Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol. 2014, 76, 845–861. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, J.A.; Szu, J.I.; Yonan, J.M.; Binder, D.K. Regulation of astrocyte glutamate transporter-1 (GLT1) and aquaporin-4 (AQP4) expression in a model of epilepsy. Exp. Neurol. 2016, 283, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, G.-P.; Xu, J.; Zhuo, F.; Sun, S.-Q.; Liu, H.; Yang, M.; Huang, J.; Lu, W.-T.; Huang, S.-Q. Loss of AQP4 polarized localization with loss of β-dystroglycan immunoreactivity may induce brain edema following intracerebral hemorrhage. Neurosci. Lett. 2015, 588, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Hue, C.D.; Cao, S.; “Dale” Bass, C.R.; Meaney, D.F.; Morrison, B., III. Repeated primary blast injury causes delayed recovery, but not additive disruption, in an in vitro blood–brain barrier model. J. Neurotrauma 2014, 31, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Merali, Z.; Huang, K.; Mikulis, D.; Silver, F.; Kassner, A. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS ONE 2017, 12, e0171558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Wang, M.; Yang, D.; Wei, X.; Li, W. Dynamics of blood brain barrier permeability and tissue microstructure following controlled cortical impact injury in rat: A dynamic contrast-enhanced magnetic resonance imaging and diffusion kurtosis imaging study. Magn. Reson. Imaging 2019, 62, 1–9. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, E.A.; Ndode-Ekane, X.E.; Lehto, L.J.; Gorter, J.A.; Andrade, P.; Aronica, E.; Gröhn, O.; Pitkänen, A. Long-lasting blood-brain barrier dysfunction and neuroinflammation after traumatic brain injury. Neurobiol. Dis. 2020, 145, 105080. [Google Scholar] [CrossRef]
- Hay, J.R.; Johnson, V.E.; Young, A.M.; Smith, D.H.; Stewart, W. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar]
- Vespa, P.; Prins, M.; Ronne-Engstrom, E.; Caron, M.; Shalmon, E.; Hovda, D.A.; Martin, N.A.; Becker, D.P. Increase in extracellular glutamate caused by reduced cerebral perfusion pressure and seizures after human traumatic brain injury: A microdialysis study. J. Neurosurg. 1998, 89, 971–982. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, H.; Guo, X.; Pluimer, B.; Zhao, Z. Blood–brain barrier dysfunction in mild traumatic brain injury: Evidence from preclinical murine models. Front. Physiol. 2020, 1030. [Google Scholar] [CrossRef]
- Piao, C.-S.; Holloway, A.L.; Hong-Routson, S.; Wainwright, M.S. Depression following traumatic brain injury in mice is associated with down-regulation of hippocampal astrocyte glutamate transporters by thrombin. J. Cereb. Blood Flow Metab. 2019, 39, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, A.; Boyko, M.; Shapira, Y.; Zlotnik, A. Blood glutamate scavenging: Insight into neuroprotection. Int. J. Mol. Sci. 2012, 13, 10041–10066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, M.A.; Jahr, C.E. Extracellular glutamate concentration in hippocampal slice. J. Neurosci. 2007, 27, 9736–9741. [Google Scholar] [CrossRef] [PubMed]
- Meur, K.L.; Galante, M.; Angulo, M.C.; Audinat, E. Tonic activation of NMDA receptors by ambient glutamate of non-synaptic origin in the rat hippocampus. J. Physiol. 2007, 580, 373–383. [Google Scholar] [CrossRef]
- Dash, M.B.; Douglas, C.L.; Vyazovskiy, V.V.; Cirelli, C.; Tononi, G. Long-term homeostasis of extracellular glutamate in the rat cerebral cortex across sleep and waking states. J. Neurosci. 2009, 29, 620–629. [Google Scholar] [CrossRef] [Green Version]
- De Bundel, D.; Schallier, A.; Loyens, E.; Fernando, R.; Miyashita, H.; Van Liefferinge, J.; Vermoesen, K.; Bannai, S.; Sato, H.; Michotte, Y. Loss of system xc− does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J. Neurosci. 2011, 31, 5792–5803. [Google Scholar] [CrossRef] [Green Version]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867S–874S. [Google Scholar] [CrossRef] [Green Version]
- Teichberg, V.; Cohen-Kashi-Malina, K.; Cooper, I.; Zlotnik, A. Homeostasis of glutamate in brain fluids: An accelerated brain-to-blood efflux of excess glutamate is produced by blood glutamate scavenging and offers protection from neuropathologies. Neuroscience 2009, 158, 301–308. [Google Scholar] [CrossRef]
- Cohen-Kashi-Malina, K.; Cooper, I.; Teichberg, V.I. Mechanisms of Glutamate Efflux at the Blood—Brain Barrier: Involvement of Glial Cells. J. Cereb. Blood Flow Metab. 2012, 32, 177–189. [Google Scholar] [CrossRef]
- Al-Sarraf, H.; Philip, L. Increased brain uptake and CSF clearance of 14C-glutamate in spontaneously hypertensive rats. Brain Res. 2003, 994, 181–187. [Google Scholar] [CrossRef]
- Zlotnik, A.; Sinelnikov, I.; Gruenbaum, B.F.; Gruenbaum, S.E.; Dubilet, M.; Dubilet, E.; Leibowitz, A.; Ohayon, S.; Regev, A.; Boyko, M. Effect of glutamate and blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome and pathohistology of the hippocampus after traumatic brain injury in rats. J. Am. Soc. Anesthesiol. 2012, 116, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruban, A.; Berkutzki, T.; Cooper, I.; Mohar, B.; Teichberg, V.I. Blood glutamate scavengers prolong the survival of rats and mice with brain-implanted gliomas. Investig. New Drugs 2012, 30, 2226–2235. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Gruenbaum, S.E.; Artru, A.A.; Rozet, I.; Dubilet, M.; Tkachov, S.; Brotfain, E.; Klin, Y.; Shapira, Y.; Teichberg, V.I. The neuroprotective effects of oxaloacetate in closed head injury in rats is mediated by its blood glutamate scavenging activity: Evidence from the use of maleate. J. Neurosurg. Anesthesiol. 2009, 21, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Campos, F.; Sobrino, T.; Ramos-Cabrer, P.; Castellanos, M.; Blanco, M.; Rodríguez-Yáñez, M.; Serena, J.; Leira, R.; Castillo, J. High blood glutamate oxaloacetate transaminase levels are associated with good functional outcome in acute ischemic stroke. J. Cereb. Blood Flow Metab. 2011, 31, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Gudmundsson, P.; Skoog, I.; Waern, M.; Blennow, K.; Zetterberg, H.; Rosengren, L.; Gustafson, D. Is there a CSF biomarker profile related to depression in elderly women? Psychiatry Res. 2010, 176, 174–178. [Google Scholar] [CrossRef]
- Gulen, B.; Serinken, M.; Eken, C.; Karcıoglu, Ö.; Kucukdagli, O.T.; Kilic, E.; Akpinar, G.; Nogay, S.; Kuh, M. Serum S100B as a surrogate biomarker in the diagnoses of burnout and depression in emergency medicine residents. Acad. Emerg. Med. 2016, 23, 786–789. [Google Scholar] [CrossRef] [Green Version]
- Arora, P.; Sagar, R.; Mehta, M.; Pallavi, P.; Sharma, S.; Mukhopadhyay, A.K. Serum S100B levels in patients with depression. Indian J. Psychiatry 2019, 61, 70. [Google Scholar]
- Kealy, J.; Greene, C.; Campbell, M. Blood-brain barrier regulation in psychiatric disorders. Neurosci. Lett. 2020, 726, 133664. [Google Scholar] [CrossRef] [Green Version]
- Sántha, P.; Veszelka, S.; Hoyk, Z.; Mészáros, M.; Walter, F.R.; Tóth, A.E.; Kiss, L.; Kincses, A.; Oláh, Z.; Seprényi, G. Restraint stress-induced morphological changes at the blood-brain barrier in adult rats. Front. Mol. Neurosci. 2016, 8, 88. [Google Scholar] [CrossRef] [Green Version]
- Menard, C.; Pfau, M.L.; Hodes, G.E.; Kana, V.; Wang, V.X.; Bouchard, S.; Takahashi, A.; Flanigan, M.E.; Aleyasin, H.; LeClair, K.B. Social stress induces neurovascular pathology promoting depression. Nat. Neurosci. 2017, 20, 1752–1760. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Kang, B.-M.; Kim, J.H.; Min, J.; Kim, H.S.; Ryu, H.; Park, H.; Bae, S.; Oh, D.; Choi, M. Real-time in vivo two-photon imaging study reveals decreased cerebro-vascular volume and increased blood-brain barrier permeability in chronically stressed mice. Sci. Rep. 2018, 8, 13064. [Google Scholar] [CrossRef] [PubMed]
- Profaci, C.P.; Munji, R.N.; Pulido, R.S.; Daneman, R. The blood-brain barrier in health and disease: Important unanswered questions. J. Exp. Med. 2020, 217, e20190062. [Google Scholar] [CrossRef] [PubMed]
- Schoknecht, K.; Shalev, H. Blood-brain barrier dysfunction in brain diseases: Clinical experience. Epilepsia 2012, 53 (Suppl. S6), 7–13. [Google Scholar] [CrossRef]
- Archie, S.R.; Al Shoyaib, A.; Cucullo, L. Blood-Brain Barrier Dysfunction in CNS Disorders and Putative Therapeutic Targets: An Overview. Pharmaceutics 2021, 13, 1779. [Google Scholar] [CrossRef] [PubMed]
- Belov Kirdajova, D.; Kriska, J.; Tureckova, J.; Anderova, M. Ischemia-Triggered Glutamate Excitotoxicity From the Perspective of Glial Cells. Front. Cell Neurosci. 2020, 14, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uprety, A.; Kang, Y.; Kim, S.Y. Blood-brain barrier dysfunction as a potential therapeutic target for neurodegenerative disorders. Arch. Pharm. Res. 2021, 44, 487–498. [Google Scholar] [CrossRef]
- Doyle, K.P.; Simon, R.P.; Stenzel-Poore, M.P. Mechanisms of ischemic brain damage. Neuropharmacology 2008, 55, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.G.; Jorge, R.E. Post-Stroke Depression: A Review. Am. J. Psychiatry 2016, 173, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Chodobski, A.; Zink, B.J.; Szmydynger-Chodobska, J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011, 2, 492–516. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Guo, J.J.; Zhan, S.; Patel, N.C. Treatment effects of antidepressants in patients with post-stroke depression: A meta-analysis. Ann. Pharm. 2006, 40, 2115–2122. [Google Scholar] [CrossRef]
- Lavoie, S.; Sechrist, S.; Quach, N.; Ehsanian, R.; Duong, T.; Gotlib, I.H.; Isaac, L. Depression in Men and Women One Year Following Traumatic Brain Injury (TBI): A TBI Model Systems Study. Front. Psychol. 2017, 8, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholten, A.C.; Haagsma, J.A.; Cnossen, M.C.; Olff, M.; van Beeck, E.F.; Polinder, S. Prevalence of and Risk Factors for Anxiety and Depressive Disorders after Traumatic Brain Injury: A Systematic Review. J. Neurotrauma 2016, 33, 1969–1994. [Google Scholar] [CrossRef] [PubMed]
- Gasca-Salas, C.; Fernandez-Rodriguez, B.; Pineda-Pardo, J.A.; Rodriguez-Rojas, R.; Obeso, I.; Hernandez-Fernandez, F.; Del Alamo, M.; Mata, D.; Guida, P.; Ordas-Bandera, C.; et al. Blood-brain barrier opening with focused ultrasound in Parkinson’s disease dementia. Nat. Commun. 2021, 12, 779. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Scalabri, F.; Pepe, G.; Castaldo, S.; Elifani, F.; Capocci, L.; De Sanctis, C.; Comerci, L.; Pompeo, F.; et al. Impairment of blood-brain barrier is an early event in R6/2 mouse model of Huntington Disease. Sci. Rep. 2017, 7, 41316. [Google Scholar] [CrossRef] [PubMed]
- Abrahao, A.; Meng, Y.; Llinas, M.; Huang, Y.; Hamani, C.; Mainprize, T.; Aubert, I.; Heyn, C.; Black, S.E.; Hynynen, K.; et al. First-in-human trial of blood-brain barrier opening in amyotrophic lateral sclerosis using MR-guided focused ultrasound. Nat. Commun. 2019, 10, 4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidari, M.E.; Nadali, J.; Parouhan, A.; Azarafraz, M.; Tabatabai, S.M.; Irvani, S.S.N.; Eskandari, F.; Gharebaghi, A. Prevalence of depression among amyotrophic lateral sclerosis (ALS) patients: A systematic review and meta-analysis. J. Affect. Disord. 2021, 287, 182–190. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Pacheco-Moises, F.P.; Macias-Islas, M.A.; Flores-Alvarado, L.J.; Mireles-Ramirez, M.A.; Gonzalez-Renovato, E.D.; Hernandez-Navarro, V.E.; Sanchez-Lopez, A.L.; Alatorre-Jimenez, M.A. Role of the blood-brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Aguera-Ortiz, L.; Garcia-Ramos, R.; Grandas Perez, F.J.; Lopez-Alvarez, J.; Montes Rodriguez, J.M.; Olazaran Rodriguez, F.J.; Olivera Pueyo, J.; Pelegrin Valero, C.; Porta-Etessam, J. Depression in Alzheimer’s Disease: A Delphi Consensus on Etiology, Risk Factors, and Clinical Management. Front. Psychiatry 2021, 12, 638651. [Google Scholar] [CrossRef]
- Madeira, C.; Vargas-Lopes, C.; Brandao, C.O.; Reis, T.; Laks, J.; Panizzutti, R.; Ferreira, S.T. Elevated Glutamate and Glutamine Levels in the Cerebrospinal Fluid of Patients with Probable Alzheimer’s Disease and Depression. Front. Psychiatry 2018, 9, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüber, T.; David, B.; Lüchters, G.; Nass, R.D.; Friedman, A.; Surges, R.; Stöcker, T.; Weber, B.; Deichmann, R.; Schlaug, G. Evidence for peri-ictal blood–brain barrier dysfunction in patients with epilepsy. Brain 2018, 141, 2952–2965. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.J.; Sharpe, L.; Hunt, C.; Gandy, M. Anxiety and depressive disorders in people with epilepsy: A meta-analysis. Epilepsia 2017, 58, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, B.F.; Sandhu, M.R.S.; Bertasi, R.A.O.; Bertasi, T.G.O.; Schonwald, A.; Kurup, A.; Gruenbaum, S.E.; Freedman, I.G.; Funaro, M.C.; Blumenfeld, H.; et al. Absence seizures and their relationship to depression and anxiety: Evidence for bidirectionality. Epilepsia 2021, 62, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Futtrup, J.; Margolinsky, R.; Benros, M.E.; Moos, T.; Routhe, L.J.; Rungby, J.; Krogh, J. Blood-brain barrier pathology in patients with severe mental disorders: A systematic review and meta-analysis of biomarkers in case-control studies. Brain Behav. Immun. Health 2020, 6, 100102. [Google Scholar] [CrossRef] [PubMed]
- Etchecopar-Etchart, D.; Korchia, T.; Loundou, A.; Llorca, P.M.; Auquier, P.; Lancon, C.; Boyer, L.; Fond, G. Comorbid Major Depressive Disorder in Schizophrenia: A Systematic Review and Meta-Analysis. Schizophr. Bull. 2021, 47, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Eftekar, M. The association between hepatic encephalopathy/minimal hepatic encephalopathy and depressive and anxiety disorders: A systematic review. Australas. Psychiatry 2020, 28, 61–65. [Google Scholar] [CrossRef]
- Huang, J.; Zeng, C.; Xiao, J.; Zhao, D.; Tang, H.; Wu, H.; Chen, J. Association between depression and brain tumor: A systematic review and meta-analysis. Oncotarget 2017, 8, 94932–94943. [Google Scholar] [CrossRef]
- Castillo, J.; Loza, M.I.; Mirelman, D.; Brea, J.; Blanco, M.; Sobrino, T.; Campos, F. A novel mechanism of neuroprotection: Blood glutamate grabber. J. Cereb. Blood Flow Metab. 2016, 36, 292–301. [Google Scholar] [CrossRef]
- Zlotnik, A.; Gurevich, B.; Cherniavsky, E.; Tkachov, S.; Matuzani-Ruban, A.; Leon, A.; Shapira, Y.; Teichberg, V.I. The contribution of the blood glutamate scavenging activity of pyruvate to its neuroprotective properties in a rat model of closed head injury. Neurochem. Res. 2008, 33, 1044–1050. [Google Scholar] [CrossRef]
- Li, Y.; Hou, X.; Qi, Q.; Wang, L.; Luo, L.; Yang, S.; Zhang, Y.; Miao, Z.; Zhang, Y.; Wang, F. Scavenging of blood glutamate for enhancing brain-to-blood glutamate efflux. Mol. Med. Rep. 2014, 9, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Boyko, M.; Melamed, I.; Gruenbaum, B.F.; Gruenbaum, S.E.; Ohayon, S.; Leibowitz, A.; Brotfain, E.; Shapira, Y.; Zlotnik, A. The effect of blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome in a rat model of subarachnoid hemorrhage. Neurotherapeutics 2012, 9, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Mato, M.; Ramos-Cabrer, P.; Sobrino, T.; Blanco, M.; Ruban, A.; Mirelman, D.; Menendez, P.; Castillo, J.; Campos, F. Human recombinant glutamate oxaloacetate transaminase 1 (GOT1) supplemented with oxaloacetate induces a protective effect after cerebral ischemia. Cell Death Dis. 2014, 5, e992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, R.; Zauner, A.; Woodward, J.J.; Myseros, J.; Choi, S.C.; Ward, J.D.; Marmarou, A.; Young, H.F. Factors affecting excitatory amino acid release following severe human head injury. J. Neurosurg. 1998, 89, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Briggs, Z.; Rink, C. Inducible glutamate oxaloacetate transaminase as a therapeutic target against ischemic stroke. Antioxid. Redox Signal. 2015, 22, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, R.; Lyons, K.; Khosla, S.; Nashatizadeh, M.; Pahwa, R. A pilot Study of oxaloacetate 100 mg capsules in Parkinson’s disease patients. J. Parkinsons Dis. Alzheimers Dis. 2016, 3, 4. [Google Scholar]
- Swerdlow, R.H.; Bothwell, R.; Hutfles, L.; Burns, J.M.; Reed, G.A. Tolerability and pharmacokinetics of oxaloacetate 100 mg capsules in Alzheimer’s subjects. BBA Clin. 2016, 5, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Tully, L.; Humiston, J.; Cash, A. Oxaloacetate reduces emotional symptoms in premenstrual syndrome (PMS): Results of a placebo-controlled, cross-over clinical trial. Obstet. Gynecol. Sci. 2020, 63, 195–204. [Google Scholar] [CrossRef]
- McGirr, A.; Berlim, M.; Bond, D.; Fleck, M.; Yatham, L.; Lam, R. A systematic review and meta-analysis of randomized, double-blind, placebo-controlled trials of ketamine in the rapid treatment of major depressive episodes. Psychol. Med. 2015, 45, 693–704. [Google Scholar] [CrossRef]
- Baeken, C.; Lefaucheur, J.-P.; Van Schuerbeek, P. The impact of accelerated high frequency rTMS on brain neurochemicals in treatment-resistant depression: Insights from 1H MR spectroscopy. Clin. Neurophysiol. 2017, 128, 1664–1672. [Google Scholar] [CrossRef]
- Gabbay, V.; Bradley, K.; Mao, X.; Ostrover, R.; Kang, G.; Shungu, D. Anterior cingulate cortex γ-aminobutyric acid deficits in youth with depression. Transl. Psychiatry 2017, 7, e1216. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.P.; Dai, H.Y.; Dai, Z.Z.; Xu, C.T.; Wu, R.H. Anterior cingulate cortex and cerebellar hemisphere neurometabolite changes in depression treatment: A 1H magnetic resonance spectroscopy study. Psychiatry Clin. Neurosci. 2014, 68, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Musazzi, L.; Treccani, G.; Mallei, A.; Popoli, M. The action of antidepressants on the glutamate system: Regulation of glutamate release and glutamate receptors. Biol. Psychiatry 2013, 73, 1180–1188. [Google Scholar] [CrossRef]
- Kraal, A.Z.; Arvanitis, N.R.; Jaeger, A.P.; Ellingrod, V.L. Could dietary glutamate play a role in psychiatric distress? Neuropsychobiology 2020, 79, 13–19. [Google Scholar] [CrossRef]
- Pal, M.M. Glutamate: The Master Neurotransmitter and Its Implications in Chronic Stress and Mood Disorders. Front. Hum. Neurosci. 2021, 15, 722323. [Google Scholar] [CrossRef] [PubMed]
- Altamura, C.A.; Mauri, M.C.; Ferrara, A.; Moro, A.R.; D’Andrea, G.; Zamberlan, F. Plasma and platelet excitatory amino acids in psychiatric disorders. Am. J. Psychiatry 1993, 150, 1731–1733. [Google Scholar]
- Zlotnik, A.; Ohayon, S.; Gruenbaum, B.F.; Gruenbaum, S.E.; Mohar, B.; Boyko, M.; Klin, Y.; Sheiner, E.; Shaked, G.; Shapira, Y. Determination of factors affecting glutamate concentrations in the whole blood of healthy human volunteers. J. Neurosurg. Anesthesiol. 2011, 23, 45–49. [Google Scholar] [CrossRef]
- Morrell, C.N.; Sun, H.; Ikeda, M.; Beique, J.-C.; Swaim, A.M.; Mason, E.; Martin, T.V.; Thompson, L.E.; Gozen, O.; Ampagoomian, D. Glutamate mediates platelet activation through the AMPA receptor. J. Exp. Med. 2008, 205, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, R.; Uehara, S.; Yatsushiro, S.; Juge, N.; Hua, Z.; Senoh, S.; Echigo, N.; Hayashi, M.; Mizoguchi, T.; Ninomiya, T. Secretion of L-glutamate from osteoclasts through transcytosis. EMBO J. 2006, 25, 4175–4186. [Google Scholar] [CrossRef] [Green Version]
- Zlotnik, A.; Gruenbaum, B.F.; Mohar, B.; Kuts, R.; Gruenbaum, S.E.; Ohayon, S.; Boyko, M.; Klin, Y.; Sheiner, E.; Shaked, G. The effects of estrogen and progesterone on blood glutamate levels: Evidence from changes of blood glutamate levels during the menstrual cycle in women. Biol. Reprod. 2011, 84, 581–586. [Google Scholar] [CrossRef]
- Chang, L.; Jiang, C.S.; Ernst, T. Effects of age and sex on brain glutamate and other metabolites. Magn. Reson. Imaging 2009, 27, 142–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyko, M.; Kuts, R.; Gruenbaum, B.F.; Melamed, I.; Gruenbaum, S.E.; Klein, M.; Shapira, Y.; Zlotnik, A. The role of hypothermia in the regulation of blood glutamate levels in naive rats. J. Neurosurg. Anesthesiol. 2013, 25, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Cottell, E.; Hutchinson, M.; Simon, J.; Harrington, M.G. Plasma glutamate levels in normal subjects and in patients with amyotrophic lateral sclerosis. Biochem. Soc. Trans. 1990, 18, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obrenovitch, T.P.; Urenjak, J. Altered glutamatergic transmission in neurological disorders: From high extracellular glutamate to excessive synaptic efficacy. Prog. Neurobiol. 1997, 51, 39–87. [Google Scholar] [CrossRef]
- Hanigan, M.; Calvert, C.; DePeters, E.; Reis, B.; Baldwin, R. Whole blood and plasma amino acid uptakes by lactating bovine mammary glands. J. Dairy Sci. 1991, 74, 2484–2490. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruenbaum, B.F.; Zlotnik, A.; Frenkel, A.; Fleidervish, I.; Boyko, M. Glutamate Efflux across the Blood–Brain Barrier: New Perspectives on the Relationship between Depression and the Glutamatergic System. Metabolites 2022, 12, 459. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12050459
Gruenbaum BF, Zlotnik A, Frenkel A, Fleidervish I, Boyko M. Glutamate Efflux across the Blood–Brain Barrier: New Perspectives on the Relationship between Depression and the Glutamatergic System. Metabolites. 2022; 12(5):459. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12050459
Chicago/Turabian StyleGruenbaum, Benjamin Fredrick, Alexander Zlotnik, Amit Frenkel, Ilya Fleidervish, and Matthew Boyko. 2022. "Glutamate Efflux across the Blood–Brain Barrier: New Perspectives on the Relationship between Depression and the Glutamatergic System" Metabolites 12, no. 5: 459. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo12050459