Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches
by
 , , and
, , and
Ramanuj Mitra
1,2,*, 
V. Srinivasa Prasannaa
3,
Bijaya K. Sahoo
1,
Nicholas R. Hutzler
4,
Minori Abe
5 and
Bhanu Pratap Das
6 1
Atomic, Molecular and Optical Physics Division, Physical Research Laboratory, Navrangpura, Ahmedabad 380009, India
2
Indian Institute of Technology Gandhinagar, Palaj, Gandhinagar 382355, India
3
Centre for Quantum Engineering, Research and Education (CQuERE), TCG CREST, Salt Lake, Kolkata 700091, India
4
Division of Physics, Mathematics, and Astronomy, California Institute of Technology, Pasadena, CA 91125, USA
5
Department of Chemistry, Tokyo Metropolitan University, 1-1, Minami-Osawa, Hachioji-city, Tokyo 192-0397, Japan
6
Department of Physics, Tokyo Institute of Technology, 2-12-1-H86 Ookayama, Meguro-ku, Tokyo 152-8550, Japan
*
Author to whom correspondence should be addressed.
Atoms 2021, 9(1), 7; https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9010007
Submission received: 27 November 2020
/
Revised: 14 January 2021
/
Accepted: 14 January 2021
/
Published: 19 January 2021
(This article belongs to the Section Quantum Chemistry, Computational Chemistry and Molecular Physics)
Abstract
:In search of suitable molecular candidates for probing the electric dipole moment (EDM) of the electron (), a property that arises due to parity and time-reversal violating (P,T-odd) interactions, we consider the triatomic mercury hydroxide (HgOH) molecule. The impetus for this proposal is based on previous works on two systems: the recently proposed ytterbium hydroxide (YbOH) experiment that demonstrates the advantages of polyatomics for such EDM searches, and the finding that mercury halides provide the highest enhancement due to compared to other diatomic molecules. We identify the ground state of HgOH as being in a bent geometry, and show that its intrinsic EDM sensitivity is comparable to the corresponding value for YbOH. Along with the theoretical results, we discuss plausible experimental schemes for an EDM measurement in HgOH. Furthermore, we provide pilot calculations of the EDM sensitivity for for HgCH and HgCF, that are natural extensions of HgOH.
1. Introduction
Probing properties of fundamental particles using atoms and molecules has become very popular in the past few decades. The electric dipole moment (EDM) of the electron () is one such property. The EDM of an electron arises due to simultaneous violations of two discrete symmetries, namely, parity (P) and time-reversal (T) (P,T-odd) [1,2]; with the latter implying that the EDM is CP-violating (C stands for charge conjugation symmetry), due to the CPT theorem [3]. This intrinsic property of the electron is extremely tiny, and its existence has not yet been confirmed experimentally; only upper bounds exist, which in turn rely upon a combination of atomic or molecular calculations with experimental results. A stringent bound on has significant implications in constraining physics that lies beyond the Standard Model (SM) of elementary particles (∼TeV-PeV energy scales) [4,5] as well as probing the underlying physics describing the matter-antimatter asymmetry (baryon asymmetry) in the universe [6,7]. Since direct measurement of is extremely challenging, atoms or molecules are used as a means of probing in non-accelerator table-top experiments. Within a molecule, the magnitude of the intrinsic electric field that an electron with an EDM experiences due to the other electrons and nuclei can be viewed as an effective electric field (). This property can only be calculated using relativistic many-body theory [8,9]. The measured energy shift in a molecule due to electron EDM is given by the negative of times . The EDM of the electron is extracted by combining the measured shift in energy and the calculated value of the effective electric field. A large value of the molecule-specific effective electric field leads to significantly improved experimental sensitivity. can be very large in certain heavy polar molecules in comparison with atoms [10,11,12,13], arising from the fact that molecules can be polarized in the lab frame. Hence, polar molecules are powerful platforms for electron EDM experiments. Theoretical studies play crucial roles in identifying suitable molecules, with enhanced , for proposing future electron EDM experiments.
For the last one and half decades, there has been tremendous growth on experimental and theoretical fronts in the search for using diatomic polar molecules. This is evident from the sheer number of ongoing experiments, such as ThO [14], HfF[15], YbF [16], and BaF [17,18]. Among these, the most stringent limit to is set by an experiment that uses ThO molecules, with e-cm. On the theoretical side, many interesting proposals have been put forth, identifying new polar molecules and molecular ions, including PtH, and HfH [19], YbRb, YbCs, YbSr, and YbBa [20], WC [21], RaF [22], TaN [23], HgX (X=F, Cl, Br, and I) [11], HgA (A=Li, Na, and K) [24], and RaH [25]. The diatomic HgX polar molecules were found to possess significantly larger effective electric fields than other proposed systems. Polyatomic molecules offer the advantages of a diatomic system, with the addition of full polarization in small fields and the ability to obtain good control over systematic effects using internal co-magnetometer states [26]. In addition, molecules such as YbOH can combine the ability to be laser cooled [26,27]. A large number of these highly polarizable molecules, prepared in the low-lying (010) vibrational state, and trapped in an optical lattice, promises a sensitivity that could exceed that of ThO by four orders of magnitude; albeit of YbOH is smaller almost by a factor of three as compared to ThO [12,28,29,30].
The two isoelectronic molecules, YbF and YbOH, both happen to have comparable values of [10,30]. In this work, we investigate the suitability of HgOH for an electron EDM experiment. HgOH is an isoelectronic triatomic counterpart of HgF. As HgF has a considerably large [11], it is expected that in HgOH can also be reasonably large for the consideration of EDM measurement. However, spectroscopic data on HgOH is very limited and the molecule has received little attention in previous studies, unlike in the case of YbOH. In fact, mention of HgOH in the literature occurs mainly in the context of investigating Hg removal mechanisms in the atmosphere, for example, Ref. [31]. It is in this backdrop that the limited information on the molecule’s geometry and properties exist in literature [32,33]. However, HgOH has not yet been undertaken in laboratories for carrying out any high-precision measurements.
In this paper, we appraise the potential of HgOH for an electron EDM experiment, based on the aforementioned considerations. In view of the limited data available, we optimize the ground state geometry of the molecule using density functional theory (DFT), and find its bond lengths and bond angle. We then obtain and the permanent electric dipole moment (PDM) of HgOH. Then, we proceed to gauge the importance of the molecule for future electron EDM measurements in plausible experimental scenarios. This involves investigating the laser-cooling possibilities in HgOH, which in turn requires calculating the Franck–Condon factors (FCF) between pairs of vibrational states across two electronic states. For this purpose, we have also optimized the geometries of higher excited states in this work. The ground vibrational state of the ground electronic state is chosen as the EDM measurement state.
2. Theory
The shift in energy level of a molecular system due to the P,T-odd electron EDM interaction can be expressed as
and the corresponding Hamiltonian is given by [34,35,36]
where c is the speed of light, is a Dirac matrix, is the product of Dirac matrices, is the momentum operator corresponding to the electron and is the number of electrons in the system. It follows from the above expression that the effective electric field is given by
Another relevant molecular property that is crucial in the sensitivity of EDM experiments is the molecular permanent electric dipole moment (PDM) and is given by
where (in atomic units (a.u.)) for the position vector of the electron from origin, is the site of the nucleus, and is the atomic number of the nucleus. Note that the left hand side is the absolute value of the expression given on the right hand side, that is, . For sake of brevity, we choose the oxygen atom as our origin and we work with the Born–Oppenheimer approximation. Therefore, in the expression for the PDM, are simply the chosen values of the bond lengths in the molecule.
3. Ground State Geometry Optimization
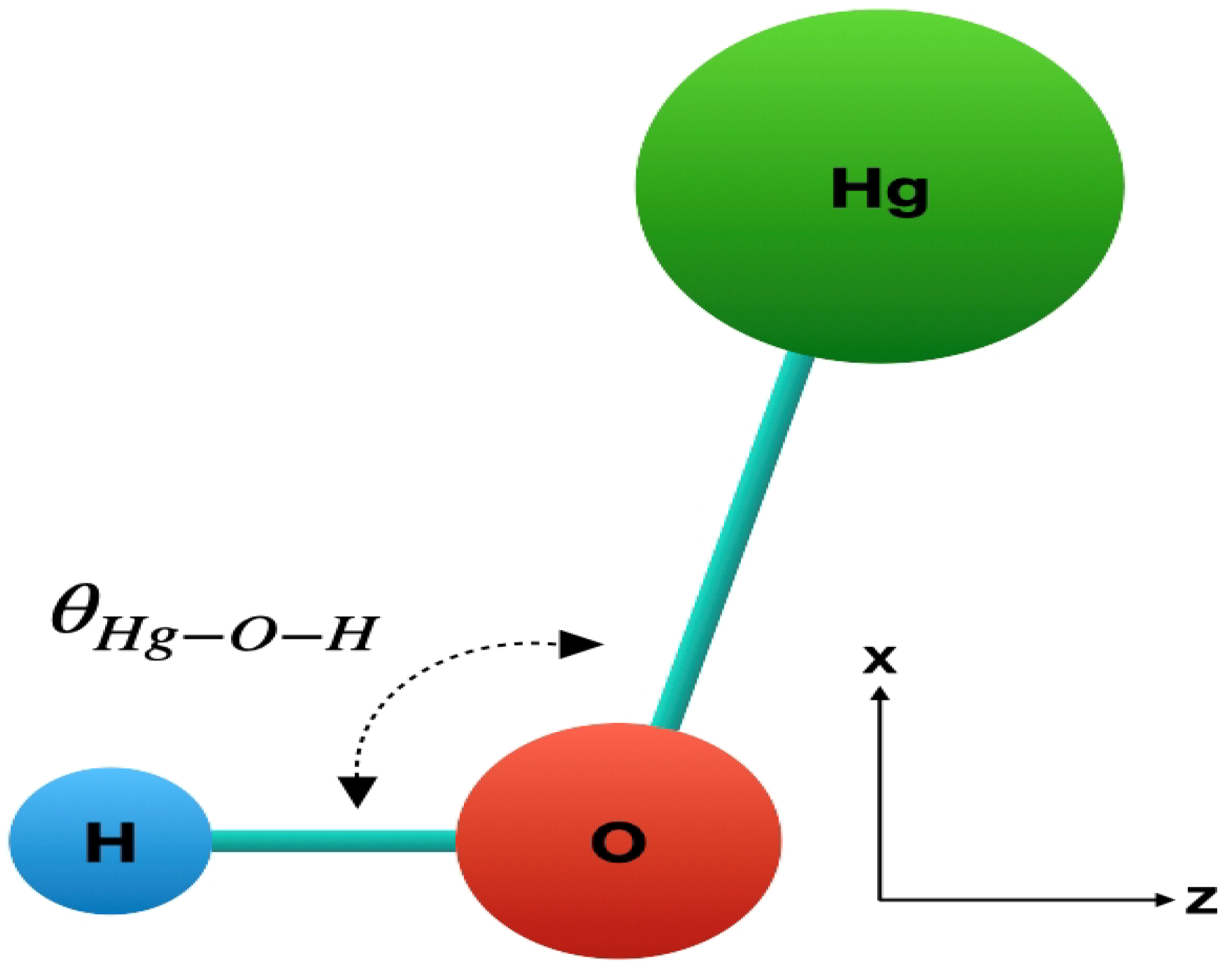
In order to obtain the aforementioned properties of HgOH, it is necessary to evaluate its many-body wave function. In the relevant equations to be solved, the ground state equilibrium geometry of the molecule is a crucial input. It includes the equilibrium bond lengths, and , and the bond angle, which is the angle formed between Hg–O and O–H, as shown in Figure 1. As stated earlier, we use the DFT approach in order to optimize the geometry of the molecule. We choose the B97xD functional, which is known to perform well in obtaining equilibrium geometries [37], and opt for the LANL2DZ [38,39,40] basis sets for Hg, while we choose cc-pVTZ [41] functions for O and H. We perform these calculations using the Gaussian 16 software [42]. We take utmost care to avoid saddle points, and among those optimized geometries without negative frequencies, we select the equilibrium bond lengths and bond angle from the configuration with the lowest energy.
In order to assess the laser coolability of HgOH, we also optimize three excited electronic states. We use time-dependent DFT for the excited state optimizations. We then calculate the Franck-Condon factors (FCFs) using the parallel approximation between the ground state and each of the three excited electronic states. The FCF for a transition from a vibrational state to another vibrational state is given by
where the integral is taken over the nuclear coordinates (denoted by ). We use the ezSpectrum 3.0 software [43] to calculate the FCF matrix elements for the transitions between the ground state and the excited states. A highly diagonal FCF matrix is a good indicator of the laser coolability of a molecule.
4. Method of Calculation
Once the equilibrium geometry is obtained, the next step is to calculate the wave function. In this work, we use relativistic coupled-cluster (RCC) theory for this purpose. In the RCC theory, the wave function of the ground state of a molecule can be expressed as
where is the Dirac–Hartree–Fock (DHF) wave function, and is called the cluster operator, which is responsible for particle-hole excitations, with subscripts denoting the levels of excitation. Each of the s are made of fermionic creation and annihilation operators representing an excitation and t the corresponding amplitude. In our calculation, we have restricted the levels of excitation to singles and doubles (RCCSD method) by defining . In the second quantization notation, they are given by
where the occupied orbitals are denoted by i, j, etc., the unoccupied (virtual) orbitals are represented by a, b, etc., is the cluster amplitude corresponding to a single excitation and is the amplitude for a double excitation. Note that by , for example, we mean , the fermionic creation operator for b. We determine these t amplitudes by solving the RCC amplitude equations,
where the Dirac–Coulomb Hamiltonian is chosen for the molecular Hamiltonian H in this work, are excited determinantal wave function with respect to and the subscript l implies that each of the terms in the expression is linked. These RCCSD amplitudes are used for evaluating the values of and PDM (). We also consider HgOH in a hypothetical linear geometry for the purposes of extrapolating the correlation effects in its bent counterpart from the linear one. Moreover, a look at the relevant properties in a linear geometry allows one to analyze the differences with a bent geometry at the DHF level of theory. For calculating the hypothetical linear geometry potential energy curve (PEC), important contributions from the triple excitations along with the RCCSD method were also included through the perturbative approach (RCCSD(T) method). All the electrons are allowed to undergo excitations by imposing a cut-off of 1000 a.u. in energy levels for the virtuals. The DHF calculations and the atomic orbital to molecular orbital integral transformations were carried out using the UTChem program [44,45], while the RCCSD calculations were run on the Dirac08 software [46]. We used the expectation value approach to calculate molecular properties. In this approach, the expectation value of any operator, O, can be evaluated as [47]
Firstly, we calculated these properties in RCC theory, keeping only the terms that are linear in T and (both of them are treated independently) of the above expression and the results from this approximation is denoted as LECC. It should be noted that only the linear terms from Equation (10) are retained in the LECC approximation, but the RCC amplitudes are still obtained with all the non-linear terms from Equation (9).
5. Results and Discussion
For performing an electron EDM experiment on HgOH, it is imperative to show that it possesses a bound ground state. Since HgOH is a triatomic, its ground state geometry need not be linear. This required us to perform geometry optimization to find out its equilibrium bond lengths and the bond angle. We followed the procedure outlined in the previous section, and found that the molecule is indeed bent in its ground electronic state ( state), unlike in the case of YbOH [30]. Although one may expect a metal-hydroxide molecule to be linear on grounds of the ionic nature of its bonding, a bent structure for HgOH indicates that the bond may possess a hint of covalent nature too, similar to the case of ZnOH [48,49]. We determined the optimized to be and to be , with the HgOH bond angle () being . These results are presented in Table 1, along with the optimized values that we obtained for some of the excited states as well. The table also shows the previously calculated optimized geometries of HgOH [32,33,50,51] in its ground state. In Ref. [32], in the context of Hg removal mechanisms in the atmosphere, the authors had employed ab initio DFT calculations with the Becke, three-parameter, Lee–Yang–Parr (B3LYP) functional and the CEP-121G basis in an ECP-based framework to obtain the optimized geometry of HgOH. The results in Ref. [51] describes bonding in Hg molecules using the normalized elimination of small component coupled-cluster theory in the singles, doubles and partial triples approximation (NESC-CCSD(T)), and en route, estimates the geometry of HgOH, among other Hg systems, using NESC-B3LYP approach. In Ref. [33], the optimized geometry of the HgOH ground electronic state was estimated in the B3LYP-ECP60MWB framework, whereas in Ref. [50], CCSD(T) computations using the aug-cc-pVQZ basis functions were performed. Our result, obtained using the B97xD functional and with the LAN2LDZ basis for Hg and cc-pVTZ basis sets for O and H, agrees well with the previous works.
We also determined the potential energy curve (PEC) for the hypothetical linear ground state geometry in the RCCSD(T) method using the two-component X2C Hamiltonian [52], by varying and keeping fixed at , using the Dirac16 program [53]. Further details can be found in Ref. [54]. As described earlier, the purpose of carrying out this exercise is to extrapolate the results from the linear geometry calculations to the actual bent geometry of the ground state of HgOH. In principle, it is possible to perform the RCC calculations using the bent geometry itself. However, it demands large computational resources.
The results of our linear geometry calculations, presented in Table 2, give an of 109.02 GV/cm. This value is somewhat similar to that of HgF [11], which is about 115 GV/cm. We also note that while correlation effects account for about 9% for HgF, it is less than 2% for HgOH in its linear geometry. This implies that it is not necessary to employ a more sophisticated method for calculating at this stage. The PDM of HgOH is 1.04 D, which is much smaller than that of HgF. This is possibly due to reasons similar to that which explains YbOH having a much smaller PDM than YbF [30]. At the DHF level of calculation (which is the dominant contribution to the total value of ), is about 3.83 times smaller than that in a hypothetical linear geometry. This gives a scaled RCCSD value of 28.47 GV/cm, as compared to the DHF value of 28.01 GV/cm. In contrast, the PDM value increases from the DHF method to the RCC calculations. After scaling the values from linear geometry to the bent geometry, we get its value to be 2.43 D. Comparing this value with the previous works, we find that our estimate value is relatively large. This could be due to two main reasons: we have used a double-zeta (DZ) quality basis sets [55,56,57] and our calculations are based on relativistic methods whereas the previous calculations were carried using non-relativistic methods. We expect that maximum error in the estimated value of to be within 10 percent, while the error in the PDM is expected can be slightly higher.
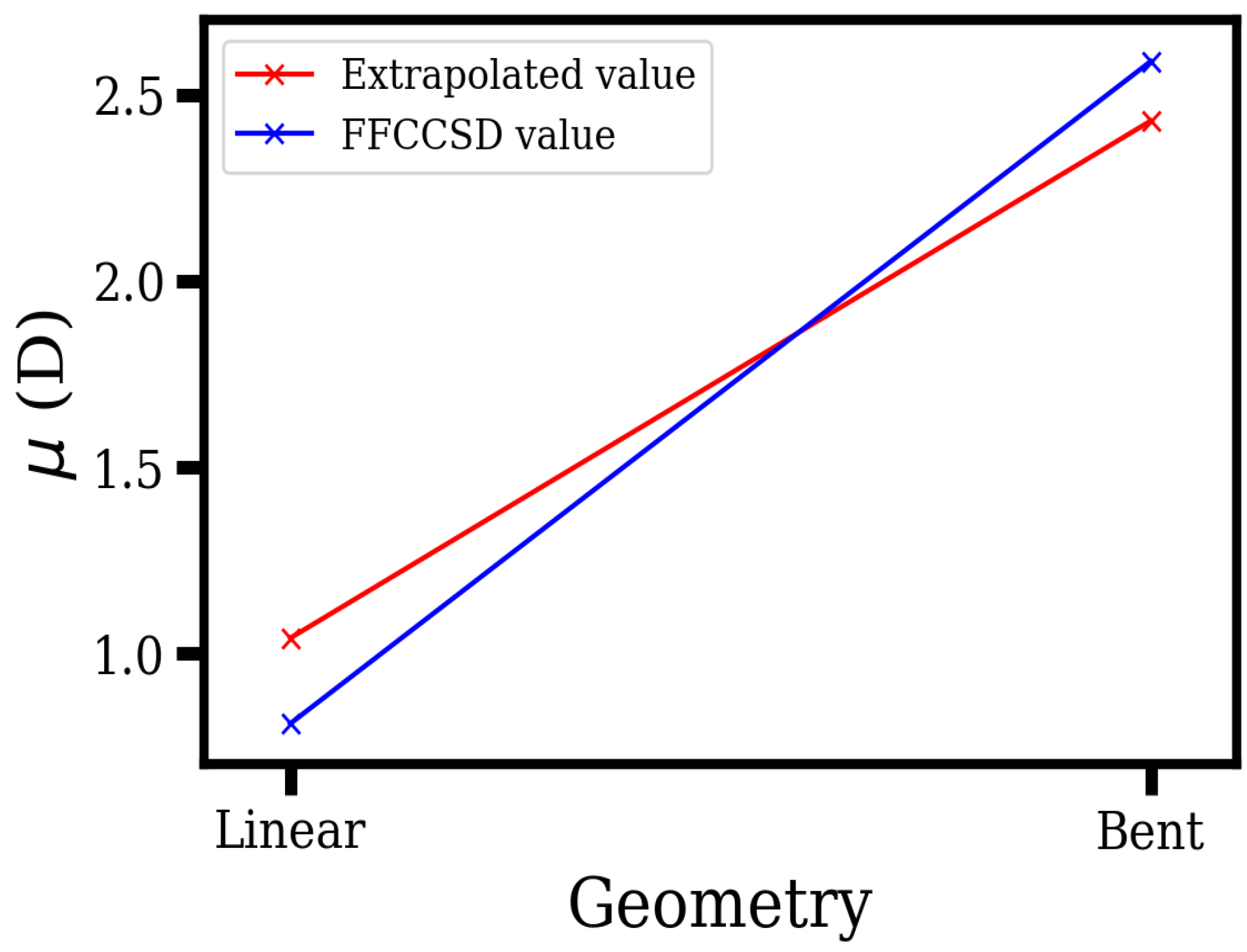
We now briefly comment on the error arising from our extrapolation. An extrapolation scheme may work if the electronic wave function does not change significantly between a linear and a bent case. This may not be the situation here, given that in the bent case, the effective field is thrice as small and the PDM is twice as large as in the linear case. As noted earlier, RCC calculations become complicated due to the bent geometry. Keeping in mind this complication, and at the same time also noting the need to address the concern on extrapolation, we performed non-relativistic finite-field coupled-cluster (FFCC) calculations of the PDM in both the hypothetical linear and the actual bent geometries (using the Dirac18 program) as shown in Figure 2, and obtained 0.81 D and 2.59 D, respectively. These points are denoted in blue color in the figure. We compare these values with our relativistic linear and bent geometry values, with the former calculated as 1.04 D and the latter obtained as 2.43 D by extrapolation. The values are depicted in red color in the plot. Based on the data (where the two values for PDM corresponding to the bent geometry differ by about 6%), and noting that the differences in the values themselves are due to relativity, we set a conservative estimate of the error in extrapolation to about 10%, for the PDM. While adopting the same extrapolation for may raise or lower the error involved in doing so, we still expect it to be a reasonable starting point and anticipate the estimate to be in the ballpark of the actual value, given that our aim is to propose a new EDM experiment. For example, starting with the extrapolated value of 28.47 GV/cm, which offers a projected sensitivity of , we arbitrarily lower and raise the value of the effective electric field by as large as 25 percent, that is, 21.36 GV/cm and 35.58 GV/cm, respectively, to find that the projected sensitivities that are still of the same order (around 6 and 3 times , respectively). In that spirit, that is, given that the proposals of the Hg polyatomic molecules for eEDM experiments are intended to be more of a proof of concept, the error that stems from our rather rudimentary extrapolation scheme may be admissible.
Table 3 gives the contributions to and from each of the terms of the RCCSD method in the LECC approximation for the linear geometry. We note that of the nine resulting terms in the LECC approximation, and its hermitian conjugate do not contribute, due to the Slater–Condon rules. We will begin the analysis of the individual terms with . It can be seen from the above table that the dominant correlation contributions come from and its hermitian conjugate terms, and there are strong cancellations of correlation effects through different RCC terms. As a result, there is a very small difference between the DHF and RCCSD values. For PDM too, term and its hermitian conjugate gives dominant correlation contributions. There is an important difference between the correlation contributions to and in that in the former quantity, they cancel out, whereas in , they add up to give a reduced value than the DHF result.
Since the DHF term contributes the most to , we intend to analyze contributions from various single-particle orbitals (especially from the heavier Hg and O atoms) to it in both the linear and bent geometry configurations. In our calculation, the DHF value is computed by
where the summations are over large (denoted by superscript L) and small (denoted by superscript S) component basis functions, is the total number of large component basis functions, and are the MO coefficients, and is the atomic orbital basis function, with the subscript n denoting the singly occupied molecular orbital. The results of this analysis are presented in Table 4. We have not provided the results for terms that arise from mixings between other orbital combinations, as they are well below error margins, and are almost zero. The table shows that the DHF term is dominated by the contributions that arise from the mixing of the s and orbitals of Hg. The rest of the terms almost completely cancel out in pairs. While the s and mixing accounts for nearly 108.14 GV/cm for the linear case, we see that it contributes only 29.30 GV/cm for the bent geometry. We also observe that although the values of the other contributions are different between the linear and bent cases, their differences turn out to be very similar. Thus, the major deciding factor for the effective electric field between the linear and bent cases is the mixing between the s and orbitals.
We propose that the electron EDM experiment be performed on the ground vibrational state of the ground electronic state of HgOH. We note that in YbOH, the chosen EDM measurement state is the low-lying vibrational state of the ground electronic state. This was necessary for YbOH, as it is in a linear geometry. The choice of a bent mode allows us to make use of the closely-spaced doublets of opposite parity as internal co-magnetometer states [26]. In such a case, there is no need to flip the external electric field in order to perform electron EDM measurements, and this feature, therefore, helps to avoid systematic effects associated with reversing electric fields. Since HgOH is permanently bent in its ground electronic state unlike YbOH, it would have these relevant parity doublets even in its ground vibrational state. We now estimate the size of such a doublet for HgOH, which is an asymmetric top. Using the computed values for the rotational constants, 623.56714, 6.39027, and 6.32545 (in GHz), we estimate the size of the K-doublet [58,59,60] to be 30 MHz, which is comparable to that in YbOH (∼ 10 MHz) [26]. It is worth adding at this point that K, the relevant quantum number, is associated with the projection of the total orbital, spin, and nuclear rotational angular momenta, onto a suitably chosen body-fixed axis of the molecule. The quantum number is normally associated with a symmetric top, and yet it is relevant in the case of HgOH as the molecule can be classified as a near-prolate asymmetric top based on its asymmetry parameter.
We now conduct a preliminary survey of the possible experimental schemes for an electron EDM measurement using the molecule. We begin with the statistical sensitivity of an electron EDM experiment with HgOH. If we were to propose a trap experiment, a preliminary requirement would be laser cooling of the molecules, as typical ‘non-perturbative’ traps are ∼ mK deep. We use the data from Table 1 to calculate FCFs from the ground electronic state to each of the four low-lying excited electronic states. We find that the diagonal FCFs are negligibly small, thus, rendering HgOH unsuitable for a trap experiment. This result suggests that, albeit one may naively expect HgOH to possess highly diagonal FCFs based on its isoelectronic counterpart, HgF, it need not be the case. A possible reason for this observation may be the presence of inner orbitals, unlike in the case of YbOH. The orbitals possibly lead to a strong coupling between electronic and vibrational degrees of freedom, thus, allowing for off-diagonal excitations. However, the precise mechanism of the orbitals in substantially lowering the diagonal nature of FCFs is unclear. A reasonable understanding of this mechanism will prove to be important in qualitatively predicting the laser coolability of other polyatomic systems in the future.
We now turn our attention to a beam experiment. The figure of merit for statistical sensitivity of a beam experiment is given by [61]
where N is the number of molecules detected per second, T is the total integration time, is the coherence time for spin precession, and is the polarization factor. We anticipate that the in the ground vibrational state will be very close to the calculated value in the absolute ground state. Photoassociation of laser-cooled Hg and magnetically trapped OH [62] to produce HgOH molecules may be a possibility [11]. The molecules could also be produced by ablating a mercury-containing compound, such as pure Hg or HgO, in the presence of a reactive gas such as to make HgOH. Though molecular beam intensities vary appreciably between species, we estimate the production of molecules in a single pulse of a slow beam based on comparison to other optimized beam sources [63]. Assuming the detection area of radius ∼1 cm to be ∼1 m away, typical values for comparable beam experiments [14,16], and using a slow beam divergence of 1 sr, we expect a total of molecules. One can also increase the number of molecules by magnetic or electrostatic guiding [64,65]. There are also possibilities that the molecular production can be chemically enhanced [66]. A distinct advantage over ThO is that HgOH is not limited by radiative decay. With a slow beam [63] of buffer gas-cooled HgOH molecules, one can hope to achieve ms (with the spin precession region set to 30 cm long (comparable to that of ThO), and the beam forward velocity to 50 m/s). We set the efficiency in state preparation/ detection to 25%, a reasonable estimate when we compare it with that for the ACME EDM experiment. Combining these estimates with an of 28.47 GV/cm and , the projected statistical sensitivity is about e-cm, which is an improvement over ThO, the system that has set the current best limit for electron EDM.
Furthermore, we would like to add a brief remark here on whether a bent ground-state geometry could introduce an asymmetry in the experiment that could mimic an EDM. Since electron EDM can arise only from the P,T-odd interactions, any other interactions cannot really contribute to its signature. For example, contributions from the PDM and a bent ground-state geometry may contribute to the energy shifts during the experiments, but their contributions effectively cancel out while inferring the signature due to the electron EDM. Therefore, there is no combination of regular molecular effects, structures, properties, etc. could intrinsically mimic a permanent EDM even in a molecule having bent geometry. Systematic effects arising from experimental imperfections, such as uncontrolled background fields, can lead to false EDM signals. However, the parity doublets in this molecule give rise to internal co-magnetometer states, similar to those which are already being used with great success for the rejection of systematic errors in the most sensitive current measurements [14,15].
We also examine the possibility of an electron EDM experiment with HgOH using the EDM proposal [18]. In this class of experiments, a molecule of interest to electron EDM searches is embedded in an inert gas matrix, while retaining the measurement schemes of a beam experiment. Since the molecules are embedded in a matrix, one can achieve large values of N and long coherence times, . Given that the molecules are already embedded, a laser cooling scheme gives no specific advantage. Since the FCFs are unfavorable for laser cooling in HgOH, we consider the possibility of an HgOH EDM experiment. A reasonably large PDM in its ground state facilitates orienting the molecule in a relatively low applied electric field. Moreover, our preliminary DFT calculations show that HgOH possesses a very large PDM of around 7 D in an excited state. A large difference between PDMs between the ground and an excited electronic state offers promise in state selective detection [18]. However, from a comagnetometry point of view, it is unclear if parity doublets can arise, in view of HgOH interacting with the inert gas atoms in the lattice. With the possible absence of this advantage, HgOH in itself may not offer any distinct advantage in an EDM experiment over HgF, although in principle, and EDM measurement with HgOH is very much possible. HgOH may offer some advantage in a clock-state EDM experiment [67] due to its polarizability, though the advantages of that approach would be most notable in a trap experiment. Hence, we conclude from our preliminary survey that a beam experiment is best suited for HgOH, given its spectroscopic properties. Moreover, we forecast that as the co-magnetometer states in polyatomic species aid extensively in controlling systematics, one can extract parameters from beyond Standard Model theories with very stringent constraints, with possible combinations like YbOH and HgOH. The more the number of such systems, the tighter the bounds. To that end, we discuss next other natural extensions of HgF and HgOH for electron EDM experiments.
6. Other Prospective Polyatomic Molecules for EDM Measurements
We now briefly look at two other molecular candidates that are natural extensions to HgF and HgOH, namely HgCH and HgCF. These systems are expected to preserve the features of K-doublets, with the additional advantage that these splittings will be even smaller [26]. We choose our geometry for HgCH from Refs. [68,69]. We obtain 75.07 GV/cm for at DHF level of theory, which is almost comparable to that of ThO, thereby giving this system an edge over HgOH. We do not expect the effective electric field to change beyond 10 percent when we include electron correlation. We find that the PDM of HgCH is about 0.47 D at the DHF level of theory and 0.44 D using DFT. This low value for PDM can be explained by observing the fact that the difference in electronegativity is very less for CH. One expects that this issue will be alleviated when CH is replaced by CF. Indeed, we observe that the value of for HgCF is 60.95 GV/cm, while its PDM is 3.33 D. We use numbers similar to those estimated for HgOH (adding that all the three species should be comparably polarizable, and noting that the production is similar to that of HgOH, except that we propose to use methanol instead of water), and the projected sensitivities thus obtained for HgCH and HgCF are e-cm and e-cm, respectively.
Table 5 presents the effective electric fields as well as upper bounds to the electron EDM from ongoing electron EDM experiments, and compares the projected sensitivities of Hg-containing polyatomics that have been considered in this work, with them. We see immediately that the projected sensitivities from HgOH, HgCH, and HgCF offer scope for sensitivities that can exceed those from the current best systems. The estimated sensitivities for HgCH and HgCF assume that they are not laser-coolable. Given the stark contrast in FCFs between HgF and HgOH, it is not inconceivable that HgCH and HgCF may offer prospects for cooling. In such a case, a trap experiment could provide much higher sensitivities comparable to YbOH.
7. Conclusions
We have investigated the HgOH molecule as a prospective candidate for the measurement of electric dipole moment due to the electric dipole moment of an electron. To demonstrate the same, we calculated the effective electric field and the molecular permanent electric dipole moment of the HgOH molecule. We also evaluated its Franck–Condon factors, which show that HgOH is not laser-coolable, although this molecule is isoelectronic to the laser-coolable HgF polar molecule. We surveyed possible experimental scenarios with HgOH, and find that a trap experiment would be very challenging. We found that HgOH might not offer specific advantages in schemes such as the EDM, except a possibility of state selective detection. We also observed that it is best suited for a beam experiment as its sensitivity is . This is an improvement over ThO, which holds the current best limit on the upper bound of the electron EDM. Furthermore, we realized from our preliminary analyses that HgCH and HgCF offer similar sensitivities.
Author Contributions
V.S.P. conceived the overall idea. R.M. and V.S.P. took the lead for the work. R.M. performed most of the computation. N.R.H. contributed to crucial ideas in the experimental sector of the manuscript. All of the authors equally participated in discussion and interpretations of the results and subsequent analyses. R.M. mainly prepared the manuscript, with V.S.P., B.K.S., N.R.H., M.A., and B.P.D. contributing equally to tuning the manuscript further to its final form. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the necessary data have been provided in the text for this theoretical work.
Acknowledgments
We would like to thank A. C. Vutha, Phelan Yu, X. Tong and Z. C. Yan for insightful discussions. All the computations were performed on the Vikram-100 super-computing cluster facility at the Physical Research Laboratory, Ahmedabad and we wish to thank Jigar Raval for his continuous support in successfully running our programs.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Landau, L. On the conservation laws for weak interactions. Nucl. Phys. 1957, 3, 127131. [Google Scholar] [CrossRef]
- Ballentine, L.E. Quantum Mechanics: A Modern Development; World Scientific: Singapore, 1998; Volume 384386, Chapter 13; pp. 372–373. [Google Scholar]
- Luders, G. Proof of the TCP Theorem. Ann. Phys. 2000, 281, 1004. [Google Scholar] [CrossRef]
- Hoogeveen, F. DESY Reports 1990; DESY: Hamburg, Germany, 1990; pp. 6–90. [Google Scholar]
- Pospelov, M.; Ritz, A. CKM benchmarks for electron electric dipole moment experiments. Phys. Rev. D 2014, 89, 056006. [Google Scholar] [CrossRef] [Green Version]
- Kazarian, A.M.; Kuzmin, S.V.; Shaposhnikov, M.E. Cosmological lower bound on the EDM of the electron. Phys. Lett. B 1992, 276, 131. [Google Scholar] [CrossRef] [Green Version]
- Fuyuto, K.; Hisano, J.; Senaha, E. Toward verification of electroweak baryogenesis by electric dipole moments. Phys. Lett. B 2016, 755, 491. [Google Scholar] [CrossRef] [Green Version]
- Schiff, L.I. Measurability of nuclear electric dipole moments. Phys. Rev. 1963, 132, 2194. [Google Scholar] [CrossRef]
- Sandars, P.G.H. The electric-dipole moments of an atom II. The contribution from an electric-dipole moment on the electron with particular reference to the hydrogen atom. J. Phys. B 1968, 1, 511. [Google Scholar] [CrossRef]
- Abe, M.; Gopakumar, G.; Hada, M.; Das, B.P.; Tatewaki, H.; Mukherjee, D. Application of relativistic coupled-cluster theory to the effective electric field in YbF. Phys. Rev. A 2014, 90, 022501. [Google Scholar] [CrossRef] [Green Version]
- Prasannaa, V.S.; Vutha, A.C.; Abe, M.; Das, B.P. Mercury monohalides: Suitability for electron electric dipole moment searches. Phys. Rev. Lett. 2015, 114, 183001. [Google Scholar] [CrossRef] [Green Version]
- Skripnikov, L.V. Combined 4-component and relativistic pseudopotential study of ThO for the electron electric dipole moment search. J. Chem. Phys. 2016, 145, 214301. [Google Scholar] [CrossRef] [Green Version]
- Skripnikov, L.V. Communication: Theoretical study of HfF+ cation to search for the T,P-odd interactions. J. Chem. Phys. 2017, 147, 021101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreev, V.; Ang, D.G.; DeMille, D.; Doyle, J.M.; Gabrielse, G.; Haefner, J.; Hutzler, N.R.; Lasner, Z.; Meisenhelder, C.; O’Leary, B.R.; et al. Improved limit on the electric dipole moment of the electron. Nature 2018, 562, 355. [Google Scholar]
- Cairncross, W.; Gresh, D.N.; Grau, M.; Cossel, K.C.; Roussy, T.S.; Ni, Y.; Zhou, Y.; Ye, J.; Cornell, E.A. Precision measurement of the electron’s electric dipole moment using trapped molecular ions. Phys. Rev. Lett. 2017, 119, 153001. [Google Scholar] [CrossRef] [Green Version]
- Kara, D.M.; Smallman, I.J.; Hudson, J.J.; Sauer, B.E.; Tarbutt, M.R.; Hinds, E.A. Measurement of the electron’s electric dipole moment using YbF molecules: Methods and data analysis. New J. Phys. 2012, 14, 103051. [Google Scholar] [CrossRef] [Green Version]
- The NL-EDM Collaboration; Aggarwal, P.; Bethlem, H.L.; Borschevsky, A.; Denis, M.; Esajas, K.; Haase, P.A.B.; Hao, Y.; Hoekstra, S.; Jungmann, K.; et al. Measuring the electric dipole moment of the electron in BaF. Eur. Phys. J. D 2018, 72, 197. [Google Scholar] [CrossRef]
- Vutha, A.C.; Horbatsch, M.; Hessels, E.A. Oriented polar molecules in a solid inert-gas matrix: A proposed method for measuring the electric dipole moment of the electron. Atoms 2018, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.R.; Bohn, J.L.; Deskevich, M.P. Candidate molecular ions for an electron electric dipole moment experiment. Phys. Rev. A 2006, 73, 062108. [Google Scholar] [CrossRef] [Green Version]
- Meyer, E.R.; Bohn, J.L. Electron electric-dipole-moment searches based on alkali-metal- or alkaline-earth-metal-bearing molecules. Phys. Rev. A 2009, 80, 042508. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Meyer, E.R.; Paudel, R.; Bohn, J.L.; Leanhardt, A.E. An electron electric dipole moment search in the X3Δ1 ground state of tungsten carbide molecules. J. Mod. Opt. 2009, 56, 2005. [Google Scholar] [CrossRef]
- Kudashov, A.D.; Petrov, A.N.; Skripnikov, L.V.; Mosyagin, N.S.; Isaev, T.A.; Berger, R.; Titov, A.V. Coupled-cluster study of radium monofluoride, RaF, as a candidate to search for P- and T,P- violation effects. Phys. Rev. A 2014, 90, 052513. [Google Scholar] [CrossRef] [Green Version]
- Skripnikov, L.V.; Petrov, A.N.; Mosyagin, N.S.; Titov, A.V.; Flambaum, V.V. TaN molecule as a candidate for the search for a T,P-violating nuclear magnetic quadrupole moment. Phys. Rev. A 2015, 92, 012521. [Google Scholar] [CrossRef] [Green Version]
- Sunaga, A.; Prasannaa, V.S.; Abe, M.; Hada, M.; Das, B.P. Enhancement factors of parity- and time-reversal-violating effects for monofluorides. Phys. Rev. A 2018, 92, 040501(R). [Google Scholar] [CrossRef] [Green Version]
- Fazil, N.M.; Prasannaa, V.S.; Latha, K.V.P.; Abe, M.; Das, B.P. RaH as a potential candidate for electron electric-dipole-moment searches. Phys. Rev. A 2019, 99, 052502. [Google Scholar] [CrossRef]
- Kozyryev, I.; Hutzler, N.R. Precision measurement of time-reversal symmetry violation with laser-cooled polyatomic molecules. Phys. Rev. Lett. 2017, 119, 133002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augenbraun, B.L.; Lasner, Z.D.; Frenett, A.; Sawaoka, H.; Miller, C.; Steimle, T.C.; Doyle, J.M. Laser-cooled polyatomic molecules for improved electron electric dipole moment searches. N. J. Phys. 2020, 22, 022003. [Google Scholar] [CrossRef]
- Gaul, K.; Berger, R. Ab initio study of parity and time-reversal violation in laser-coolable triatomic molecules. arXiv 2018, arXiv:1811.05749. [Google Scholar] [CrossRef] [Green Version]
- Denis, M.; Haase, P.A.B.; Timmermans, R.G.E.; Eliav, E.; Hutzler, N.R.; Borschevsky, A. Enhancement factor for the electric dipole moment of the electron in the BaOH and YbOH molecules. Phys. Rev. A 2019, 99, 042512. [Google Scholar] [CrossRef] [Green Version]
- Prasannaa, V.S.; Shitara, N.; Sakurai, A.; Abe, M.; Das, B.P. Enhanced sensitivity of the electron electric dipole moment from YbOH: The role of theory. Phys. Rev. A 2019, 99, 062502. [Google Scholar] [CrossRef] [Green Version]
- Calvert, J.G.; Lindberg, S.E. Mechanisms of mercury removal by O3 and OH in the atmosphere. Atm. Environ. 2005, 39, 3355. [Google Scholar] [CrossRef]
- Goodsite, M.E.; Plane, J.M.C.; Skov, H. A theoretical study of the oxidation of HgO to HgBr2 in the troposphere. Environ. Sci. Technol. 2004, 38, 1772. [Google Scholar] [CrossRef]
- Ezarfi, N.; Benjelloun, A.T.; Sabor, S.; Benzakour, M.; Mcharfi, M. Theoretical investigations of structural, thermal properties and stability of the group 12 metal M(XH) isomers in atmosphere: M=(Zn, Cd, Hg) and XH=(OH, SH). Theory Chem. Acc. 2019, 138, 109. [Google Scholar] [CrossRef]
- Lindroth, E.; Lynn, B.W.; Sandars, P.G.H. Order α2 theory of the atomic electric dipole moment due to an electric dipole moment on the electron. J. Phys. B At. Mol. Opt. Phys. 1989, 22, 559. [Google Scholar] [CrossRef]
- Das, B.P. Aspects of Many-Body Effects in Molecules and Extended Systems; Mukherjee, D., Ed.; Springer: Berlin/Heidelberg, Germany, 1989; p. 411. [Google Scholar]
- Kozlov, M.G. New limit on the scalar P,T-odd electron-nucleus interaction. Phys. Lett. A 1988, 130, 426. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations: Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations: Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations: Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 16; Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mozhayskiy, V.A.; Krylov, A.I. ezSpectrum. Available online: http://iopenshell.usc.edu/downloads (accessed on 14 October 2020).
- Yanai, T.; Nakano, H.; Nakajima, T.; Tsuneda, T.; Hirata, S.; Kawashima, Y.; Nakao, Y.; Kamiya, M.; Sekino, H.; Hirao, K. UTCHEM—A Program for ab initio Quantum Chemistry; Goos, G., Hartmanis, J., van Leeuwen, J., Eds.; Lecture Notes in Computer Science; Springer: Berlin/Heidelberg, Germany, 2003; Volume 2660, p. 84. [Google Scholar]
- Yanai, T.; Nakajima, T.; Ishikawa, Y.; Hirao, K. A new computational scheme for the Dirac-Hartree-Fock method employing an efficient integral algorithm. J. Chem. Phys. 2001, 114, 6526–6538. [Google Scholar] [CrossRef]
- Visscher, L.; Lee, T.J.; Dyall, K.G. Formulation and implementation of a relativistic unrestricted coupled-cluster method including noniterative connected triples. J. Chem. Phys. 1996, 105, 8769. [Google Scholar] [CrossRef] [Green Version]
- Cizek, J. Correlation Effects in Atoms and Molecules; Advances in Chemical Physics; Lefebvre, W.C., Moser, C., Eds.; Interscience Publishers: New York, NY, USA, 1969. [Google Scholar]
- Zack, L.N.; Sun, M.; Bucchino, M.P.; Clouthier, D.J.; Ziurys, L.M. Gas-phase synthesis and structure of monomeric ZnOH: A model species for metalloenzymes and catalytic surfaces. J. Phys. Chem. A 2012, 116, 1542. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Andaloussi, M.B.D.; Nagashima, U.; Jensen, P. Electronic structure and rovibrational properties of ZnOH in the A′ electronic state: A computational molecular spectroscopy study. J. Chem. Phys. 2014, 141, 094308. [Google Scholar] [CrossRef] [PubMed]
- Saiz-Lopez, A.; Acuna, U.; Trabelsi, T.; Carmona-Garcia, J.; Davalos, J.Z.; Rivero, D.; Cuevas, C.A.; Kinnison, D.E.; Sitkiewicz, S.P.; Roca-Sanjuan, D.; et al. Gas-phase photolysis of Hg(I) radical species: A new atmospheric mercury reduction process. J. Am. Chem. Soc. 2019, 141, 8698. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Kraka, E.; Filatov, M. Bonding in mercury molecules described by the normalized elimination of the small component and coupled cluster theory. ChemPhysChem 2008, 9, 2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saue, T. Relativistic hamiltonians for chemistry: A primer. ChemPhysChem 2011, 12, 3077. [Google Scholar] [CrossRef]
- Jensen, H.J.A.; Bast, R.; Saue, T.; Visscher, l. A Relativistic Ab Initio Electronic Structure Program. Available online: https://www.researchgate.net/publication/315699001DIRAC16DIRACarelativisticabinitioelectronicstructureprogramReleaseDIRAC162016 (accessed on 14 October 2020).
- Mitra, R.; Prasannaa, V.S.; Sahoo, B.K.; Tong, X.; Abe, M.; Das, B.P. Mercury hydroxide as a promising triatomic molecule to probe P,T-odd interactions. arXiv 2019, arXiv:1908.07360, unpublished. [Google Scholar]
- Dyall, K.G. Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the 5d elements Hf–Hg. Theor. Chem. Acc. 2004, 112, 403. [Google Scholar] [CrossRef]
- Dyall, K.G.; Gomes, A.S.P. Revised relativistic basis sets for the 5d elements Hf-Hg. Theory Chem. Acc. 2010, 125, 97. [Google Scholar] [CrossRef]
- Dyall, K.G. Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the light elements H-Ar. Theory Chem. Acc. 2016, 135, 128. [Google Scholar] [CrossRef]
- Wang, S.C. On the asymmetrical top in quantum mechanics. Phys. Rev. 1929, 34, 243. [Google Scholar] [CrossRef]
- Kivelson, D. A (K + 2)nd order formula for asymmetry doublets in rotational spectra. J. Chem. Phys. 1953, 21, 536. [Google Scholar] [CrossRef]
- Polo, S.R. Energy levels of slightly asymmetric top molecules. Can. J. Phys. 1957, 35, 8. [Google Scholar] [CrossRef]
- Khriplovich, I.B.; Lamoreaux, S.K. CP Violation Without Strangeness; Springer: Berlin/Heidelberg, Germany, 1997. [Google Scholar]
- Sawyer, B.C.; Lev, B.L.; Hudson, E.R.; Stuhl, B.K.; Lara, M.; Bohn, J.L.; Ye, J. Magnetoelectrostatic trapping of ground state OH molecules. Phys. Rev. Lett. 2007, 98, 253002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutzler, N.R.; Lu, H.; Doyle, J.M. The buffer gas beam: An intense, cold, and slow source for atoms and molecules. Chem. Rev. 2012, 112, 4803. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Han, Z.; Chow, J.; Ang, D.G.; Meisenhelder, C.; Panda, C.D.; West, E.P.; Gabrielse, G.; Doyle, J.M.; DeMille, D. The metastable Q3Δ2 state of ThO: A new resource for the ACME electron EDM search. New J. Phys. 2020, 22, 023013. [Google Scholar] [CrossRef]
- Grasdijk, O.; Timgren, O.; Kastelic, J.; Wright, T.; Lamoreaux, S.; DeMille, D.; Wenz, K.; Aitken, M.; Zelevinsky, T.; Winick, T.; et al. CeNTREX: A new search for time-reversal symmetry violation in the 205Tl nucleus. arXiv 2020, arXiv:2010.01451v1. [Google Scholar]
- Jadbabaie, A.; Pilgram, N.H.; Klos, J.; Kotochigova, S.; Hutzler, N.R. Enhanced molecular yield from a cryogenic buffer gas beam source via excited state chemistry. New J. Phys. 2020, 22, 022002. [Google Scholar] [CrossRef]
- Verma, M.; Jayich, A.M.; Vutha, A.C. Electron electric dipole moment searches using clock transitions in ultracold molecules. Phys. Rev. Lett. 2020, 125, 153201. [Google Scholar] [CrossRef]
- Filatov, M.; Cremer, D. Relativistically corrected hyperfine structure constants calculated with the regular approximation applied to correlation corrected ab initio theory. J. Chem. Phys. 2004, 121, 5618. [Google Scholar] [CrossRef]
- Filatov, M.; Zou, W.; Cremer, D. Calculation of response properties with the normalized elimination of the small component method. Int. J. Quant. Chem. 2014, 114, 993. [Google Scholar] [CrossRef]
Figure 1.
Pictorial representation of the bent geometry of the ground state of HgOH. Our finding shows .
Figure 1.
Pictorial representation of the bent geometry of the ground state of HgOH. Our finding shows .

Figure 2.
The extrapolation scheme from a linear to a bent geometry, for the permanent electric dipole moment (PDM) of HgOH. The data points in blue show the finite-field coupled-cluster (FFCC) values of the PDM in a non-relativistic framework, while for those in red, the point corresponding to the linear geometry is calculated, and the one pertaining to the bent geometry is the extrapolated value. We add that the distance that we have set between the points corresponding to the linear and bent geometries on the X-axis is arbitrary.
Figure 2.
The extrapolation scheme from a linear to a bent geometry, for the permanent electric dipole moment (PDM) of HgOH. The data points in blue show the finite-field coupled-cluster (FFCC) values of the PDM in a non-relativistic framework, while for those in red, the point corresponding to the linear geometry is calculated, and the one pertaining to the bent geometry is the extrapolated value. We add that the distance that we have set between the points corresponding to the linear and bent geometries on the X-axis is arbitrary.


{kind=link}
{kind=link}
Table 1.
List of the optimized geometry of the ground electronic state and three low-lying excited states of HgOH from various works. The unit of bond-lengths is angstrom (Å), while that of the bond angle is degrees.
Table 1.
List of the optimized geometry of the ground electronic state and three low-lying excited states of HgOH from various works. The unit of bond-lengths is angstrom (Å), while that of the bond angle is degrees.
| State | Reference | |||
|---|---|---|---|---|
| Ground | 2.091 | 0.966 | 104.1 | Ref. [50] |
| 2.25 | 0.99 | 106.8 | Ref. [32] | |
| 2.181 | - | - | Ref. [51] | |
| 2.2079 | 0.9691 | 103.6 | Ref. [33] | |
| 2.2294 | 0.9633 | 104.83 | This work | |
| First-excited | 3.1458 | 0.9563 | 180 | This work |
| Second-excited | 2.0766 | 0.9615 | 102.5 | This work |
| Third-excited | 3.5482 | 1.0095 | 81.93 | This work |
Table 2.
Table showing the calculated (in GV/cm) and (in D) values in HgOH by assuming its hypothetical linear and the actual bent geometry ground state using the Dirac–Hartree–Fock (DHF) and RCCSD methods. We also give values from the previous calculations using density functional theory (DFT).
Table 2.
Table showing the calculated (in GV/cm) and (in D) values in HgOH by assuming its hypothetical linear and the actual bent geometry ground state using the Dirac–Hartree–Fock (DHF) and RCCSD methods. We also give values from the previous calculations using density functional theory (DFT).
| Geometry | ||||
|---|---|---|---|---|
| DHF | LECC | DHF | LECC | |
| From this work | ||||
| Linear | 107.24 | 109.02 | 1.57 | 1.04 |
| Bent | 28.01 | 28.47 | 3.67 | 2.43 |
| From other works | ||||
| 1.89 [32] | ||||
| 1.92 [51] | ||||
| 1.96 [33] | ||||
† Scaled results from the DHF and LECC values of the linear geometry calculations.
Table 3.
Contributions from the individual relativistic coupled-cluster (RCC) terms to (in GV/cm), (in D), and (in kHz) from both the linear and bent geometries of HgOH. O denotes the operator corresponding to the properties and h.c. means hermitian conjugate. Note that for the PDM, the term corresponding to the DHF contribution also accounts for the nuclear contribution in it.
Table 3.
Contributions from the individual relativistic coupled-cluster (RCC) terms to (in GV/cm), (in D), and (in kHz) from both the linear and bent geometries of HgOH. O denotes the operator corresponding to the properties and h.c. means hermitian conjugate. Note that for the PDM, the term corresponding to the DHF contribution also accounts for the nuclear contribution in it.
| Term | (GV/cm) | (D) |
|---|---|---|
| O (DHF) | 107.24 | 1.57 |
| h.c. | 9.50 | −0.42 |
| −2.76 | −0.15 | |
| h.c. | −0.38 | 0.12 |
| −4.58 | −0.11 |
Table 4.
Contributions from different atomic orbital (AO) mixing to the DHF value of (in GV/cm), where the AO in the left hand side is a small component AO and that in the right hand side is a large component AO. Non-zero contributions may come only from odd-parity AO mixings where superscript S, and L stand for small component and large component AOs respectively. Results are given for both the linear and the actual bent geometry HgOH molecule.
Table 4.
Contributions from different atomic orbital (AO) mixing to the DHF value of (in GV/cm), where the AO in the left hand side is a small component AO and that in the right hand side is a large component AO. Non-zero contributions may come only from odd-parity AO mixings where superscript S, and L stand for small component and large component AOs respectively. Results are given for both the linear and the actual bent geometry HgOH molecule.
| Atom | AOs | Linear | Bent |
|---|---|---|---|
| Hg | 378.40 | 100.11 | |
| −270.26 | −71.81 | ||
| −31.40 | −8.07 | ||
| 30.19 | 7.77 | ||
| 0.79 | 0.19 | ||
| −0.78 | −0.18 | ||
| O | 2.78 | 1.44 | |
| −2.77 | −1.44 |
Table 5.
Comparison of measured (ThO, HfF, and YbF) and projected sensitivities (from appropriate references as given in the table) offered by different molecules for EDM experiments. For molecules where measurements are not available, the sensitivity is estimated with appropriate N, T, , and values. The unit chosen for is e-cm, while the effective electric field is given in GV/cm.
Table 5.
Comparison of measured (ThO, HfF, and YbF) and projected sensitivities (from appropriate references as given in the table) offered by different molecules for EDM experiments. For molecules where measurements are not available, the sensitivity is estimated with appropriate N, T, , and values. The unit chosen for is e-cm, while the effective electric field is given in GV/cm.
| Molecule | Reference(s) | ||
|---|---|---|---|
| ThO | 79.9 [12] | Ref. [14] | |
| HfF | 22.5 [13] | Ref. [15] | |
| YbF | 23.1 [10] | Ref. [16] | |
| HgOH | 28.47 | This work | |
| HgCH | 75.07 | This work | |
| HgCF | 60.95 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mitra, R.; Prasannaa, V.S.; Sahoo, B.K.; Hutzler, N.R.; Abe, M.; Das, B.P. Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches. Atoms 2021, 9, 7. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9010007
AMA Style
Mitra R, Prasannaa VS, Sahoo BK, Hutzler NR, Abe M, Das BP. Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches. Atoms. 2021; 9(1):7. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9010007
Chicago/Turabian StyleMitra, Ramanuj, V. Srinivasa Prasannaa, Bijaya K. Sahoo, Nicholas R. Hutzler, Minori Abe, and Bhanu Pratap Das. 2021. "Study of HgOH to Assess Its Suitability for Electron Electric Dipole Moment Searches" Atoms 9, no. 1: 7. https://0-doi-org.brum.beds.ac.uk/10.3390/atoms9010007
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.