Metabolic Consequences of TGFb Stimulation in CulturedPrimary Mouse Hepatocytes Screened from Transcript Data with ModeScore

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
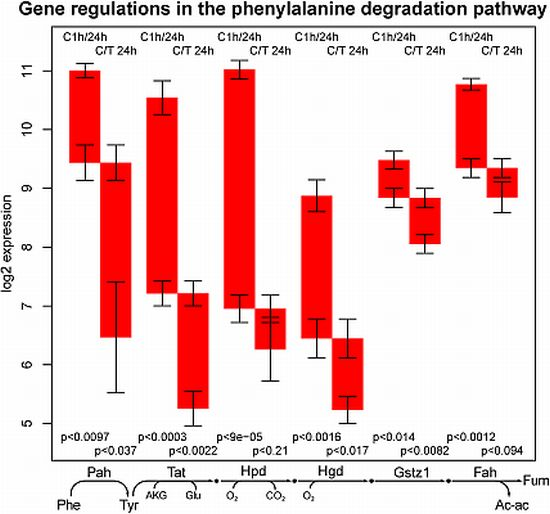
2.1. General Observations
2.2. ModeScore Analysis

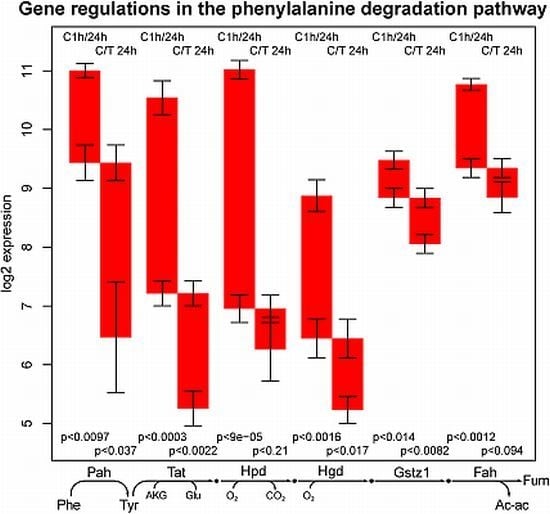
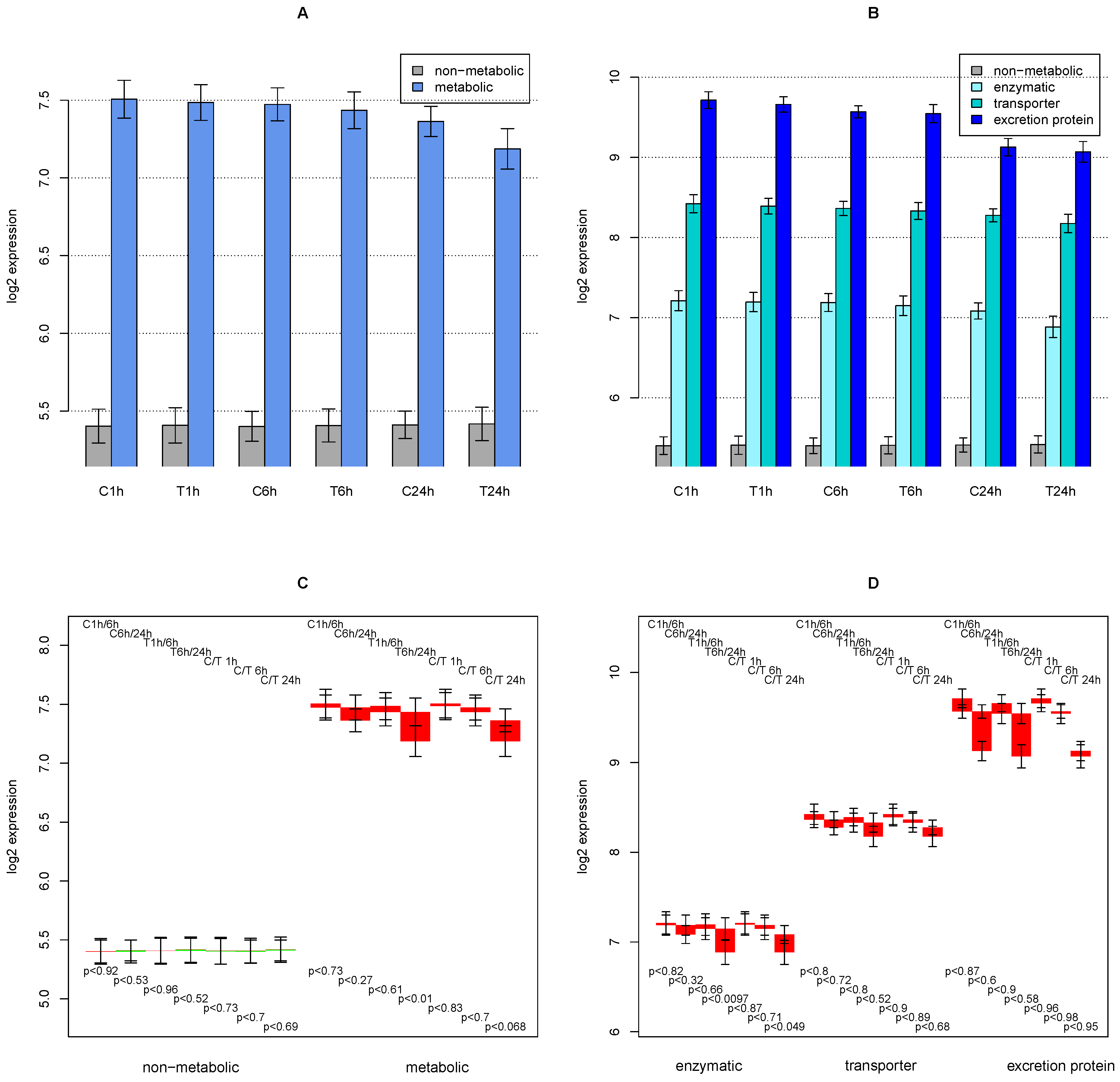
2.3. Tyrosine Degradation

2.4. Collagen Regulation
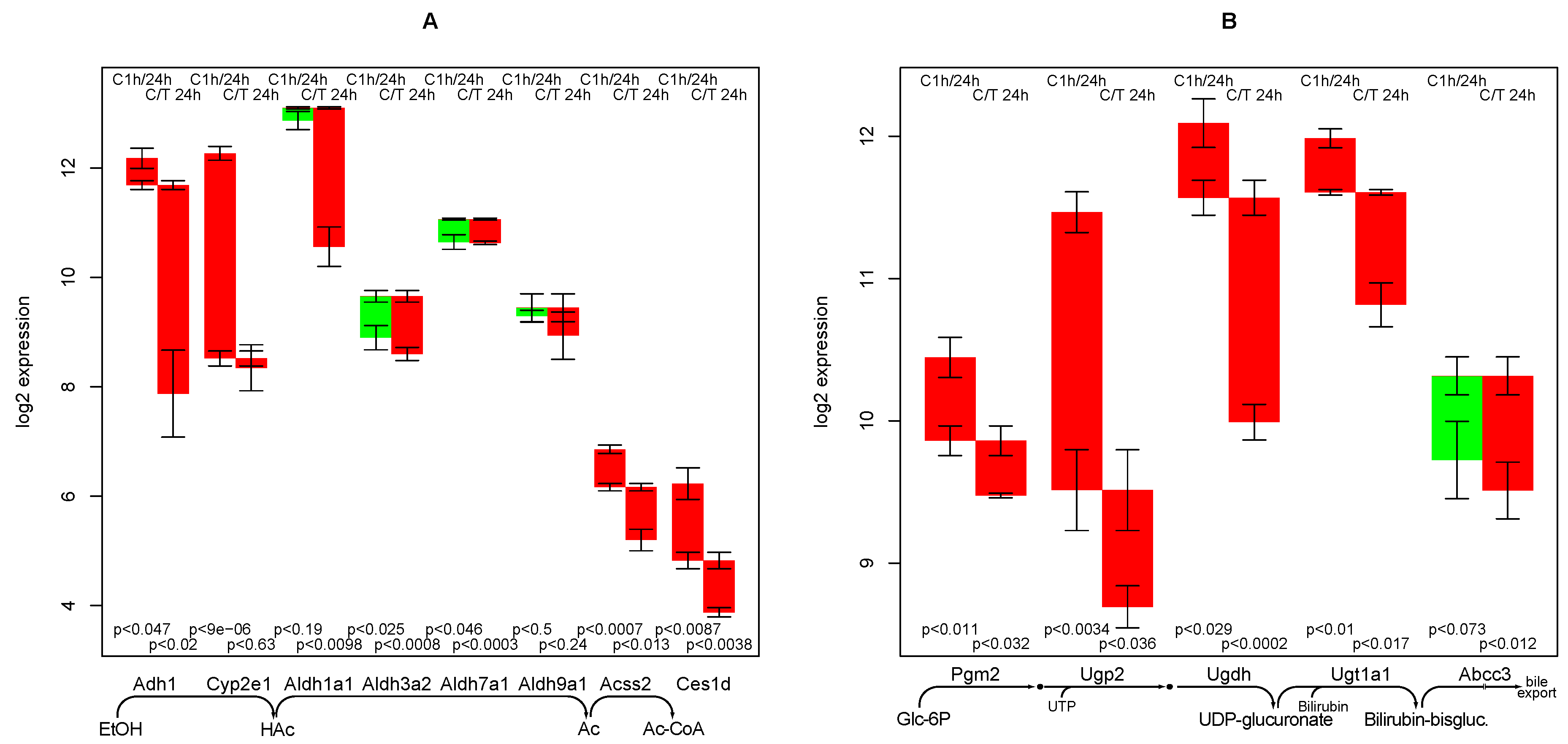
2.5. Ethanol Degradation

2.6. Bilirubin Conjugation
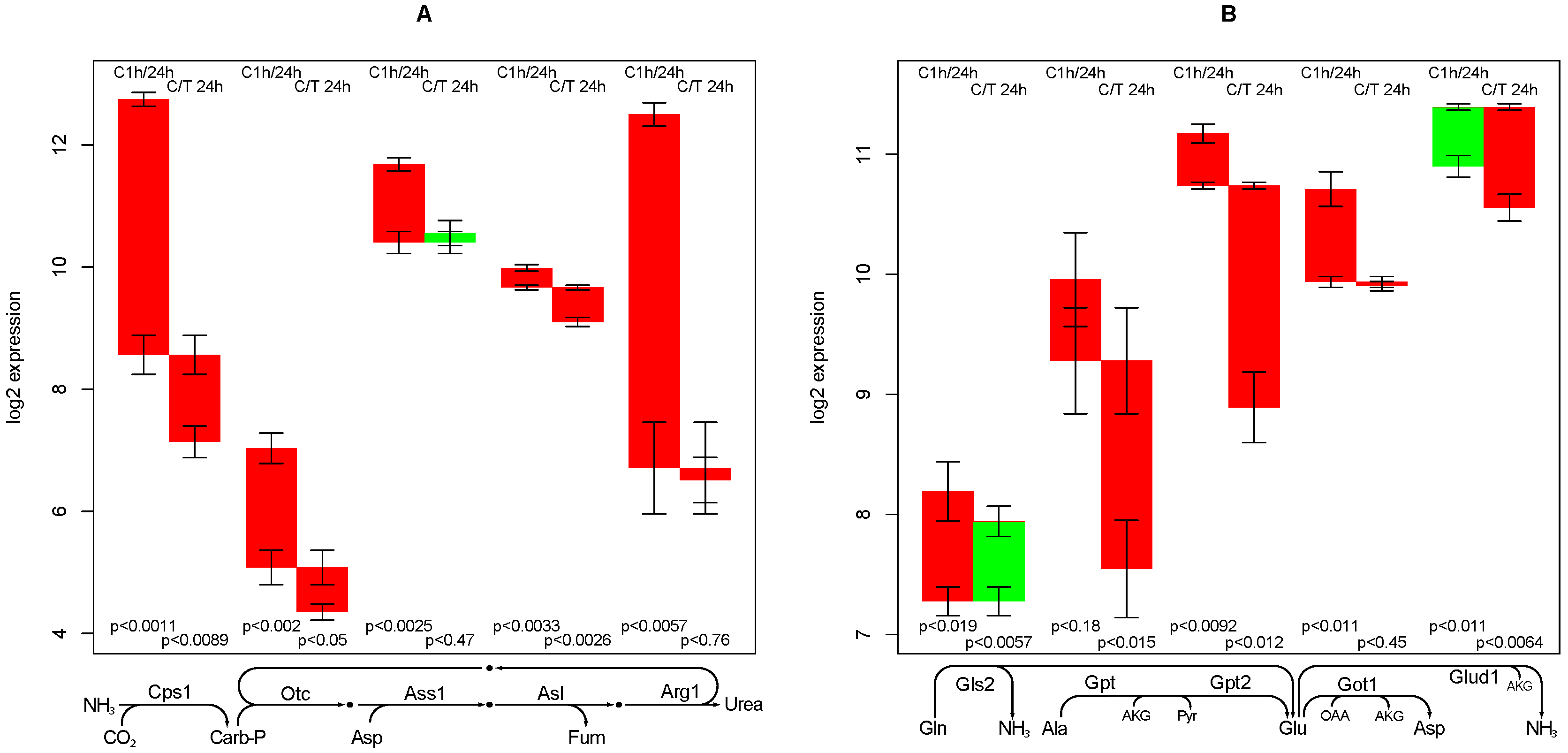
2.7. Urea Synthesis

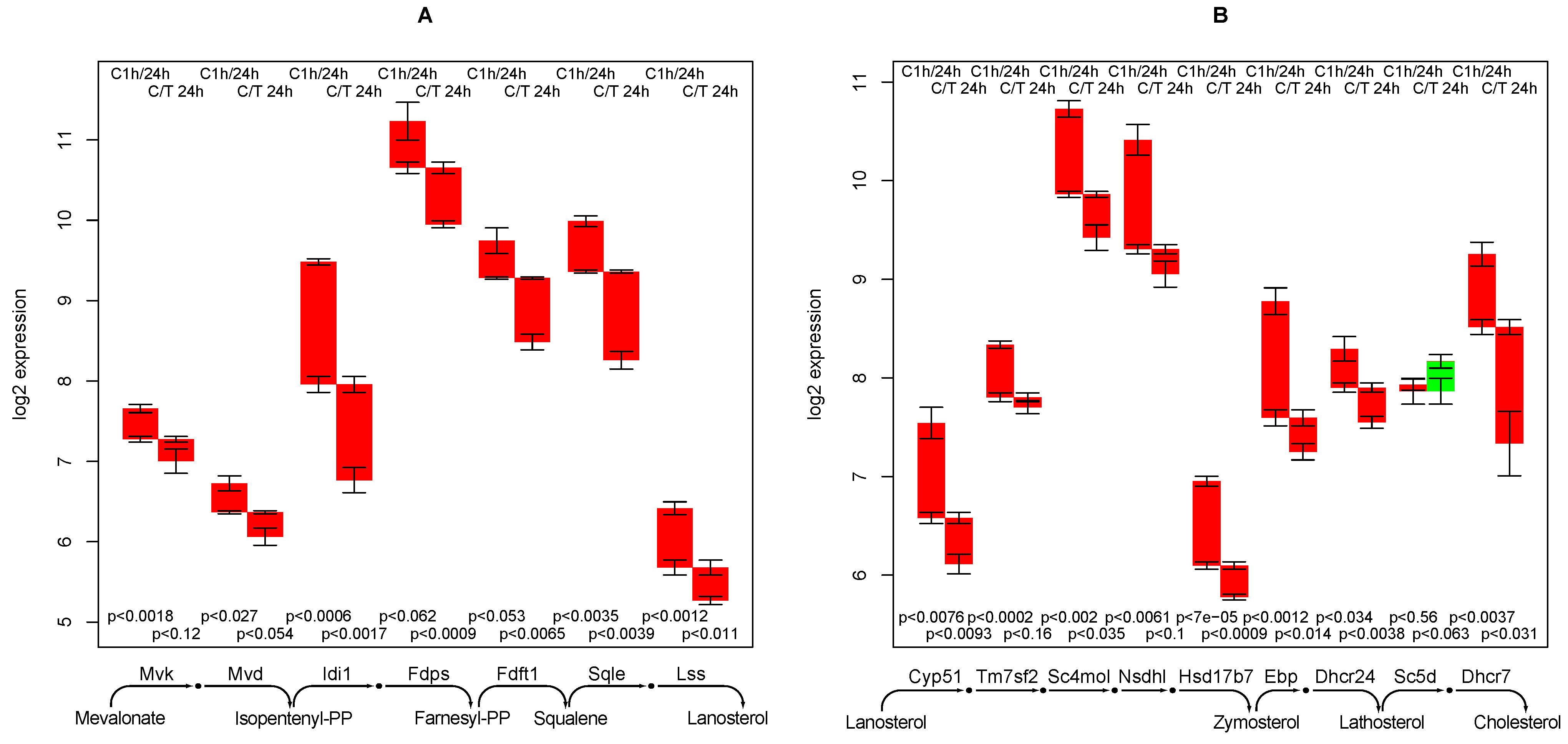
2.8. Cholesterol Synthesis
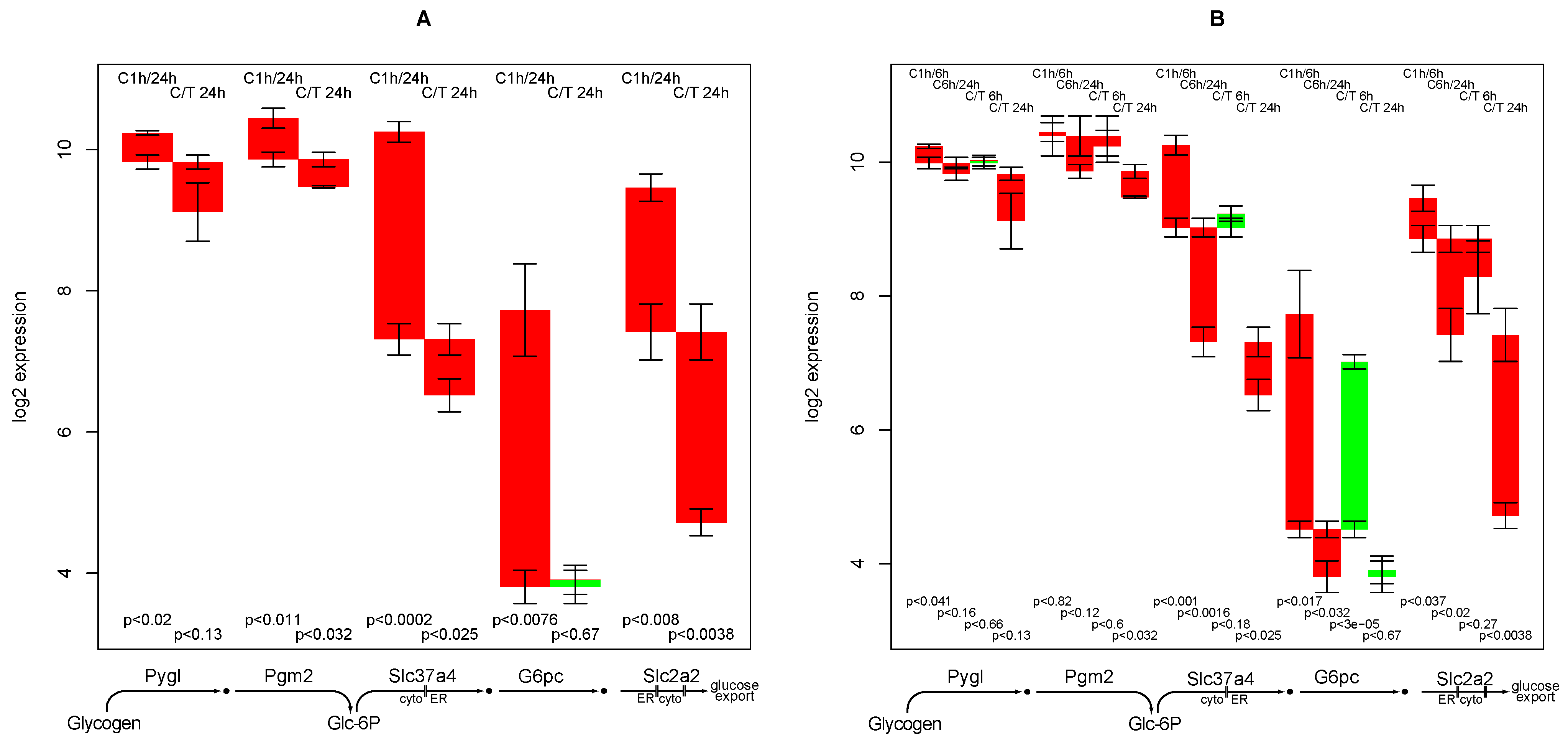
2.9. Glucose Release from Glycogen


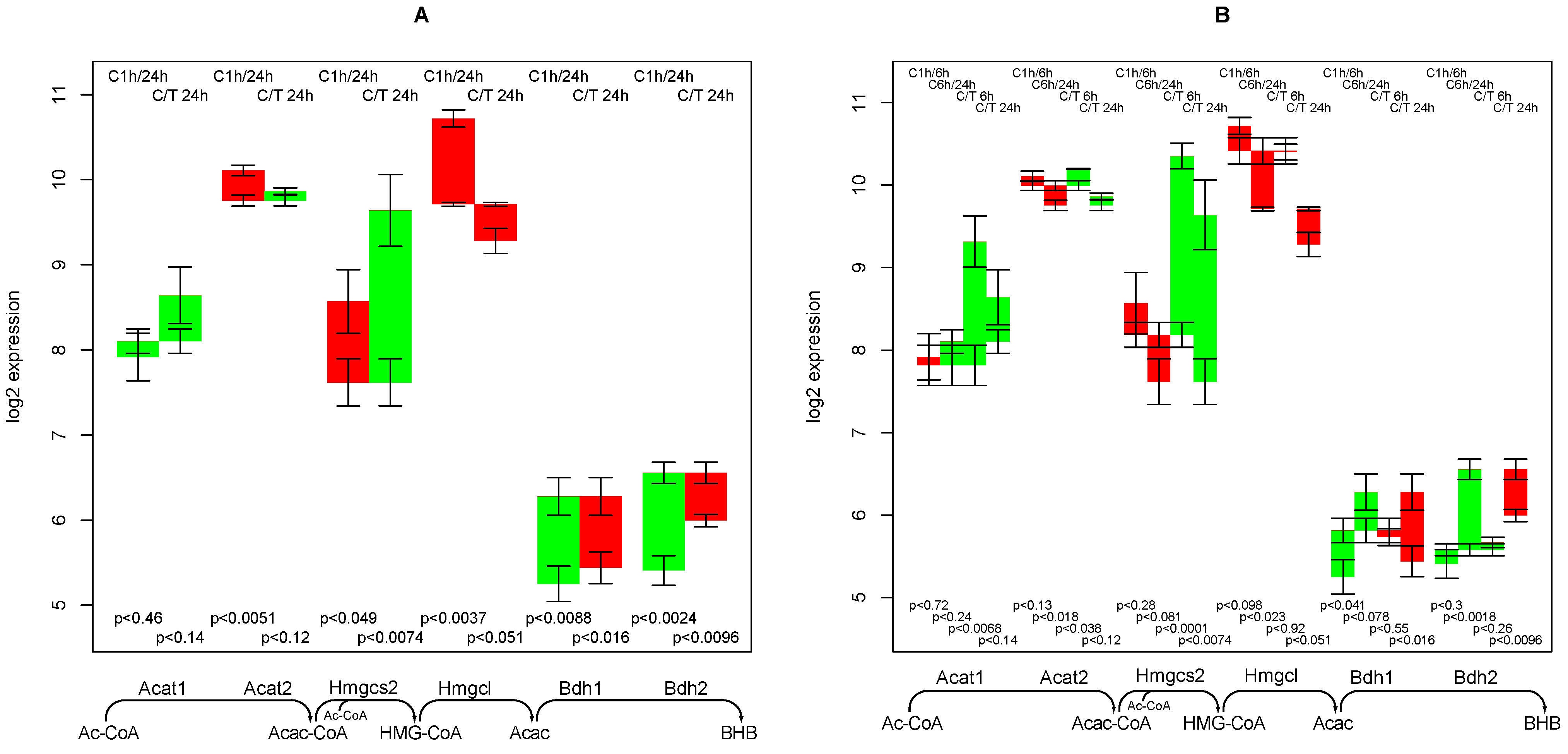
2.10. Supply of β-hydroxybutyrate
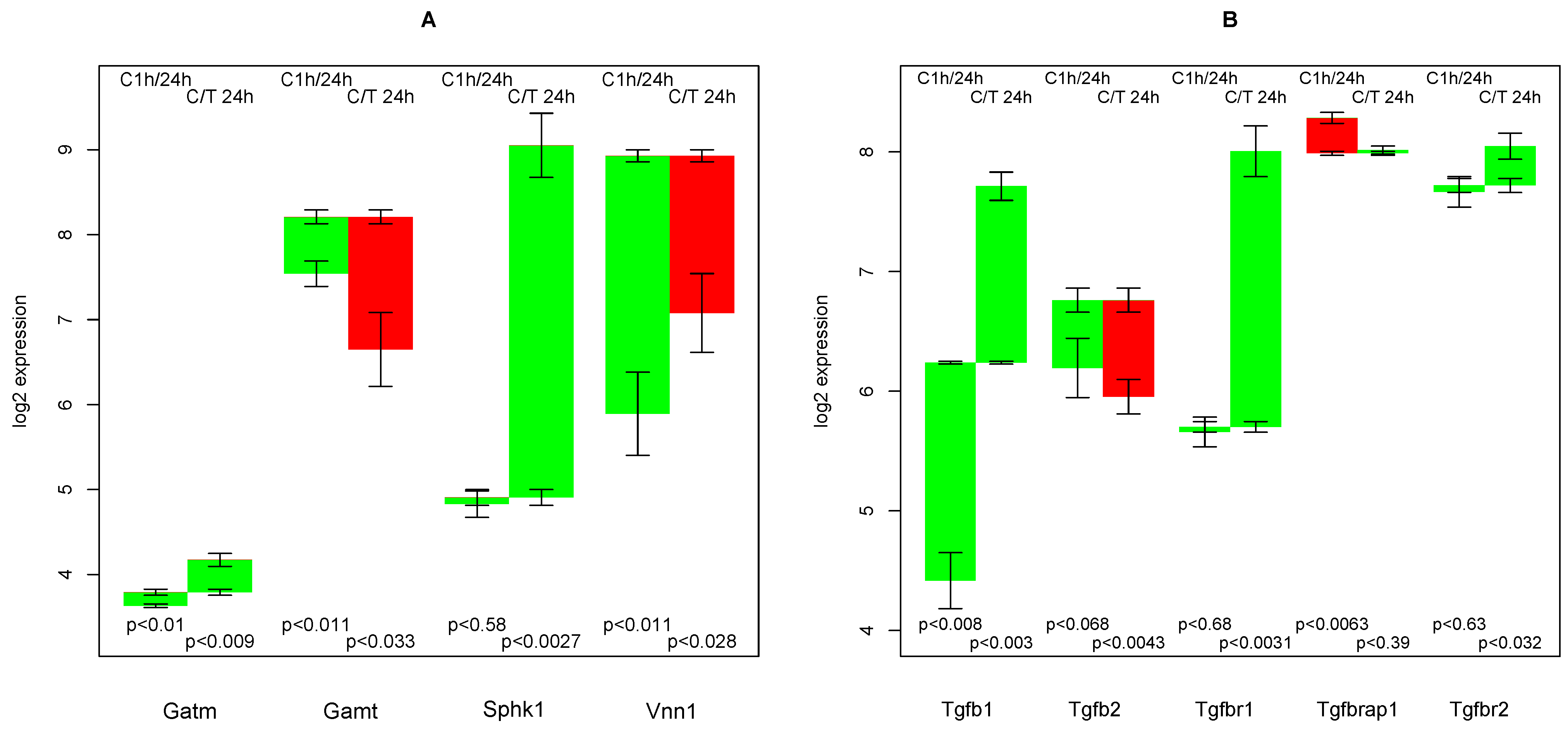
2.11. Creatine Synthesis


2.12. Solitude Genes with a Remarkable Pattern
2.13. Endogenous TGFβ Production
3. Discussion
4. Methods and Materials
4.1. Affymetrix Chip Experiment
4.2. Network and Gene Assignments
4.3. Reference Solutions
4.4. ModeScore Method



4.5. ModeScore Analysis
5. Conclusions
- the high sensitivity of the phenylalanine/tyrosine degradation capacity to TGFβ. Even though there is a strong down-regulation in the control culture, the large additional impact of TGFβ makes a specific effect very likely. The inhibition of TGFβ production or its signaling pathway could be a starting point for treatment of an imminent danger of brain failure in patients with critical liver diseases [20].
- the strong negative impact of TGFβ on ethanol degradation capacity, a fact that has been experimentally confirmed [11]. As it is not found in the control culture, a specific mechanism is highly likely.
- the collagens that are up-regulated most by TGFβ treatment (XXVIIα1, XVα1,Iα1, VIIα1,Vα2). They can subsequently be related to the predominant fiber proteins in liver fibrosis and lead to a therapeutic starting point to estimate how much a particular cirrhotic disease process is related to hepatocyte dysfunction and sensitive to TGFβ,
- down-regulation of glucose export is postponed by TGFβ while for most other functions TGFβ accelerates the down-regulation,
- creatine synthesis, glucuronization capacity, urea synthesis, and cholesterol synthesis are negatively affected by TGFβ,
- an early and short-term up-regulating response to TGFβ regarding the synthesis capacity of ketone bodies
- that TGFβ suppresses the strong culture stress induced up-regulation of Vanin, and
- that TGFβ induces the re-formation of ceramides and sphingomyelin.
Acknowledgments
Conflicts of Interest
References
- Liska, V.; Treska, V.; Mirka, H.; Kobr, J.; Sykora, R.; Skalicky, T.; Sutnar, A.; Vycital, O.; Bruha, J.; Pitule, P.; et al. Inhibition of transforming growth factor beta-1 augments liver regeneration after partial portal vein ligation in a porcine experimental model. Hepatogastroenterology 2012, 59, 235–240. [Google Scholar]
- Dooley, S.; ten Dijke, P. TGFβin progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar]
- Gerjevic, L.N.; Liu, N.; Lu, S.; Harrison-Findik, D.D. Alcohol activates TGF-beta but inhibits BMP receptor-mediated Smad signaling and Smad4 binding to hepcidin promoter in the liver. Int. J. Hepatol. 2012, 2012, 459278. [Google Scholar]
- Klingmüller, U.; Bauer, A.; Bohl, S.; Nickel, P.J.; Breitkopf, K.; Dooley, S.; Zellmer, S.; Kern, C.; Merfort, I.; Sparna, T.; et al. Primary mouse hepatocytes for systems biology approaches: A standardized in vitro system for modelling of signal transduction pathways. Syst. Biol. 2006, 153, 433–447. [Google Scholar] [CrossRef]
- Zeilinger, K.; Auth, S.; Unger, J.; Grebe, A.; Mao, L.; Petrik, M.; Holland, G.; Appel, K.; Nüssler, A.; Neuhaus, P.; et al. Liver cell culture in bioreactors for in vitro drug studies as an alternative to animal testing. ALTEX 2000, 17, 3–10. [Google Scholar]
- Risal, P.; Cho, B.H.; Sylvester, K.G.; Kim, J.C.; Kim, H.T.; Jeong, Y.J. The establishment and characterization of immortal hepatocyte cell lines from a mouse liver injury model. In Vitro Cell. Dev. Biol.-Anim. 2011, 47, 526–534. [Google Scholar] [CrossRef]
- Knobeloch, D.; Ehnert, S.; Schyschka, L.; Büchler, P.; Schoenberg, M.; Kleeff, J.; Thasler, W.E.; Nussler, N.C.; Godoy, P.; Hengstler, J.; et al. Human hepatocytes: Isolation, culture, and quality procedures. Methods Mol. Biol. 2012, 806, 99–120. [Google Scholar] [CrossRef]
- Guthke, R.; Zeilinger, K.; Sickinger, S.; Schmidt-Heck, W.; Buentemeyer, H.; Iding, K.; Lehmann, J.; Pfaff, M.; Pless, G.; Gerlach, J.C. Dynamics of amino acid metabolism of primary human liver cells in 3D bioreactors. Bioprocess. Biosyst. Eng. 2006, 28, 331–340. [Google Scholar] [CrossRef]
- Godoy, P.; Hengstler, J.G.; Ilkavets, I.; Meyer, C.; Bachmann, A.; Müller, A.; Tuschl, G.; Mueller, S.O.; Dooley, S. Extracellular matrix modulates sensitivity of hepatocytes to fibroblastoid dedifferentiation and transforming growth factor beta-induced apoptosis. Hepatology 2009, 49, 2031–2043. [Google Scholar] [CrossRef]
- Dooley, S.; Hamzavi, J.; Ciuclan, L.; Godoy, P.; Ilkavets, I.; Ehnert, S.; Ueberham, E.; Gebhardt, R.; Kanzler, S.; Geier, A.; et al. Hepatocyte-specific Smad7 expression attenuates TGF-beta-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008, 135, 642–659. [Google Scholar]
- Ciuclan, L.; Ehnert, S.; Ilkavets, I.; Weng, H.L.; Gaitantzi, H.; Tsukamoto, H.; Ueberham, E.; Meindl-Beinker, N.M.; Singer, M.V.; Breitkopf, K.; et al. TGF-beta enhances alcohol dependent hepatocyte damage via down-regulation of alcohol dehydrogenase I. J. Hepatol. 2010, 52, 407–416. [Google Scholar]
- Hoppe, A. What mRNA abundances can tell us about metabolism. Metabolites 2012, 2, 614–631. [Google Scholar] [CrossRef]
- Becker, S.A.; Palsson, B.Ø. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef]
- Shlomi, T.; Cabili, M.N.; Herrgård, M.J.; Palsson, B. Network-based prediction of human tissue-specific metabolism. Nat. Biotechnol. 2008, 26, 1003–1010. [Google Scholar] [CrossRef]
- Hoppe, A.; Holzhütter, H.G. ModeScore: A Method to Infer Changed Activity of Metabolic Function from Transcript Profiles. In Proceedings of GCB’12 German Conference on Bioinformatics, Jena, Germany, 20-22 September 2012.
- Hoffmann, S.; Hoppe, A.; Holzhütter, H.G. Composition of metabolic flux distributions by functionally interpretable minimal flux modes (MinModes). Genome Inform. 2006, 17, 195–207. [Google Scholar]
- Gille, C.; Bölling, C.; Hoppe, A.; Bulik, S.; Hoffmann, S.; Hübner, K.; Karlstädt, A.; Ganeshan, R.; König, M.; Rother, K.; et al. HepatoNet1: A comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol. Syst. Biol. 2010, 6, 411. [Google Scholar]
- Welch, B.L. The generalisation of student’s problems when several different population variances are involved. Biometrika 1947, 34, 28–35. [Google Scholar]
- Hoppe, A.; Hoffmann, S.; Gerasch, A.; Gille, C.; Holzhütter, H.G. FASIMU: Flexible software for flux-balance computation series in large metabolic networks. BMC Bioinform. 2011, 12, 28. [Google Scholar] [CrossRef]
- Fulenwider, J.T.; Nordlinger, B.M.; Faraj, B.A.; Ivey, G.L.; Rudman, D. Deranged tyrosine metabolism in cirrhosis. Yale J. Biol. Med. 1978, 51, 625–633. [Google Scholar]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845–17850. [Google Scholar] [CrossRef]
- Boot-Handford, R.P.; Tuckwell, D.S.; Plumb, D.A.; Rock, C.F.; Poulsom, R. A novel and highly conserved collagen (pro(alpha)1(XXVII)) with a unique expression pattern and unusual molecular characteristics establishes a new clade within the vertebrate fibrillar collagen family. J. Biol. Chem. 2003, 278, 31067–31077. [Google Scholar]
- Jenkins, E.; Moss, J.B.; Pace, J.M.; Bridgewater, L.C. The new collagen gene COL27A1 contains SOX9-responsive enhancer elements. Matrix Biol. 2005, 24, 177–184. [Google Scholar] [CrossRef]
- Liétard, J.; Théret, N.; Rehn, M.; Musso, O.; Dargère, D.; Pihlajaniemi, T.; Clément, B. The promoter of the long variant of collagen XVIII, the precursor of endostatin, contains liver-specific regulatory elements. Hepatology 2000, 32, 1377–1385. [Google Scholar] [CrossRef]
- Oh, S.P.; Kamagata, Y.; Muragaki, Y.; Timmons, S.; Ooshima, A.; Olsen, B.R. Isolation and sequencing of cDNAs for proteins with multiple domains of Gly-Xaa-Yaa repeats identify a distinct family of collagenous proteins. Proc. Natl. Acad. Sci. USA 1994, 91, 4229–4233. [Google Scholar] [CrossRef]
- Clément, B.; Musso, O.; Liétard, J.; Théret, N. Homeostatic control of angiogenesis: A newly identified function of the liver? Hepatology 1999, 29, 621–623. [Google Scholar] [CrossRef]
- Dogrul, A.B.; Colakoglu, T.; Kosemehmetoglu, K.; Birben, E.; Yaman, E.; Gedikoglu, G.; Abbasoglu, O. Antiangiogenic response after 70% hepatectomy and its relationship with hepatic regeneration and angiogenesis in rats. Surgery 2010, 147, 288–294. [Google Scholar] [CrossRef]
- Zhuge, J.; Cederbaum, A.I. Increased toxicity by transforming growth factor-beta 1 in liver cells overexpressing CYP2E1. Free Radic Biol. Med. 2006, 41, 1100–1112. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, H.; Kong, X.; Wang, K.; Mao, X.; Yan, X.; Wang, Y.; Liu, S.; Zhang, X.; Li, J.; et al. Proteomics analysis reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E287–E295. [Google Scholar] [CrossRef]
- Shibakusa, T.; Mizunoya, W.; Okabe, Y.; Matsumura, S.; Iwaki, Y.; Okuno, A.; Shibata, K.; Inoue, K.; Fushiki, T. Transforming growth factor-beta in the brain is activated by exercise and increases mobilization of fat-related energy substrates in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1851–R1861. [Google Scholar] [CrossRef]
- Ishikawa, T.; Mizunoya, W.; Shibakusa, T.; Inoue, K.; Fushiki, T. Transforming growth factor-beta in the brain regulates fat metabolism during endurance exercise. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1151–E1159. [Google Scholar] [CrossRef]
- Deminice, R.; da Silva, R.P.; Lamarre, S.G.; Brown, C.; Furey, G.N.; McCarter, S.A.; Jordao, A.A.; Kelly, K.B.; King-Jones, K.; Jacobs, R.L.; et al. Creatine supplementation prevents the accumulation of fat in the livers of rats fed a high-fat diet. J. Nutr. 2011, 141, 1799–1804. [Google Scholar] [CrossRef]
- Kaskow, B.J.; Proffit, J.M.; Blangero, J.; Moses, E.K.; Abraham, L.J. Diverse biological activities of the vascular non-inflammatory molecules-the Vanin pantetheinases. Biochem. Biophys. Res. Commun. 2012, 417, 653–658. [Google Scholar] [CrossRef]
- Zimran, A.; Forman, L.; Suzuki, T.; Dale, G.L.; Beutler, E. In vivo aging of red cell enzymes: Study of biotinylated red blood cells in rabbits. Am. J. Hematol. 1990, 33, 249–254. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: tool for the unification of biology.The Gene Ontology Consortium. Nat. Genet 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG database. Novartis Found. Symp. 2002, 247, 91–101. [Google Scholar]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar]
- De Figueiredo, L.F.; Podhorski, A.; Rubio, A.; Kaleta, C.; Beasley, J.E.; Schuster, S.; Planes, F.J. Computing the shortest elementary flux modes in genome-scale metabolic networks. Bioinformatics 2009, 25, 3158–3165. [Google Scholar] [CrossRef]
- Holzhütter, H.G. The principle of flux minimization and its application to estimate stationary fluxes in metabolic networks. Eur. J. Biochem. 2004, 271, 2905–2922. [Google Scholar] [CrossRef]
- Jerby, L.; Shlomi, T.; Ruppin, E. Computational reconstruction of tissue-specific metabolic models: Application to human liver metabolism. Mol. Syst. Biol. 2010, 6, 401. [Google Scholar]
- Duarte, N.C.; Becker, S.A.; Jamshidi, N.; Thiele, I.; Mo, M.L.; Vo, T.D.; Srivas, R.; Palsson, B.Ø. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc. Natl. Acad. Sci. USA 2007, 104, 1777–1782. [Google Scholar]
- Shlomi, T. Metabolic network-based interpretation of gene expression data elucidates human cellular metabolism. Biotechnol. Genet Eng. Rev. 2010, 26, 281–296. [Google Scholar] [CrossRef]
- Kharchenko, P.; Vitkup, D.; Church, G.M. Filling gaps in a metabolic network using expression information. Bioinformatics 2004, 20 (Suppl 1), i178–i185. [Google Scholar] [CrossRef]
- Manichaikul, A.; Ghamsari, L.; Hom, E.F.Y.; Lin, C.; Murray, R.R.; Chang, R.L.; Balaji, S.; Hao, T.; Shen, Y.; Chavali, A.K.; et al. Metabolic network analysis integrated with transcript verification for sequenced genomes. Nat. Methods 2009, 6, 589–592. [Google Scholar]
- Hoppe, A.; Hoffmann, S.; Holzhütter, H.G. Including metabolite concentrations into flux balance analysis: Thermodynamic realizability as a constraint on flux distributions in metabolic networks. BMC Syst. Biol. 2007, 1, 23. [Google Scholar]
- ArrayExpress — a database of functional genomics experiments. Available online: http://www. ebi.ac.uk/arrayexpress/ (Accessed on 12 November 2012).
- BioModels Database — a database of annotated published models. Available online: http:// BioModels.net/ (Accessed on 12 November 2012).
Supplementary Files
Definition of the simulations and solution summary (PDF, 792 KB)
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hoppe, A.; Ilkavets, I.; Dooley, S.; Holzhütter, H.-G. Metabolic Consequences of TGFb Stimulation in CulturedPrimary Mouse Hepatocytes Screened from Transcript Data with ModeScore . Metabolites 2012, 2, 983-1003. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo2040983
Hoppe A, Ilkavets I, Dooley S, Holzhütter H-G. Metabolic Consequences of TGFb Stimulation in CulturedPrimary Mouse Hepatocytes Screened from Transcript Data with ModeScore . Metabolites. 2012; 2(4):983-1003. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo2040983
Chicago/Turabian StyleHoppe, Andreas, Iryna Ilkavets, Steven Dooley, and Hermann-Georg Holzhütter. 2012. "Metabolic Consequences of TGFb Stimulation in CulturedPrimary Mouse Hepatocytes Screened from Transcript Data with ModeScore " Metabolites 2, no. 4: 983-1003. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo2040983