Modulation of Guanylate Cyclase Activating Protein 1 (GCAP1) Dimeric Assembly by Ca2+ or Mg2+: Hints to Understand Protein Activity

 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Human GCAP1 Expression and Purification
2.2. Circular Dichroism
2.3. Analytical Size Exclusion Chromatography
2.4. Microscale Thermophoresis (MST)
2.5. Dynamic Light Scattering
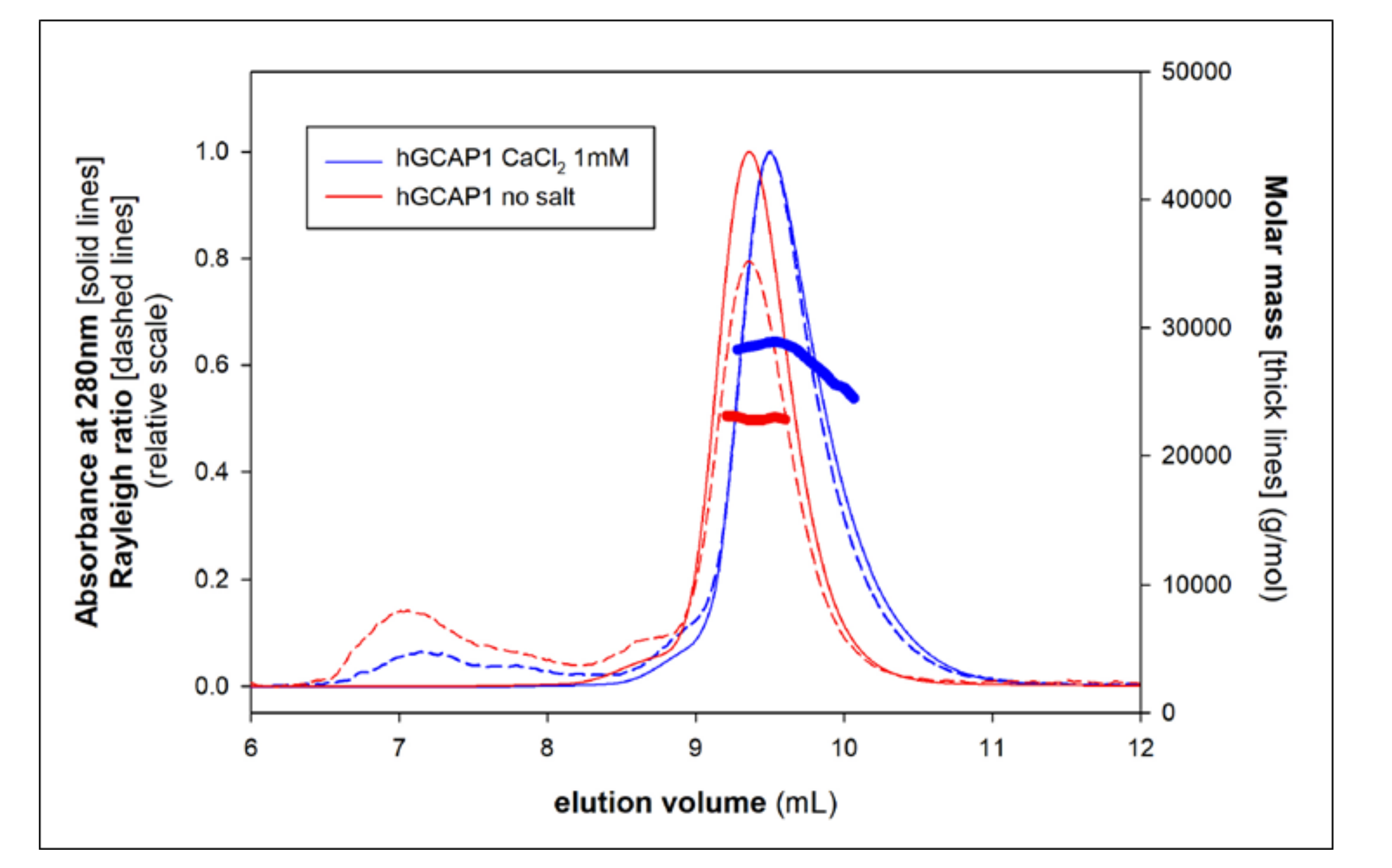
2.6. SEC-MALS
2.7. Trp Fluorescence
2.8. SAXS Data Collection
2.9. Model of hGCAP1 Dimeric Assembly and Molecular Dynamics Simulations
3. Results
3.1. hGCAP1 Expression and Purification
3.2. Thermal Denaturation of hGCAP1
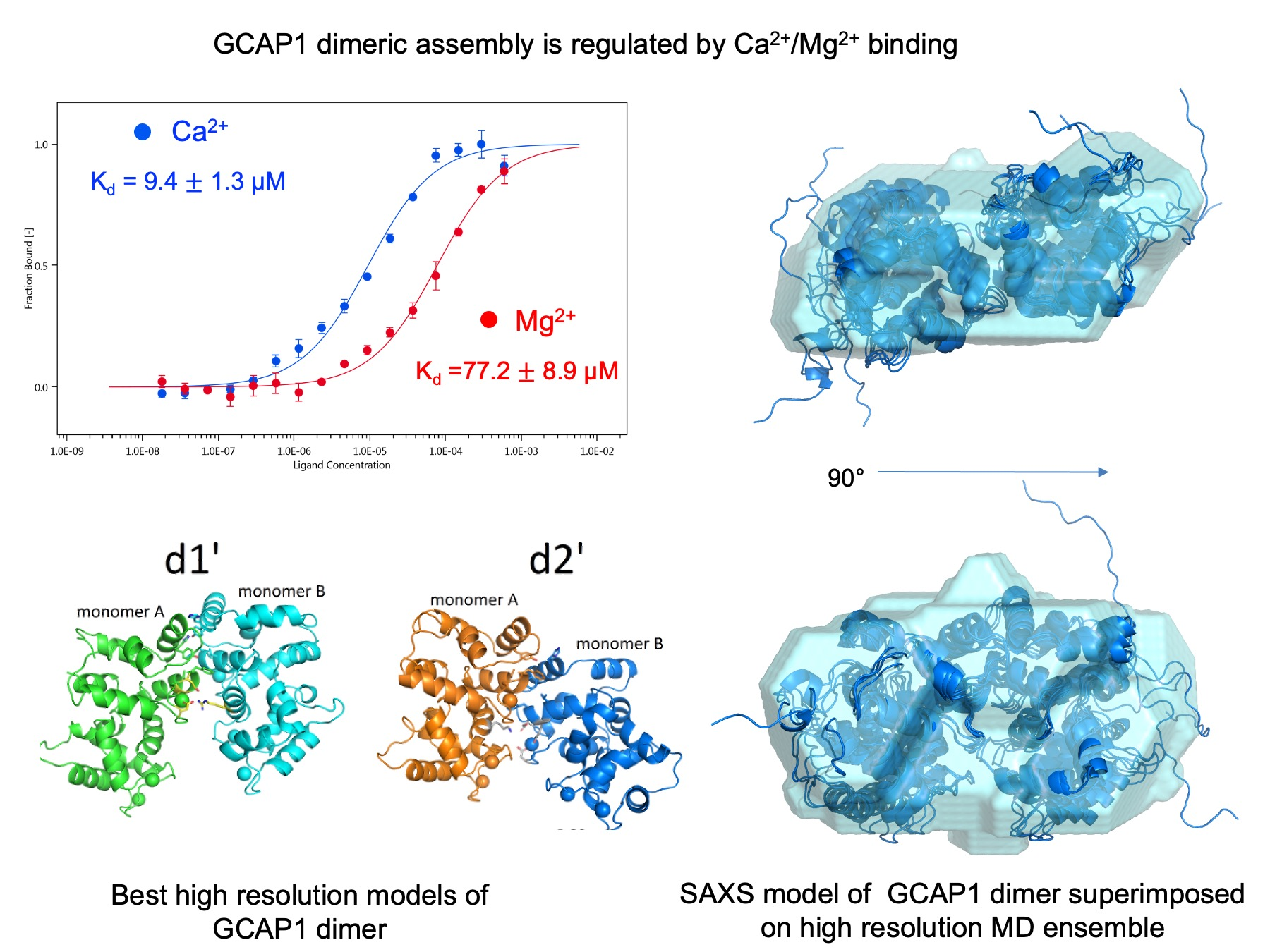
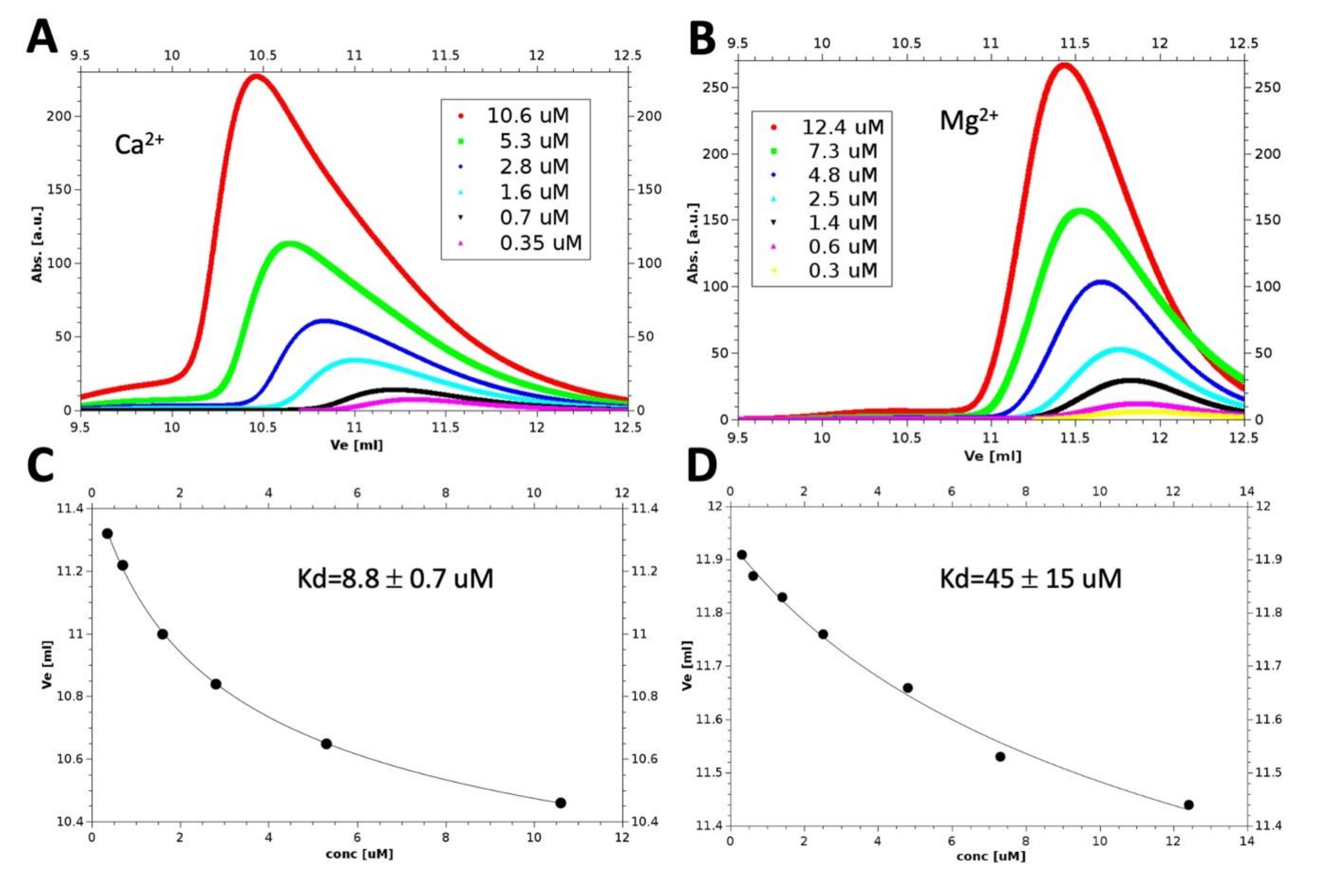
3.3. Monomer-Dimer Equilibrium of hGCAP1 in the Presence of Ca2+ or Mg2+
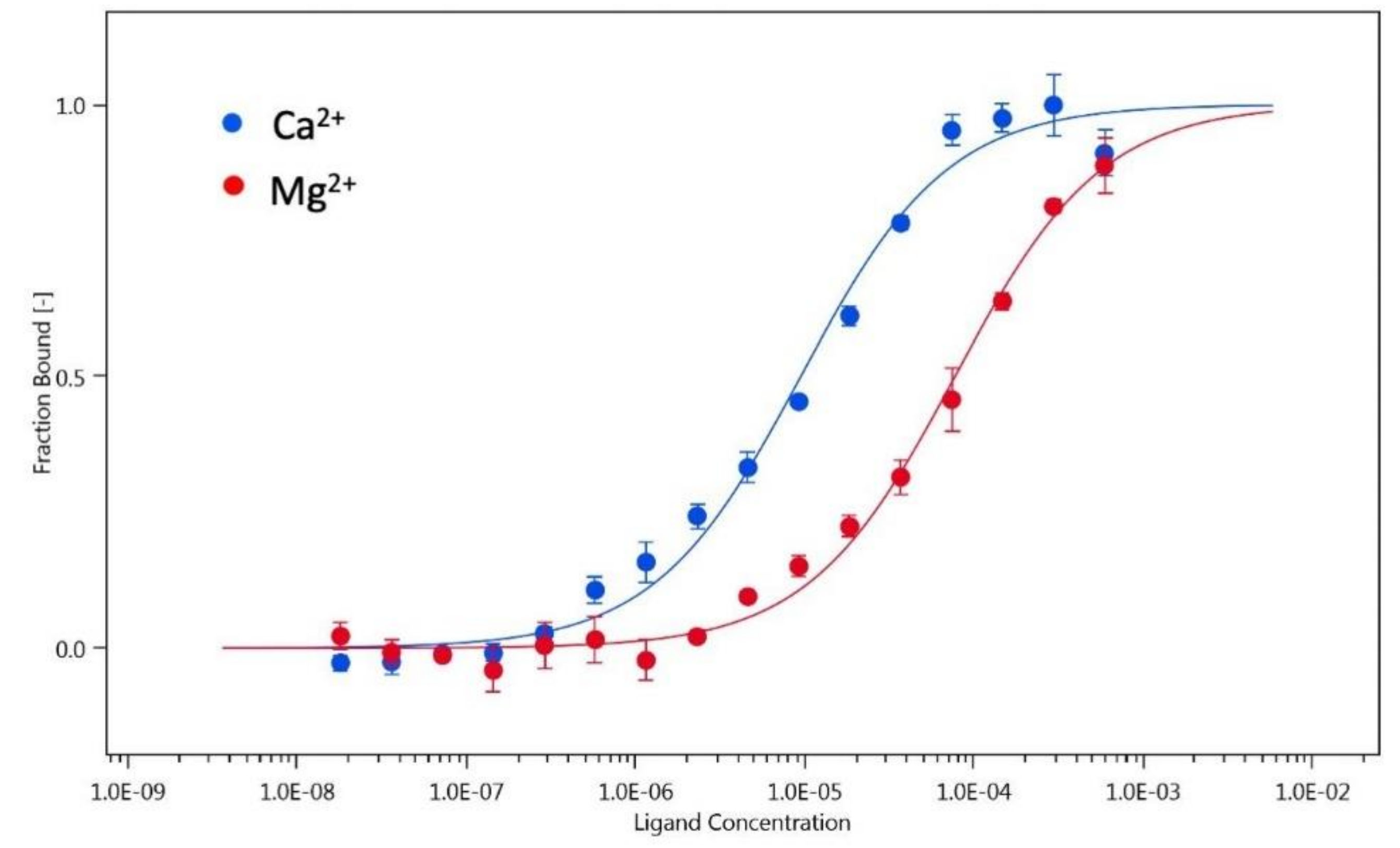
3.4. Monomer-Dimer Equilibrium of hGCAP1 Analysed with Microscale Thermophoresis
3.5. Monomer-Dimer Equilibrium Is Affected by Ionic Strength
3.6. hGCAP1 Trp Fluorescence in the Different Ion Binding States
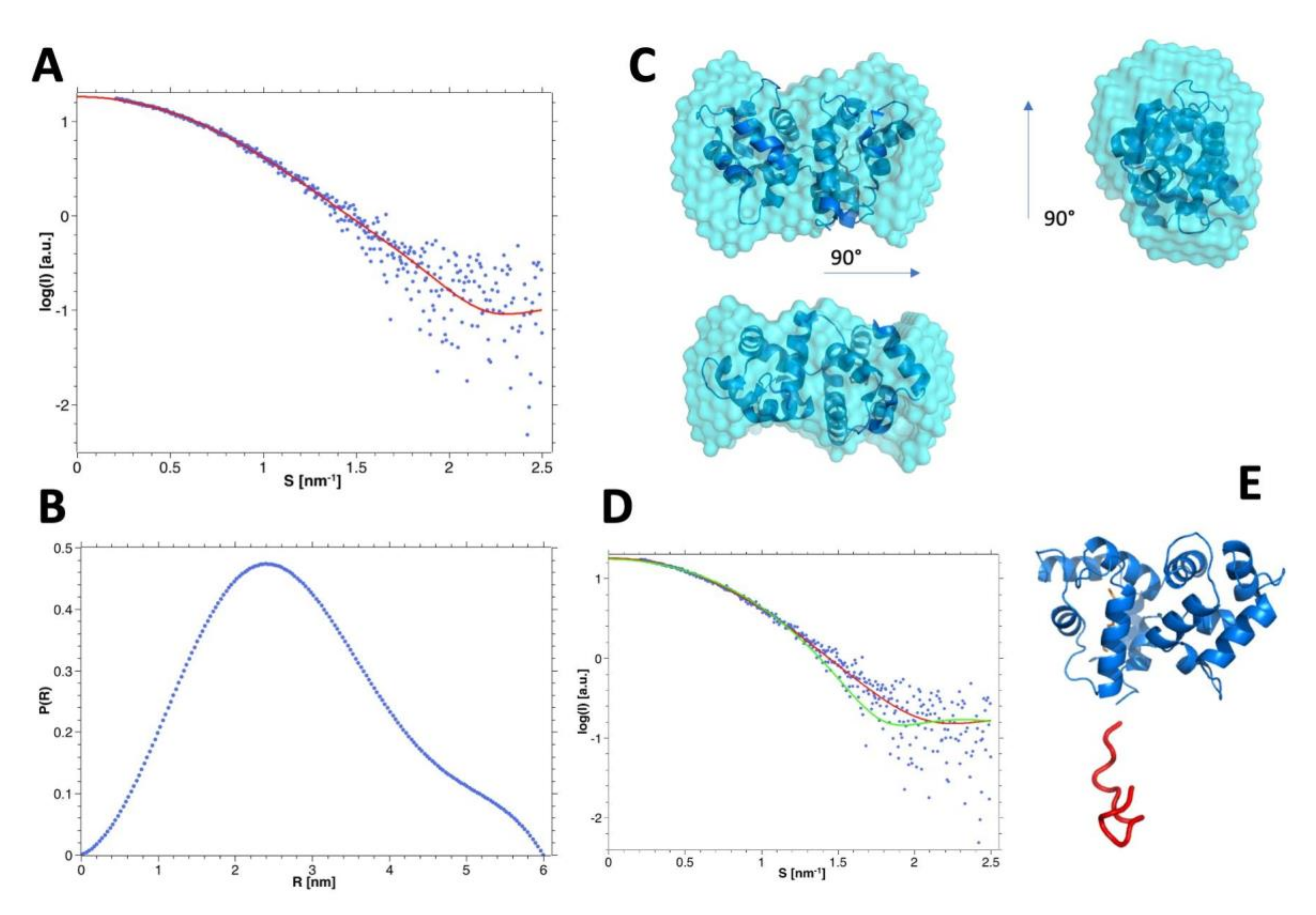
3.7. SAXS Analysis of the hGCAP1 Monomer
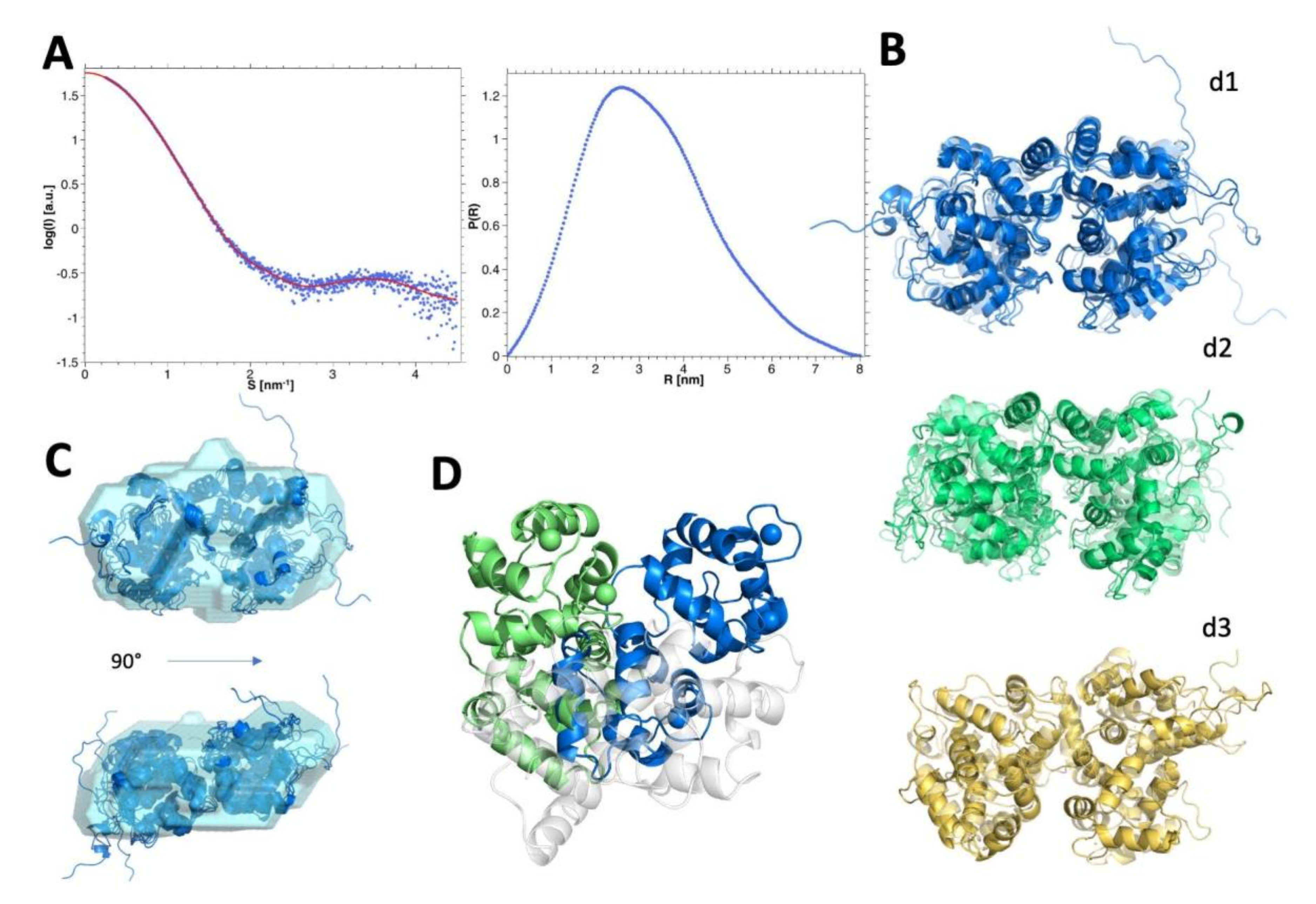
3.8. SEC-SAXS Evidence of the Monomer-Dimer Equilibrium
3.9. Modelling and Selection of Possible Dimeric Assemblies
3.10. Computational Analysis of Mutations in hGCAP1 Dimeric Assembly
3.11. Insights into the Molecular Interface of the Most Likely Dimers d1 and d2
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koch, K.-W.; Edell’Orco, D. Protein and Signaling Networks in Vertebrate Photoreceptor Cells. Front. Mol. Neurosci. 2015, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peshenko, I.V.; Cideciyan, A.V.; Sumaroka, A.; Olshevskaya, E.V.; Scholten, A.; Abbas, S.; Koch, K.-W.; Jacobson, S.G.; Dizhoor, A.M. A G86R mutation in the calcium-sensor protein GCAP1 alters regulation of retinal guanylyl cyclase and causes dominant cone-rod degeneration. J. Biol. Chem. 2019, 294, 3476–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Peshenko, I.V.; Olshevskaya, E.V.; Dizhoor, A.M.; Ames, J.B. Structure of Guanylyl Cyclase Activator Protein 1 (GCAP1) Mutant V77E in a Ca2+-free/Mg2+-bound Activator State. J. Biol. Chem. 2015, 291, 4429–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, V.; Sulmann, S.; Koch, K.-W.; Dell’Orco, D. Structural effects of Mg2+ on the regulatory states of three neuronal calcium sensors operating in vertebrate phototransduction. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1853, 2055–2065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermilov, A.N.; Olshevskaya, E.V.; Dizhoor, A.M. Instead of Binding Calcium, One of the EF-hand Structures in Guanylyl Cyclase Activating Protein-2 Is Required for Targeting Photoreceptor Guanylyl Cyclase. J. Biol. Chem. 2001, 276, 48143–48148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto-Bruc, A.; Buczyłko, J.; Surgucheva, I.; Subbaraya, I.; Rudnicka-Nawrot, M.; Crabb, J.W.; Arendt, A.; Hargrave, P.A.; Baehr, A.W.; Palczewski, K. Functional Reconstitution of Photoreceptor Guanylate Cyclase with Native and Mutant Forms of Guanylate Cyclase-Activating Protein 1. Biochemistry 1997, 36, 4295–4302. [Google Scholar] [CrossRef]
- Viviano, J. Evolutionary Interrelationships and Insights into Molecular Mechanisms of Functional Divergence: An Analysis of Neuronal Calcium Sensor Proteins. J. Phylogenetics Evol. Biol. 2013, 1, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Manes, G.; Mamouni, S.; Hérald, E.; Richard, A.-C.; Sénéchal, A.; Aouad, K.; Bocquet, B.; Meunier, I.; Hamel, C.P. Cone dystrophy or macular dystrophy associated with novel autosomal dominant GUCA1A mutations. Mol. Vis. 2017, 23, 198–209. [Google Scholar]
- Payne, A.M.; Downes, S.M.; Bessant, D.A.; Taylor, R.; Holder, G.E.; Warren, M.J.; Bird, A.C.; Bhattacharya, S.S. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum. Mol. Genet. 1998, 7, 273–277. [Google Scholar] [CrossRef]
- Behnen, P.; Dell’Orco, D.; Koch, K.-W. Involvement of the calcium sensor GCAP1 in hereditary cone dystrophies. Biol. Chem. 2010, 391, 631–637. [Google Scholar] [CrossRef]
- Marino, V.; Scholten, A.; Koch, K.-W.; Dell’Orco, D. Two retinal dystrophy-associated missense mutations in GUCA1A with distinct molecular properties result in a similar aberrant regulation of the retinal guanylate cyclase. Hum. Mol. Genet. 2015, 24, 6653–6666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vocke, F.; Weisschuh, N.; Malfatti, S.; Marino, V.; Jacobson, S.G.; Reiff, C.M.; Dell’Orco, D.; Koch, K.-W. Dysfunction of cGMP signalling in photoreceptors by a macular dystrophy-related mutation in the calcium sensor GCAP1. Hum. Mol. Genet. 2016, 26, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.; Marino, V.; Weisschuh, N.; Kieninger, S.; Solaki, M.; Dell’Orco, D.; Koch, K.-W. Neuronal Calcium Sensor GCAP1 Encoded by GUCA1A Exhibits Heterogeneous Functional Properties in Two Cases of Retinitis Pigmentosa. ACS Chem. Neurosci. 2020, 11, 1458–1470. [Google Scholar] [CrossRef]
- Olshevskaya, E.V. Dimerization of Guanylyl Cyclase-activating Protein and a Mechanism of Photoreceptor Guanylyl Cyclase Activation. J. Biol. Chem. 1999, 274, 25583–25587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Roseman, G.; Peshenko, I.; Manchala, G.; Cudia, D.; Dizhoor, A.M.; Millhauser, G.; Ames, J.B. Retinal guanylyl cyclase activating protein 1 forms a functional dimer. PLoS ONE 2018, 13, e0193947. [Google Scholar] [CrossRef] [Green Version]
- Ames, J.B. Dimerization of Neuronal Calcium Sensor Proteins. Front. Mol. Neurosci. 2018, 11, 397. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Konarev, P.V.; Volkov, V.; Sokolova, A.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Marino, V.; Dell’Orco, D. Allosteric communication pathways routed by Ca2+/Mg2+ exchange in GCAP1 selectively switch target regulation modes. Sci. Rep. 2016, 6, 34277. [Google Scholar] [CrossRef]
- Pierce, B.G.; Hourai, Y.; Weng, Z. Accelerating Protein Docking in ZDOCK Using an Advanced 3D Convolution Library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef]
- Abraham, M.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, V.; Dell’Orco, D. Evolutionary-Conserved Allosteric Properties of Three Neuronal Calcium Sensor Proteins. Front. Mol. Neurosci. 2019, 12, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL– a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Li, L.; Weng, Z. ZDOCK: An initial-stage protein-docking algorithm. Proteins Struct. Funct. Bioinform. 2003, 52, 80–87. [Google Scholar] [CrossRef]
- Dell’Orco, D.; De Benedetti, P.G.; Fanelli, F. In silico screening of mutational effects on enzyme-proteic inhibitor affinity: A docking-based approach. BMC Struct. Biol. 2007, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Tiberti, M.; Invernizzi, G.; Lambrughi, M.; Inbar, Y.; Schreiber, G.; Papaleo, E. PyInteraph: A Framework for the Analysis of Interaction Networks in Structural Ensembles of Proteins. J. Chem. Inf. Model. 2014, 54, 1537–1551. [Google Scholar] [CrossRef]
- Flinta, C.; Persson, B.; Jörnvall, H.; Von Heijne, G. Sequence determinants of cytosolic N-terminal protein processing. JBIC J. Biol. Inorg. Chem. 1986, 154, 193–196. [Google Scholar] [CrossRef]
- Marino, V.; Cortivo, G.D.; Oppici, E.; Maltese, P.E.; D’Esposito, F.; Manara, E.; Ziccardi, L.; Falsini, B.; Magli, A.; Bertelli, M.; et al. A novel p.(Glu111Val) missense mutation in GUCA1A associated with cone-rod dystrophy leads to impaired calcium sensing and perturbed second messenger homeostasis in photoreceptors. Hum. Mol. Genet. 2018, 27, 4204–4217. [Google Scholar] [CrossRef]
- Vivian, J.T.; Callis, P.R. Mechanisms of Tryptophan Fluorescence Shifts in Proteins. Biophys. J. 2001, 80, 2093–2109. [Google Scholar] [CrossRef] [Green Version]
- Sokal, I.; Otto-Bruc, A.E.; Surgucheva, I.; Verlinde, C.L.; Wang, C.-K.; Baehr, W.; Palczewski, K. Conformational Changes in Guanylyl Cyclase-activating Protein 1 (GCAP1) and Its Tryptophan Mutants as a Function of Calcium Concentration. J. Biol. Chem. 1999, 274, 19829–19837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orthaber, D.; Bergmann, A.; Glatter, O. SAXS experiments on absolute scale with Kratky systems using water as a secondary standard. J. Appl. Crystallogr. 2000, 33, 218–225. [Google Scholar] [CrossRef]
- Svergun, D.I.; Semenyuk, A.V.; Feigin, L.A. Small-angle-scattering-data treatment by the regularization method. Acta Crystallogr. Sect. A Found. Crystallogr. 1988, 44, 244–250. [Google Scholar] [CrossRef]
- Svergun, D.I. Restoring Low Resolution Structure of Biological Macromolecules from Solution Scattering Using Simulated Annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef] [Green Version]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in theATSASprogram package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapidab-initioshape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [Green Version]
- Stephen, R.; Bereta, G.; Golczak, M.; Palczewski, K.; Sousa, M.C. Stabilizing Function for Myristoyl Group Revealed by the Crystal Structure of a Neuronal Calcium Sensor, Guanylate Cyclase-Activating Protein 1. Structure 2007, 15, 1392–1402. [Google Scholar] [CrossRef] [Green Version]
- Krissinel, E.; Henrick, K. Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Dindo, M.; Oppici, E.; Dell’Orco, D.; Montone, R.; Cellini, B. Correlation between the molecular effects of mutations at the dimer interface of alanine–glyoxylate aminotransferase leading to primary hyperoxaluria type I and the cellular response to vitamin B6. J. Inherit. Metab. Dis. 2017, 41, 263–275. [Google Scholar] [CrossRef]
- Rudnicka-Nawrot, M.; Surgucheva, I.; Hulmes, J.D.; Haeseleer, F.; Sokal, I.; Crabb, J.W.; Baehr, A.W.; Palczewski, K. Changes in Biological Activity and Folding of Guanylate Cyclase-Activating Protein 1 as a Function of Calcium. Biochemistry 1998, 37, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Marino, V.; Borsatto, A.; Vocke, F.; Koch, K.-W.; Dell’Orco, D. CaF2 nanoparticles as surface carriers of GCAP1, a calcium sensor protein involved in retinal dystrophies. Nanoscale 2017, 9, 11773–11784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, K.; Oobatake, M.; Goto, Y. Salt-dependent monomer–dimer equilibrium of bovine β-lactoglobulin at pH 3. Protein Sci. 2008, 10, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.-Y.; Lange, C.; Helten, A.; Hoppner-Heitmann, D.; Duda, T.; Sharma, R.K.; Koch, K.-W. Regulatory modes of rod outer segment membrane guanylate cyclase differ in catalytic efficiency and Ca2+-sensitivity. JBIC J. Biol. Inorg. Chem. 2003, 270, 3814–3821. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer | Conc. [μM] a | Rg [Å] | I0 | Mw (I0) c [kDa] | % mon. | Dmax [Å] | Mw (Vc) [kDa] | Mw (qmax) [kDa] |
|---|---|---|---|---|---|---|---|---|
| EGTA | 10.9 | 21.5 | 18.2 | 21.1 | 100 | 60 | 29.4 | 28.2 |
| Mg2+ | 59.4 | 22.4 | 32.3 | 35.9 | 43 | 75 | 29.0 | 28.8 |
| Ca2+/Mg2+ | 25.3 | 24.1 | 31.6 | 35.2 | 46 | 76 | 27.4 | 27.1 |
| Ca2+/Mg2+ | 51.1 | 24.5 | 39.2 | 43.7 | 9 | 81 | 35.2 | 38.0 |
| Ca2+/Mg2+ | ~76 b | 24.5 | - | - | - | 81 | 40.8 | 45.6 |
| Assembly | ‘Static’ χ2 with SAXS a | ‘Dynamic’ χ2 with SAXS b | ZD-s c | Native-Like Poses c | Best Ranked Poses d |
|---|---|---|---|---|---|
| d1′ | 2.42 | 1.16 | 54.4 ± 0.8 | 22 | 1 |
| d2′ | 1.95 | 1.17 | 44.5 ± 0.9 | 24 | 16 |
| d3′ | 2.08 | 2.37 | 44.9 ± 0.7 | 16 | 1 |
| Assembly | Mutant | Native-Like Poses | ΔZD-s | ΔΔG0 * [kcal/mol] |
|---|---|---|---|---|
| D1′ | F73E | 15 | −13.14 | 5.12 |
| H19R | 12 | −14.75 | 5.75 | |
| V77E | 21 | −11.62 | 4.53 | |
| Y22D | 17 | −12.20 | 4.76 | |
| D2′ | F73E | 12 | −2.65 | 1.04 |
| H19R | 8 | −4.85 | 1.89 | |
| V77E | 9 | −4.15 | 1.62 | |
| Y22D | 9 | −4.70 | 1.84 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonì, F.; Marino, V.; Bidoia, C.; Mastrangelo, E.; Barbiroli, A.; Dell’Orco, D.; Milani, M. Modulation of Guanylate Cyclase Activating Protein 1 (GCAP1) Dimeric Assembly by Ca2+ or Mg2+: Hints to Understand Protein Activity. Biomolecules 2020, 10, 1408. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101408
Bonì F, Marino V, Bidoia C, Mastrangelo E, Barbiroli A, Dell’Orco D, Milani M. Modulation of Guanylate Cyclase Activating Protein 1 (GCAP1) Dimeric Assembly by Ca2+ or Mg2+: Hints to Understand Protein Activity. Biomolecules. 2020; 10(10):1408. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101408
Chicago/Turabian StyleBonì, Francesco, Valerio Marino, Carlo Bidoia, Eloise Mastrangelo, Alberto Barbiroli, Daniele Dell’Orco, and Mario Milani. 2020. "Modulation of Guanylate Cyclase Activating Protein 1 (GCAP1) Dimeric Assembly by Ca2+ or Mg2+: Hints to Understand Protein Activity" Biomolecules 10, no. 10: 1408. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10101408