Serum Steroid Profiling by Liquid Chromatography–Tandem Mass Spectrometry for the Rapid Confirmation and Early Treatment of Congenital Adrenal Hyperplasia: A Neonatal Case Report

 ,
,
Abstract
:
{kind=link}
{kind=link}
1. Introduction
2. Case Report
2.1. Clinical Presentation
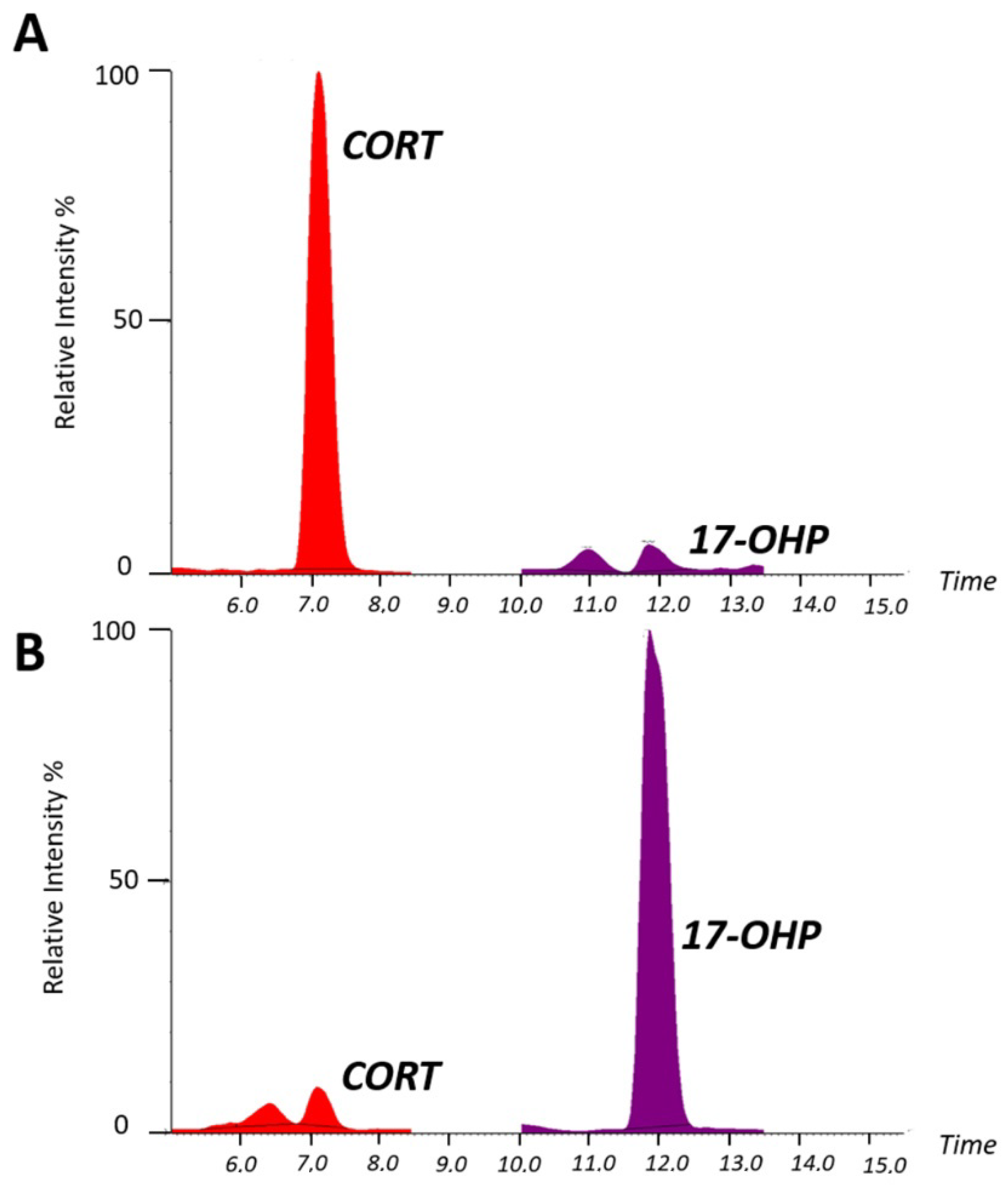
2.2. Serum Steroid Profiling by LC–MS/MS for Diagnostic Confirmation of Congenital Adrenal Hyperplasia
Analytical Performance of LC–MS/MS Analysis for Serum Steroid Determination
2.3. Molecular Testing Confirmation of Congenital Adrenal Hyperplasia
3. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rauh, M. Steroid measurement with LC–MS/MS in pediatric endocrinology. Mol. Cell. Endocrinol. 2009, 301, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Kushnir, M.M.; Rockwood, A.L.; Roberts, W.L.; Pattison, E.G.; Owen, W.E.; Bunker, A.M.; Meikle, A.W. Development and Performance Evaluation of a Tandem Mass Spectrometry Assay for 4 Adrenal Steroids. Clin. Chem. 2006, 52, 1559–1567. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Calton, L.; Hammond, G.; Brown, H.A.; Wallace, A.M.; Sacchetta, P.; Morris, M. Serum steroid profiling for Congenital Adrenal Hyperplasia using liquid chromatography–tandem mass spectrometry. Clin. Chim. Acta 2010, 411, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Janzen, N.; Peter, M.; Steuerwald, U.; Terhardt, M.; Holtkamp, U.; Sander, S. Newborn Screening for Congenital Adrenal Hyperplasia: Additional Steroid Profile using Liquid Chromatography-Tandem Mass Spectrometry. J. Clin. Endocrinol. Metab. 2007, 92, 2581–2589. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, K.; Engels, M.; Bennecke, E.; Bouvattier, C.; Falhammar, H.; Kreukels, B.P.C.; Nordenstrom, A.; Reisch, N.; Gehrmann, N.; Stikkelbroeck, N.M.M.L.; et al. Sexuality in Males With Congenital Adrenal Hyperplasia Resulting From 21-Hydroxylase Deficiency. J. Endocr. Soc. 2019, 3, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.G.; Bachega, T.A.; Mendonca, B.B. Classic congenital adrenal hyperplasia and its impact on reproduction. Fertil. Steril. 2019, 111, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Wudy, S.A.; Schuler, G.; Sanchez-Guijo, A.; Hartmann, M.F. The art of measuring steroids: Principles and practice of current hormonal steroid analysis. J. Steroid Biochem. Mol. Biol. 2018, 179, 88–103. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Calton, L.; Brown, H.A.; Gillingwater, S.; Wallace, A.M.; Petrucci, F.; Ciavardelli, D.; Urbani, A.; Sacchetta, P.; Morris, M. Confirmation of congenital adrenal hyperplasia by adrenal steroid profiling of filter paper dried blood samples using ultra-performance liquid chromatography-tandem mass spectrometry. Clin. Chem. Lab. Med. 2011, 49, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Vogeser, M.; Seger, C. A decade of HPLC–MS/MS in the routine clinical laboratory—Goals for further developments. Clin. Biochem. 2008, 41, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Rossi, C.; Cicalini, I.; Zucchelli, M.; Di Ioia, M.; Onofrj, M.; Federici, L.; Del Boccio, P.; Pieragostino, D. Metabolomic Signature in Sera of Multiple Sclerosis Patients during Pregnancy. Int. J. Mol. Sci. 2018, 19, 3589. [Google Scholar] [CrossRef]
- Pieragostino, D.; Agnifili, L.; Cicalini, I.; Calienno, R.; Zucchelli, M.; Mastropasqua, L.; Sacchetta, P.; Del Boccio, P.; Rossi, C. Tear Film Steroid Profiling in Dry Eye Disease by Liquid Chromatography Tandem Mass Spectrometry. Int. J. Mol. Sci. 2017, 18, 1349. [Google Scholar] [CrossRef]
- Németh, S.; Riedl, S.; Kriegshäuser, G.; Baumgartner-Parzer, S.; Concolino, P.; Neocleous, V.; Phylactou, L.A.; Borucka-Mankiewicz, M.; Onay, H.; Tukun, A.; et al. Reverse-hybridization assay for rapid detection of common CYP21A2 mutations in dried blood spots from newborns with elevated 17-OH progesterone. Clin. Chim. Acta 2012, 414, 211–214. [Google Scholar] [CrossRef]
- Lacey, J.M.; Minutti, C.Z.; Magera, M.J.; Tauscher, A.L.; Casetta, B.; McCann, M.; Lymp, J.; Hahn, S.H.; Rinaldo, P.; Matern, D. Improved Specificity of Newborn Screening for Congenital Adrenal Hyperplasia by Second-Tier Steroid Profiling Using Tandem Mass Spectrometry. Clin. Chem. 2004, 50, 621–625. [Google Scholar] [CrossRef]
- Speiser, P.W.; White, P.C. Congenital adrenal hyperplasia. N. Engl. J. Med. 2003, 349, 776–788. [Google Scholar] [CrossRef]
- Minutti, C.Z.; Lacey, J.M.; Magera, M.J.; Hahn, S.H.; McCann, M.; Schulze, A.; Cheillan, D.; Dorche, C.; Chace, D.H.; Lymp, J.F.; et al. Steroid Profiling by Tandem Mass Spectrometry Improves the Positive Predictive Value of Newborn Screening for Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2004, 89, 3687–3693. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.B.; Hoffman, G.L.; Fitzpatrick, P.; Laessig, R.; Maby, S.; Slyper, A. Improved precision of newborn screening for congenital adrenal hyperplasia using weight-adjusted criteria for 17-hydroxyprogesterone levels. J. Pediatr. 1997, 130, 128–133. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicalini, I.; Tumini, S.; Guidone, P.I.; Pieragostino, D.; Zucchelli, M.; Franchi, S.; Lisi, G.; Lelli Chiesa, P.; Stuppia, L.; De Laurenzi, V.; et al. Serum Steroid Profiling by Liquid Chromatography–Tandem Mass Spectrometry for the Rapid Confirmation and Early Treatment of Congenital Adrenal Hyperplasia: A Neonatal Case Report. Metabolites 2019, 9, 284. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo9120284
Cicalini I, Tumini S, Guidone PI, Pieragostino D, Zucchelli M, Franchi S, Lisi G, Lelli Chiesa P, Stuppia L, De Laurenzi V, et al. Serum Steroid Profiling by Liquid Chromatography–Tandem Mass Spectrometry for the Rapid Confirmation and Early Treatment of Congenital Adrenal Hyperplasia: A Neonatal Case Report. Metabolites. 2019; 9(12):284. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo9120284
Chicago/Turabian StyleCicalini, Ilaria, Stefano Tumini, Paola Irma Guidone, Damiana Pieragostino, Mirco Zucchelli, Sara Franchi, Gabriele Lisi, Pierluigi Lelli Chiesa, Liborio Stuppia, Vincenzo De Laurenzi, and et al. 2019. "Serum Steroid Profiling by Liquid Chromatography–Tandem Mass Spectrometry for the Rapid Confirmation and Early Treatment of Congenital Adrenal Hyperplasia: A Neonatal Case Report" Metabolites 9, no. 12: 284. https://0-doi-org.brum.beds.ac.uk/10.3390/metabo9120284