Natural Products Attenuating Biosynthesis, Processing, and Activity of Ras Oncoproteins: State of the Art and Future Perspectives


Abstract
:1. Introduction
- (i)
- here is a strong need for targeted therapies to treat Ras-dependent diseases, such as most cancers, in the perspective of precision medicine. At the moment, there are few drugs, even synthetic ones, which have proved promisingly effective in clinical trials and are limited to targeting specific pathological mutants of Ras, which, although important, represent only a small percentage of those involved in human pathologies;
- (ii)
- NPs can be successfully applied to different direct and indirect strategies of inhibition of Ras activity;
- (iii)
- NPs represent an unlimited and still little-explored resource of chemical structures that are currently accessible thanks to the new platforms for isolation, purification, and characterization that can warrant the quality, safety, and efficacy of the active compounds.
- (iv)
- the advancement of knowledge of the structure/function relationships of Ras proteins and their pathological variants allowed to identifiy novel druggable pockets of the proteins to be considered in the virtual/experimental screening of novel inhibitors.
2. Ras Proteins
3. Ras Processing and Subcellular Localization
4. Ras Signaling in Mammalian Cells
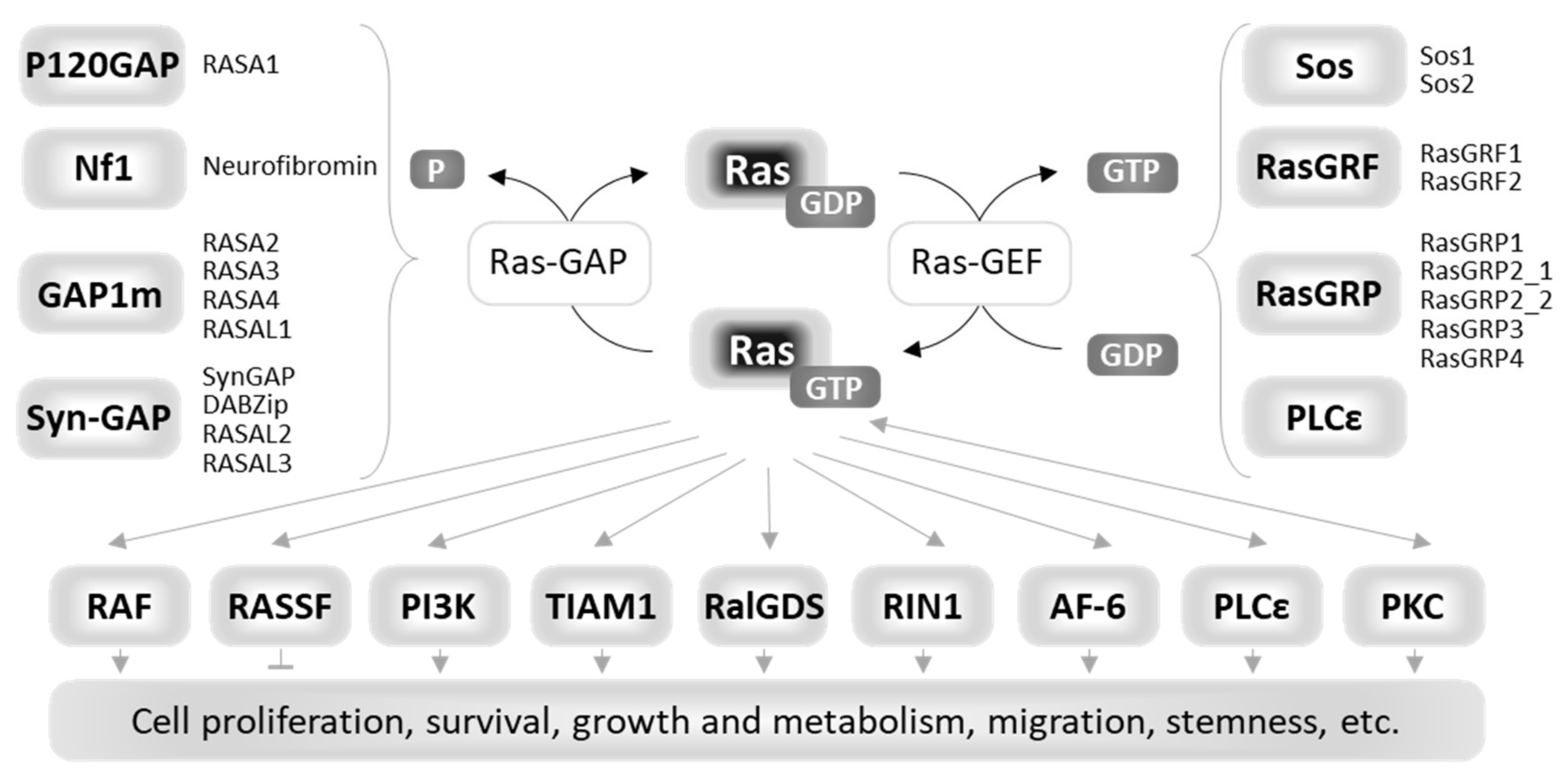
4.1. Upstream Regulators of Ras
4.2. Downstream Effectors of Ras
5. Ras Mutants in Human Diseases
6. Strategies for Inhibiting Ras Oncoproteins Biosynthesis, Processing, Activity, and Signaling in Cancer Therapy
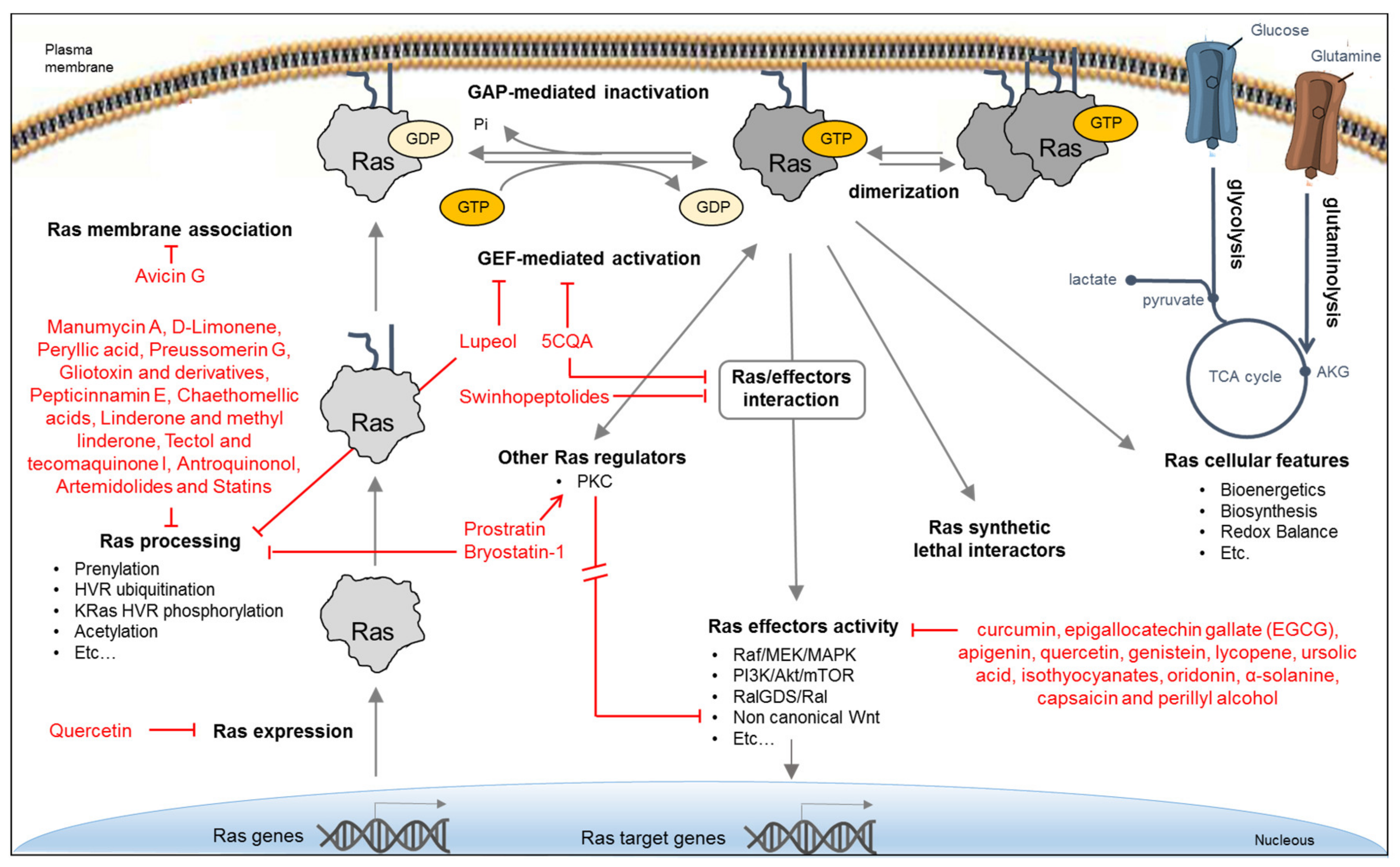
6.1. Indirect Strategies
6.2. Direct Strategies: Ras Proteins as Pharmacological Targets
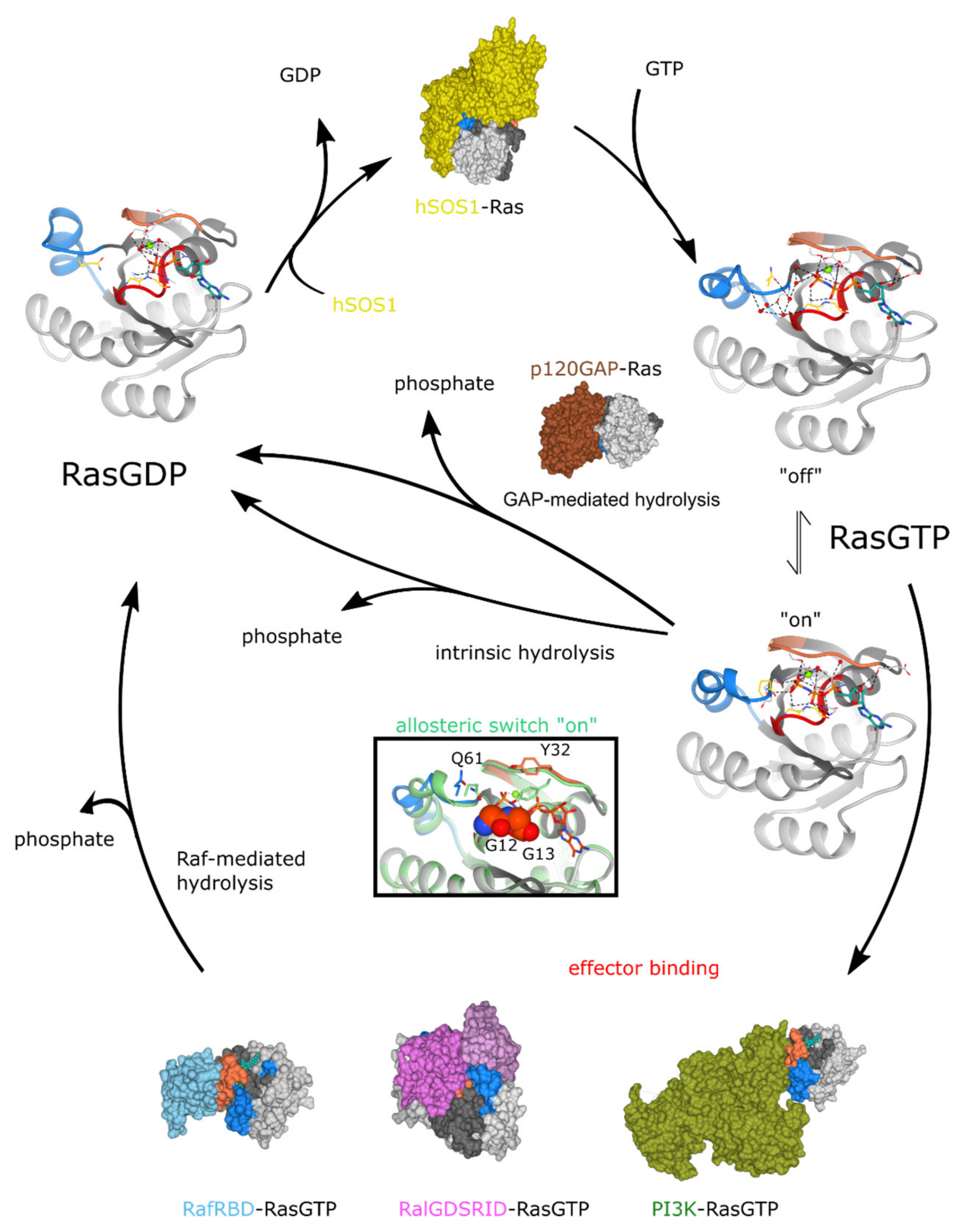
6.2.1. Molecular Issues in Targeting Ras
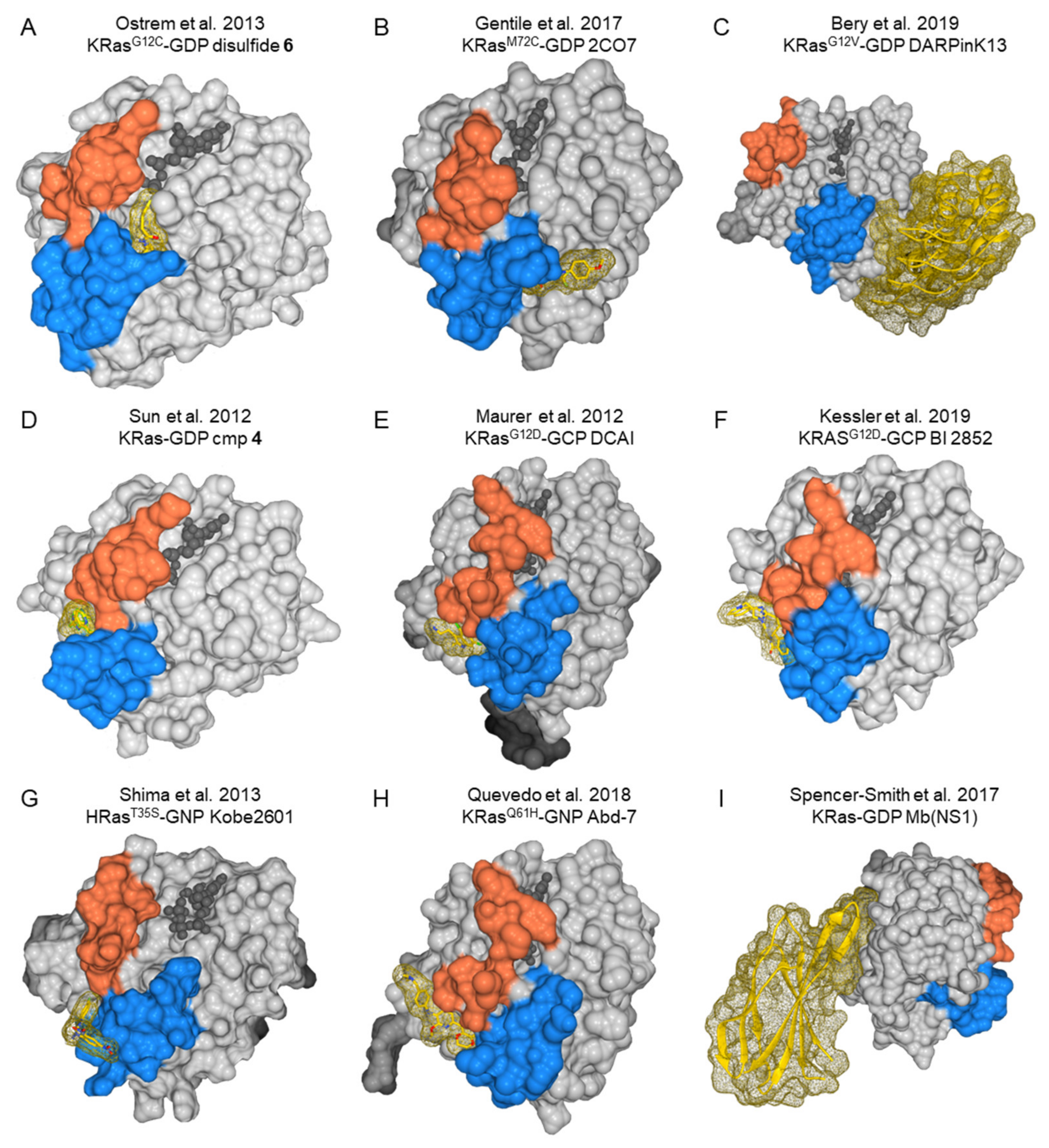
6.2.2. Druggable Pockets in Ras Proteins
7. Natural Products Targeting Biosynthesis, Processing, and Activity of Ras Oncoproteins
7.1. NPs Indirectly Targeting Ras Function
7.1.1. NP Inhibiting Ras Expression
7.1.2. NPs Inhibiting Ras Regulation and Membrane Association
7.1.3. NPs Targeting Ras Processing
7.2. NPs Inhibiting Ras Effectors
7.3. NPs Targeting Ras Activity Directly
8. Conclusion and Perspectives: Natural Products as a Source of Selective Inhibitors of a Ras Oncoproteins
- (i)
- (ii)
- the simultaneous development of experimental and computational approaches for their high-throughput screening (HTS) on targets of clinical relevance;
- (iii)
- the availability of virtual screening allowing to identify the structurally most promising compounds for a target of interest, thereby reducing the research costs.
- (iv)
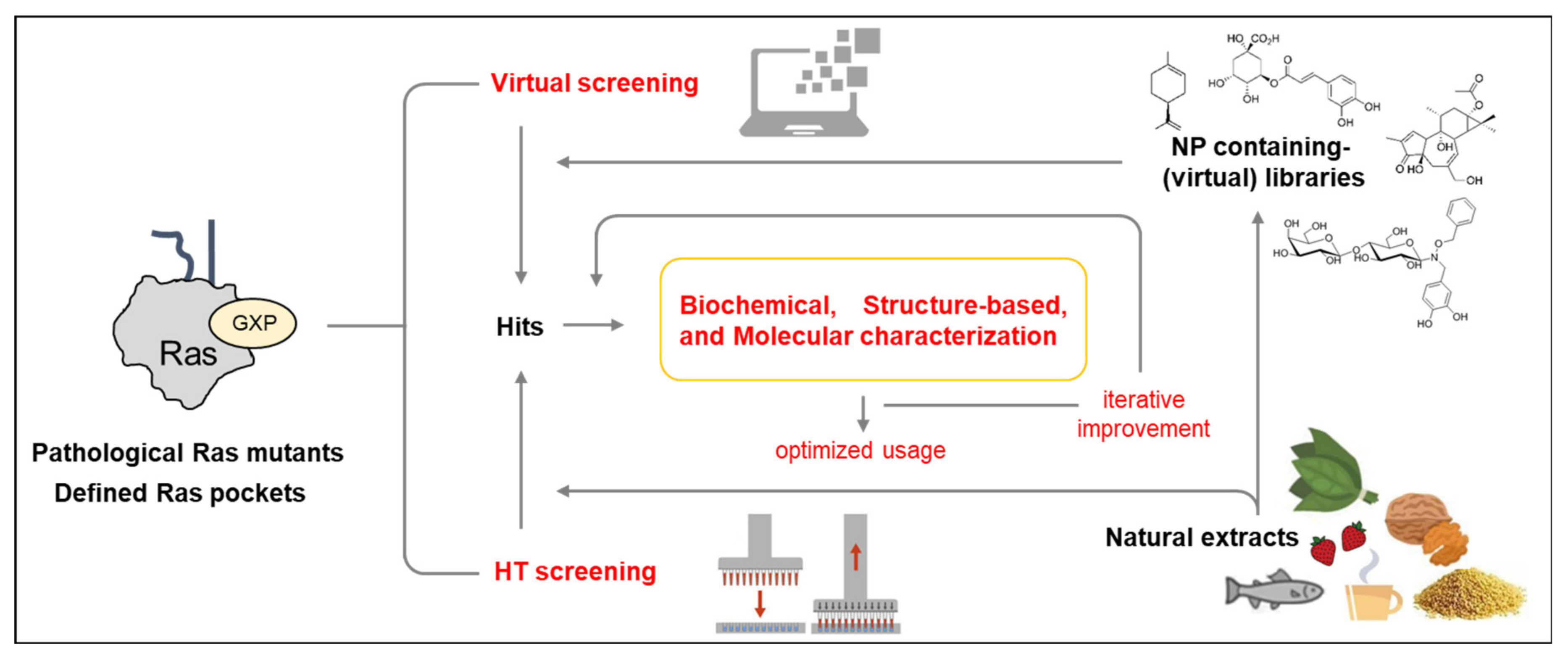
- Both structure-based virtual screenings and HTS approaches with Ras oncoproteins as targets will now be able to take advantage of the newly discovered druggable pockets available in specific oncogenic Ras isoforms and mutant proteins to isolate, characterize, and iteratively improve Ras-specific inhibitors (Figure 5).
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmüller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadian, M.R.; Stege, P.; Scheffzek, K.; Wittinghofer, A. Confirmation of the arginine-finger hypothesis for the GAP-stimulated GTP-hydrolysis reaction of Ras. Nat. Struct. Biol. 1997, 4, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical Elements in the Control of Small G Proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.J. Ras effectors. Curr. Opin. Cell Biol. 1996, 8, 197–204. [Google Scholar] [CrossRef]
- Nakhaeizadeh, H.; Amin, E.; Nakhaei-Rad, S.; Dvorsky, R.; Ahmadian, M.R. The RAS-effector interface: Isoform-specific differences in the effector binding regions. PLoS ONE 2016, 11, e0167145. [Google Scholar] [CrossRef]
- Omerovic, J.; Laude, A.J.; Prior, I.A. Ras proteins: Paradigms for compartmentalised and isoform-specific signalling. Cell. Mol. Life Sci. 2007, 64, 2575–2589. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Tsai, C.J.; Chakrabarti, M.; Jang, H. A new view of ras isoforms in cancers. Cancer Res. 2016, 76, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Gorfe, A.A.; Grant, B.J.; McCammon, J.A. Mapping the Nucleotide and Isoform-Dependent Structural and Dynamical Features of Ras Proteins. Structure 2008, 16, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahearn, I.; Zhou, M.; Philips, M.R. Posttranslational Modifications of RAS Proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a031484. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J.; Philips, M.R. Targeting RAS membrane association: Back to the future for anti-RAS drug discovery? Clin. Cancer Res. 2015, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, J.F.; Magee, A.I.; Childs, J.E.; Marshall, C.J. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell 1989, 57, 1167–1177. [Google Scholar] [CrossRef]
- Chavan, T.S.; Muratcioglu, S.; Marszalek, R.; Jang, H.; Keskin, O.; Gursoy, A.; Nussinov, R.; Gaponenko, V. Plasma membrane regulates Ras signaling networks. Cell. Logist. 2015, 5, e1136374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inouye, K.; Mizutani, S.; Koide, H.; Kaziro, Y. Formation of the Ras dimer is essential for Raf-1 activation. J. Biol. Chem. 2000, 275, 3737–3740. [Google Scholar] [CrossRef] [Green Version]
- Muratcioglu, S.; Chavan, T.S.; Freed, B.C.; Jang, H.; Khavrutskii, L.; Natasha Freed, R.; Dyba, M.A.; Stefanisko, K.; Tarasov, S.G.; Gursoy, A.; et al. GTP-Dependent K-Ras Dimerization. Structure 2015, 23, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Ambrogio, C.; Köhler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868. [Google Scholar] [CrossRef]
- Zhou, Y.; Prakash, P.; Gorfe, A.A.; Hancock, J.F. Ras and the Plasma Membrane: A Complicated Relationship. Cold Spring Harb. Perspect. Med. 2018, 8, a031831. [Google Scholar] [CrossRef] [Green Version]
- Sarkar-Banerjee, S.; Sayyed-Ahmad, A.; Prakash, P.; Cho, K.J.; Waxham, M.N.; Hancock, J.F.; Gorfe, A.A. Spatiotemporal Analysis of K-Ras Plasma Membrane Interactions Reveals Multiple High Order Homo-oligomeric Complexes. J. Am. Chem. Soc. 2017, 139, 13466–13475. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Li, L.; Prasad, S.; Koide, A.; Koide, S.; O’Bryan, J.P. Targeting the α4-α5 interface of RAS results in multiple levels of inhibition. Small GTPases 2019, 10, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Fang, Z.; Enomoto, M.; Gasmi-Seabrook, G.; Zheng, L.; Koide, S.; Ikura, M.; Marshall, C.B. Two Distinct Structures of Membrane-Associated Homodimers of GTP- and GDP-Bound KRAS4B Revealed by Paramagnetic Relaxation Enhancement. Angew. Chemie Int Ed Engl. 2020, 59. [Google Scholar] [CrossRef]
- Chiu, V.K.; Bivona, T.; Hach, A.; Sajous, J.B.; Silletti, J.; Wiener, H.; Johnson, R.L.; Cox, A.D.; Philips, M.R. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 2002, 4. [Google Scholar] [CrossRef] [PubMed]
- Omerovic, J.; Prior, I.A. Compartmentalized signalling: Ras proteins and signalling nanoclusters. FEBS J. 2009, 276, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saikumar, P.; Ulsh, L.S.; Clanton, D.J.; Huang, K.P. Novel phosphorylation of c-ras p21 by protein kinases. Oncogene Res. 1988, 3, 213–222. [Google Scholar] [PubMed]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef]
- Sung, P.J.; Tsai, F.D.; Vais, H.; Court, H.; Yang, J.; Fehrenbacher, N.; Foskett, J.K.; Philips, M.R. Phosphorylated K-Ras limits cell survival by blocking Bcl-xL sensitization of inositol trisphosphate receptors. Proc. Natl. Acad. Sci. USA 2013, 110, 20593–20598. [Google Scholar] [CrossRef] [Green Version]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M.; et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef]
- Abdelkarim, H.; Banerjee, A.; Grudzien, P.; Leschinsky, N.; Abushaer, M.; Gaponenko, V. The hypervariable region of k-ras4b governs molecular recognition and function. Int. J. Mol. Sci. 2019, 20, 5718. [Google Scholar] [CrossRef] [Green Version]
- Buday, L.; Downward, J. Many faces of Ras activation. Biochim. Biophys. Acta Rev. Cancer 2008, 1786, 178–187. [Google Scholar] [CrossRef]
- Boguski, M.S.; McCormick, F. Proteins regulating Ras and its relatives. Nature 1993, 366, 643–654. [Google Scholar] [CrossRef]
- Rojas, J.M.; Oliva, J.L.; Santos, E. Mammalian son of sevenless guanine nucleotide exchange factors: Old concepts and new perspectives. Genes Cancer 2011, 2, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Zhao, C.; Du, G.; Skowronek, K.; Frohman, M.A.; Bar-Sagi, D. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 2007, 9, 707–712. [Google Scholar] [CrossRef]
- Sacco, E.; Farina, M.; Greco, C.; Lamperti, S.; Busti, S.; DeGioia, L.; Alberghina, L.; Liberati, D.; Vanoni, M. Regulation of hSos1 activity is a system-level property generated by its multi-domain structure. Biotechnol. Adv. 2012, 30, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Gureasko, J.; Kuchment, O.; Makino, D.L.; Sondermann, H.; Bar-Sagi, D.; Kuriyan, J. Role of the histone domain in the autoinhibition and activation of the Ras activator Son of Sevenless. Proc. Natl. Acad. Sci. USA 2010, 107, 3430–3435. [Google Scholar] [CrossRef] [Green Version]
- Gureasko, J.; Galush, W.J.; Boykevisch, S.; Sondermann, H.; Bar-Sagi, D.; Groves, J.T.; Kuriyan, J. Membrane-dependent signal integration by the Ras activator Son of sevenless. Nat. Struct. Mol. Biol. 2008, 15, 452. [Google Scholar] [CrossRef]
- Yadav, K.K.; Bar-Sagi, D. Allosteric gating of Son of sevenless activity by the histone domain. Proc. Natl. Acad. Sci. USA 2010, 107, 3436–3440. [Google Scholar] [CrossRef] [Green Version]
- Margarit, S.M.; Sondermann, H.; Hall, B.E.; Nagar, B.; Hoelz, A.; Pirruccello, M.; Bar-Sagi, D.; Kuriyan, J. Structural evidence for feedback activation by Ras·GTP of the Ras-specific nucleotide exchange factor SOS. Cell 2003, 112, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Sondermann, H.; Soisson, S.M.; Boykevisch, S.; Yang, S.S.; Bar-Sagi, D.; Kuriyan, J. Structural analysis of autoinhibition in the Ras activator son of sevenless. Cell 2004, 119, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Medarde, A.; Santos, E. The RasGrf family of mammalian guanine nucleotide exchange factors. Biochim. Biophys. Acta Rev. Cancer 2011, 1815, 170–188. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.C. Regulation and function of the rasGRP family of ras activators in blood cells. Genes Cancer 2011, 2, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Kelley, G.G.; Reks, S.E.; Smrcka, A.V. Hormonal regulation of phospholipase Cε through distinct and overlapping pathways involving G12 and Ras family G-proteins. Biochem. J. 2004, 378. [Google Scholar] [CrossRef] [PubMed]
- Mitin, N.; Rossman, K.L.; Der, C.J. Signaling interplay in ras superfamily function. Curr. Biol. 2005, 15, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Maertens, O.; Cichowski, K. An expanding role for RAS GTPase activating proteins (RAS GAPs) in cancer. Adv. Biol. Regul. 2014, 55, 1–14. [Google Scholar] [CrossRef]
- Cichowski, K.; Jacks, T. NF1 Tumor Suppressor Gene Function. Cell 2001, 104, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Wortzel, I.; Seger, R. The ERK cascade: Distinct functions within various subcellular organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Desideri, E.; Cavallo, A.L.; Baccarini, M. Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell 2015, 161, 967–970. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, H.; Therrien, M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef]
- Krygowska, A.A.; Castellano, E. PI3K: A crucial piece in the RAS signaling puzzle. Cold Spring Harb. Perspect. Med. 2018, 8, a031450. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Does Ras Activate Raf and PI3K Allosterically? Front. Oncol. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Neel, N.F.; Martin, T.D.; Stratford, J.K.; Zand, T.P.; Reiner, D.J.; Der, C.J. The RalGEF-ral effector signaling network: The road less traveled for anti-ras drug discovery. Genes Cancer 2011, 2, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Boettner, B.; Van Aelst, L. The RASputin effect. Genes Dev. 2002, 16, 2033–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, L.A. Ral-GTPases: Approaching their 15 minutes of fame. Trends Cell Biol. 2003, 13, 419–425. [Google Scholar] [CrossRef]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [Green Version]
- Symonds, J.M.; Ohm, A.M.; Carter, C.J.; Heasley, L.E.; Boyle, T.A.; Franklin, W.A.; Reyland, M.E. Protein kinase C δ is a downstream effector of oncogenic K-ras in lung tumors. Cancer Res. 2011, 71, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Meco, M.T.; Lozano, J.; Municio, M.M.; Berra, E.; Frutos, S.; Sanz, L.; Moscat, J. Evidence for the in vitro and in vivo interaction of Ras with protein kinase C zeta. J. Biol. Chem. 1994, 269, 31706–31710. [Google Scholar] [PubMed]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras Promotes Tumorigenicity through Suppression of Non-canonical Wnt Signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Mlodzik, M. Wnt-Frizzled/Planar Cell Polarity Signaling: Cellular Orientation by Facing the Wind (Wnt). Annu. Rev. Cell Dev. Biol. 2015, 31, 623–646. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Wong, D.; Dhaka, A.; Afar, D.; White, M.; Xie, W.; Herschman, H.; Witte, O.; Colicelli, J. Protein binding and signaling properties of RIN1 suggest a unique effector function. Proc. Natl. Acad. Sci. USA 1997, 94, 4954–4959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Bunney, T.D.; Harris, R.; Gandarillas, N.L.; Josephs, M.B.; Roe, S.M.; Sorli, S.C.; Paterson, H.F.; Rodrigues-Lima, F.; Esposito, D.; Ponting, C.P.; et al. Structural and mechanistic insights into ras association domains of phospholipase C epsilon. Mol. Cell 2006, 21, 495–507. [Google Scholar] [CrossRef]
- Dammann, R.; Li, C.; Yoon, J.H.; Chin, P.L.; Bates, S.; Pfeifer, G.P. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000, 25, 315–319. [Google Scholar] [CrossRef]
- Burbee, D.G.; Forgacs, E.; Zöchbauer-Müller, S.; Shivakumar, L.; Fong, K.; Gao, B.; Randle, D.; Kondo, M.; Virmani, A.; Bader, S.; et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J. Natl. Cancer Inst. 2001, 93, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Donninger, H.; Vos, M.D.; Clark, G.J. The RASSF1A tumor suppressor. J. Cell Sci. 2007, 120, 3163–3172. [Google Scholar] [CrossRef] [Green Version]
- Donninger, H.; Schmidt, M.L.; Mezzanotte, J.; Barnoud, T.; Clark, G.J. Ras signaling through RASSF proteins. Semin. Cell Dev. Biol. 2016, 58, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Boettner, B.; Herrmann, C.; Van Aelst, L. Ras and Rap1 interaction with AF-6 effector target. Methods Enzymol. 2001, 332, 151–168. [Google Scholar] [CrossRef]
- Kuriyama, M.; Harada, N.; Kuroda, S.; Yamamoto, T.; Nakafuku, M.; Iwamatsu, A.; Yamamoto, D.; Prasad, R.; Croce, C.; Canaani, E.; et al. Identification of AF-6 and Canoe as putative targets for Ras. J. Biol. Chem. 1996, 271, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Matheny, S.A.; Chen, C.; Kortum, R.L.; Razidlo, G.L.; Lewis, R.E.; White, M.A. Ras regulates assembly of mitogenic signalling complexes through the effector protein IMP. Nature 2004, 427, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.R.H.; Clark, G.J. Pumping the brakes on RAS-negative regulators and death effectors of RAS. J. Cell Sci. 2020, 133, jcs238865. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Duan, F.; Lu, H.; Abdulkadhim Dragh, M.; Xia, Y.; Liang, H.; Hong, L. UBIAD1 suppresses the proliferation of bladder carcinoma cells by regulating H-Ras intracellular trafficking via interaction with the C-terminal domain of H-Ras. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.H.; et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Daniels, M.A.; Teixeiro, E.; Gill, J.; Hausmann, B.; Roubaty, D.; Holmberg, K.; Werlen, G.; Holländer, G.A.; Gascoigne, N.R.J.; Palmer, E. Thymic selection threshold defined by compartmentalization of Ras/MAPK signalling. Nature 2006, 444, 724–729. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Sabatier, C.; McCormick, F. Signaling Specificity by Ras Family GTPases Is Determined by the Full Spectrum of Effectors They Regulate. Mol. Cell. Biol. 2004, 24, 4943–4954. [Google Scholar] [CrossRef] [Green Version]
- Bentires-Alj, M.; Kontaridis, M.I.; Neel, B.G. Stops along the RAS pathway in human genetic disease. Nat. Med. 2006, 12, 283–285. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Rauen, K.A. Pathogenetics of the RASopathies. Hum. Mol. Genet. 2016, 25, R123–R132. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A comparative analysis of individual ras mutations in cancer biology. Front. Oncol. 2019, 9, 1088. [Google Scholar] [CrossRef] [Green Version]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.J.; Neel, B.G.; Ikura, M. NMR-based functional profiling of RASopathies and oncogenic RAS mutations. Proc. Natl. Acad. Sci. USA 2013, 110, 4574–4579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmioli, A.; Sacco, E.; Airoldi, C.; Di Nicolantonio, F.; D’Urzo, A.; Shirasawa, S.; Sasazuki, T.; Di Domizio, A.; De Gioia, L.; Martegani, E.; et al. Selective cytotoxicity of a bicyclic Ras inhibitor in cancer cells expressing K-RasG13D. Biochem. Biophys. Res. Commun. 2009, 386, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.W.; Lin, Y.J.; Reid, D.; Parker, J.; Pavlopoulos, S.; Dischinger, P.; Graveel, C.; Aguirre, A.J.; Steensma, M.; Haigis, K.M.; et al. Isoform-Specific Destabilization of the Active Site Reveals a Molecular Mechanism of Intrinsic Activation of KRas G13D. Cell Rep. 2019, 28, 1538–1550.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaradonna, F.; Sacco, E.; Manzoni, R.; Giorgio, M.; Vanoni, M.; Alberghina, L. Ras-dependent carbon metabolism and transformation in mouse fibroblasts. Oncogene 2006, 25, 5391–5404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Deberardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [Green Version]
- De Sanctis, G.; Spinelli, M.; Vanoni, M.; Sacco, E. K-ras activation induces differential sensitivity to sulfur amino acid limitation and deprivation and to oxidative and anti-oxidative stress in mouse fibroblasts. PLoS ONE 2016, 11, e0163790. [Google Scholar] [CrossRef] [Green Version]
- Baracca, A.; Chiaradonna, F.; Sgarbi, G.; Solaini, G.; Alberghina, L.; Lenaz, G. Mitochondrial Complex I decrease is responsible for bioenergetic dysfunction in K-ras transformed cells. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 314–323. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Gorfe, A.A.; Cho, K.J. Approaches to inhibiting oncogenic K-Ras. Small GTPases 2019, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, D.; Li, X.; He, X.; Zhang, H.; Zhang, J.; Lu, S. Drugging K-RasG12C through covalent inhibitors: Mission possible? Pharmacol. Ther. 2019, 202, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hillig, R.C.; Sautier, B.; Schroeder, J.; Moosmayer, D.; Hilpmann, A.; Stegmann, C.M.; Werbeck, N.D.; Briem, H.; Boemer, U.; Weiske, J.; et al. Discovery of potent SOS1 inhibitors that block RAS activation via disruption of the RAS–SOS1 interaction. Proc. Natl. Acad. Sci. USA 2019, 116, 2551–2560. [Google Scholar] [CrossRef] [Green Version]
- Hocker, H.J.; Cho, K.J.; Chen, C.Y.K.; Rambahal, N.; Sagineedu, S.R.; Shaari, K.; Stanslas, J.; Hancock, J.F.; Gorfe, A.A. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proc. Natl. Acad. Sci. USA 2013, 110, 10201–10206. [Google Scholar] [CrossRef] [Green Version]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011, 7, 585–587. [Google Scholar] [CrossRef] [Green Version]
- Evelyn, C.R.; Duan, X.; Biesiada, J.; Seibel, W.L.; Meller, J.; Zheng, Y. Rational Design of Small Molecule Inhibitors Targeting the Ras GEF, SOS1. Chem. Biol. 2014, 21, 1618–1628. [Google Scholar] [CrossRef] [Green Version]
- Gray, J.L.; von Delft, F.; Brennan, P.E. Targeting the Small GTPase Superfamily through Their Regulatory Proteins. Angew. Chemie Int. Ed. 2020, 59, 6342–6366. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Kambayashi, Y.; Ohuchi, K.; Muto, Y.; Aiba, S. Treatment of advanced melanoma: Past, present and future. Life 2020, 10, 208. [Google Scholar] [CrossRef]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Liu, H.; Nazmun, N.; Hassan, S.; Liu, X.; Yang, J. BRAF mutation and its inhibitors in sarcoma treatment. Cancer Med. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Iida, M.; Harari, P.M.; Wheeler, D.L.; Toulany, M. Targeting AKT/PKB to improve treatment outcomes for solid tumors. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2020, 819–820, 111690. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Wang, J.; Chang, S.; Liu, M.; Pang, X. The greedy nature of mutant RAS: A boon for drug discovery targeting cancer metabolism? Acta Biochim. Biophys. Sin. 2015, 48, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downward, J. RAS synthetic lethal screens revisited: Still seeking the elusive prize? Clin. Cancer Res. 2015, 21. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 2017, 168, 890–903. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A Genome-wide RNAi Screen Identifies Multiple Synthetic Lethal Interactions with the Ras Oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Melnik, B.C.; John, S.M.; Carrera-Bastos, P.; Schmitz, G. MicroRNA-21-enriched exosomes as epigenetic regulators in melanomagenesis and melanoma progression: The impact of western lifestyle factors. Cancers 2020, 12, 2111. [Google Scholar] [CrossRef]
- Tokumaru, Y.; Takabe, K.; Yoshida, K.; Akao, Y. Effects of MIR143 on rat sarcoma signaling networks in solid tumors: A brief overview. Cancer Sci. 2020, 111, 1076. [Google Scholar] [CrossRef]
- Baranyi, M.; Buday, L.; Hegedűs, B. K-Ras prenylation as a potential anticancer target. Cancer Metastasis Rev. 2020. [Google Scholar] [CrossRef]
- Gajewski, T.F.; Salama, A.K.S.; Niedzwiecki, D.; Johnson, J.; Linette, G.; Bucher, C.; Blaskovich, M.A.; Sebti, S.M.; Haluska, F. Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104). J. Transl. Med. 2012, 10, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luger, S.; Wang, V.X.; Paietta, E.; Ketterling, R.P.; Rybka, W.; Lazarus, H.M.; Litzow, M.R.; Rowe, J.M.; Larson, R.A.; Appelbaum, F.R.; et al. Tipifarnib As Maintenance Therapy in Acute Myeloid Leukemia (AML) Improves Survival in a Subgroup of Patients with High Risk Disease. Results of the Phase III Intergroup Trial E2902. Blood 2015, 126, 1308. [Google Scholar] [CrossRef]
- Adjei, A.A.; Mauer, A.; Bruzek, L.; Marks, R.S.; Hillman, S.; Geyer, S.; Hanson, L.J.; Wright, J.J.; Erlichman, C.; Kaufmann, S.H.; et al. Phase II study of the farnesyl transferase inhibitor R115777 in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Karasic, T.B.; Chiorean, E.G.; Sebti, S.M.; O’Dwyer, P.J. A Phase I Study of GGTI-2418 (Geranylgeranyl Transferase I Inhibitor) in Patients with Advanced Solid Tumors. Target. Oncol. 2019, 14, 613–618. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Burns, M.C.; Howes, J.E.; Sun, Q.; Little, A.J.; Camper, D.M.V.; Abbott, J.R.; Phan, J.; Lee, T.; Waterson, A.G.; Rossanese, O.W.; et al. High-throughput screening identifies small molecules that bind to the RAS:SOS:RAS complex and perturb RAS signaling. Anal. Biochem. 2018, 548, 44–52. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Palmioli, A.; Sacco, E.; Abraham, S.; Thomas, C.J.; Di Domizio, A.; De Gioia, L.; Gaponenko, V.; Vanoni, M.; Peri, F. First experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand. Bioorganic Med. Chem. Lett. 2009, 19, 4217–4222. [Google Scholar] [CrossRef]
- Sacco, E.; Spinelli, M.; Vanoni, M. Approaches to Ras signaling modulation and treatment of Ras-dependent disorders: A patent review (2007 present). Expert Opin. Ther. Pat. 2012, 22, 1263–1287. [Google Scholar] [CrossRef]
- Schöpel, M.; Jockers, K.F.G.; Düppe, P.M.; Autzen, J.; Potheraveedu, V.N.; Ince, S.; Yip, K.T.; Heumann, R.; Herrmann, C.; Scherkenbeck, J.; et al. Bisphenol a binds to Ras proteins and competes with guanine nucleotide exchange: Implications for GTPase-selective antagonists. J. Med. Chem. 2013, 56, 9664–9672. [Google Scholar] [CrossRef]
- Maurer, T.; Garrenton, L.S.; Oh, A.; Pitts, K.; Anderson, D.J.; Skelton, N.J.; Fauber, B.P.; Pan, B.; Malek, S.; Stokoe, D.; et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc. Natl. Acad. Sci. USA 2012, 109, 5299–5304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Burke, J.P.; Phan, J.; Burns, M.C.; Olejniczak, E.T.; Waterson, A.G.; Lee, T.; Rossanese, O.W.; Fesik, S.W. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew. Chemie Int. Ed. 2012, 51, 6140–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, S.; Palmioli, A.; Airoldi, C.; Tisi, R.; Fantinato, S.; Olivieri, S.; De Gioia, L.; Martegani, E.; Peri, F. Structure-Activity Studies on Arylamides and Arysulfonamides Ras Inhibitors. Curr. Cancer Drug Targets 2010, 10, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Colombo, S.; Peri, F.; Tisi, R.; Nicotra, F.; Martegani, E. Design and characterization of a new class of inhibitors of ras activation. Ann. N. Y. Acad. Sci. 2004, 1030, 52–61. [Google Scholar] [CrossRef]
- Ganguly, A.K.; Sen Wang, Y.; Pramanik, B.N.; Doll, R.J.; Snow, M.E.; Taveras, A.G.; Remiszewski, S.; Cesarz, D.; Del Rosario, J.; Vibulbhan, B.; et al. Interaction of a novel GDP exchange inhibitor with the Ras protein. Biochemistry 1998, 37, 15631–15637. [Google Scholar] [CrossRef]
- Sacco, E.; Abraham, S.J.; Palmioli, A.; Damore, G.; Bargna, A.; Mazzoleni, E.; Gaponenko, V.; Vanoni, M.; Peri, F. Binding properties and biological characterization of new sugar-derived Ras ligands. MedChemComm 2011, 2, 396–401. [Google Scholar] [CrossRef]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [Green Version]
- Quevedo, C.E.; Cruz-Migoni, A.; Bery, N.; Miller, A.; Tanaka, T.; Petch, D.; Bataille, C.J.R.; Lee, L.Y.W.; Fallon, P.S.; Tulmin, H.; et al. Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [Green Version]
- Pálfy, G.; Menyhárd, D.K.; Perczel, A. Dynamically encoded reactivity of Ras enzymes: Opening new frontiers for drug discovery. Cancer Metastasis Rev. 2020. [Google Scholar] [CrossRef]
- Buhrman, G.; Holzapfel, G.; Fetics, S.; Mattos, C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 4931–4936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargi, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Shima, F.; Muraoka, S.; Liao, J.; Okamoto, H.; Yamamoto, M.; Tamura, A.; Yagi, N.; Ueki, T.; Kataoka, T. Crystal structure of M-Ras reveals a GTP-bound “off” state conformation of Ras family small GTPases. J. Biol. Chem. 2005, 280, 31267–31275. [Google Scholar] [CrossRef] [Green Version]
- Kalbitzer, H.R.; Spoerner, M.; Ganser, P.; Hozsa, C.; Kremer, W. Fundamental link between folding states and functional states of proteins. J. Am. Chem. Soc. 2009, 131. [Google Scholar] [CrossRef]
- Muraoka, S.; Shima, F.; Araki, M.; Inoue, T.; Yoshimoto, A.; Ijiri, Y.; Seki, N.; Tamura, A.; Kumasaka, T.; Yamamoto, M.; et al. Crystal structures of the state 1 conformations of the GTP-bound H-Ras protein and its oncogenic G12V and Q61L mutants. FEBS Lett. 2012, 586. [Google Scholar] [CrossRef]
- Gentile, D.R.; Rathinaswamy, M.K.; Jenkins, M.L.; Moss, S.M.; Siempelkamp, B.D.; Renslo, A.R.; Burke, J.E.; Shokat, K.M. Ras Binder Induces a Modified Switch-II Pocket in GTP and GDP States. Cell Chem. Biol. 2017, 24, 1455–1466.e14. [Google Scholar] [CrossRef]
- Palmioli, A.; Ciaramelli, C.; Tisi, R.; Spinelli, M.; De Sanctis, G.; Sacco, E.; Airoldi, C. Natural Compounds in Cancer Prevention: Effects of Coffee Extracts and Their Main Polyphenolic Component, 5-O-Caffeoylquinic Acid, on Oncogenic Ras Proteins. Chem. Asian J. 2017, 12, 2457–2466. [Google Scholar] [CrossRef]
- Bery, N.; Legg, S.; Debreczeni, J.; Breed, J.; Embrey, K.; Stubbs, C.; Kolasinska-Zwierz, P.; Barrett, N.; Marwood, R.; Watson, J.; et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer-Smith, R.; Koide, A.; Zhou, Y.; Eguchi, R.R.; Sha, F.; Gajwani, P.; Santana, D.; Gupta, A.; Jacobs, M.; Herrero-Garcia, E.; et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat. Chem. Biol. 2017, 13, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.A.; Kim, J.Y.; Lee, J.Y.; Kang, C.M.; Kwon, H.J.; Yoo, Y.D.; Kim, T.W.; Lee, Y.S.; Lee, S.J. Induction of cell cycle arrest and apoptosis in human breast cancer cells by quercetin. Int. J. Oncol. 2001, 19. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.Q.; Liu, M.; Li, W.; Che, J.P.; Wang, G.C.; Zheng, J.H. Combination of quercetin and hyperoside inhibits prostate cancer cell growth and metastasis via regulation of microRNA-21. Mol. Med. Rep. 2015, 11, 1085–1092. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Guo, Q.; Chen, J.; Chen, Z. Quercetin enhances cisplatin sensitivity of human osteosarcoma cells by modulating microRNA-217-KRAS axis. Mol. Cells 2015, 38, 638. [Google Scholar] [CrossRef] [Green Version]
- Ranelletti, F.O.; Maggiano, N.; Serra, F.G.; Ricci, R.; Larocca, L.M.; Lanza, P.; Scambia, G.; Fattorossi, A.; Capelli, A.; Piantelli, M. Quercetin inhibits p21-ras expression in human colon cancer cell lines and in primary colorectal tumors. Int. J. Cancer 2000, 85, 438–445. [Google Scholar] [CrossRef]
- Garrido, C.M.; Henkels, K.M.; Rehl, K.M.; Liang, H.; Zhou, Y.; Gutterman, J.U.; Cho, K.J. Avicin G is a potent sphingomyelinase inhibitor and blocks oncogenic K- and H-Ras signaling. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and Structure of Bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Kortmansky, J.; Schwartz, G.K. Bryostatin-1: A Novel PKC Inhibitor in Clinical Development. Cancer Investig. 2003, 21, 924–936. [Google Scholar] [CrossRef]
- Raghuvanshi, R.; Bharate, S.B. Preclinical and Clinical Studies on Bryostatins, A Class of Marine-Derived Protein Kinase C Modulators: A Mini-Review. Curr. Top. Med. Chem. 2020, 20, 1124–1135. [Google Scholar] [CrossRef]
- Bharate, S.; Singh, B.; Vishwakarma, R.A. Modulation of k-Ras Signaling by Natural Products. Curr. Med. Chem. 2012, 19, 2273–2291. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Sun, Y.; Zhang, T.; Ming, Y.; Hongwei, G. An overview on natural farnesyltransferase inhibitors for efficient cancer therapy. J. Enzyme Inhib. Med. Chem. 2020, 35, 1027–1044. [Google Scholar] [CrossRef] [PubMed]
- Angamuthu, V.; Shanmugavadivu, M.; Nagarajan, G.; Velmurugan, B.K. Pharmalogical activities of antroquinonol- Mini review. Chem. Biol. Interact. 2019, 297, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Nandi, D. Farnesyltransferase inhibitors reduce Ras activation and ameliorate acetaminophen-induced liver injury in mice. Hepatology 2009, 50, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Nagase, T.; Kawata, S.; Tamura, S.; Matsuda, Y.; Inui, Y.; Yamasaki, E.; Ishiguro, H.; Ito, T.; Miyagawa, J.; Mitsui, H.; et al. Manumycin and gliotoxin derivative KT7595 block Ras farnesylation and cell growth but do not disturb lamin farnesylation and localization in human tumour cells. Br. J. Cancer 1997, 76, 1001–1010. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Akasaka, K.; Akinaga, S.; Okabe, M.; Nakano, H.; Gomez, R.; Wood, D.; Uh, M.; Tamanoi, F. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc. Natl. Acad. Sci. USA 1993, 90, 2281–2285. [Google Scholar] [CrossRef] [Green Version]
- Lantry, L.E.; Zhang, Z.; Crist, K.A.; Wang, Y.; Hara, M.; Zeeck, A.; Lubet, R.A.; You, M. Chemopreventive efficacy of promising farnesyltransferase inhibitors. Exp. Lung Res. 2000, 26, 773–790. [Google Scholar] [CrossRef]
- Kouchi, H.; Nakamura, K.; Fushimi, K.; Sakaguchi, M.; Miyazaki, M.; Ohe, T.; Namba, M. Manumycin A, inhibitor of ras farnesyltransferase, inhibits proliferation and migration of rat vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1999, 264, 915–920. [Google Scholar] [CrossRef]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef]
- Chowdhury, R.; Webber, J.P.; Gurney, M.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015, 6, 715. [Google Scholar] [CrossRef]
- Chen, X.G.; Shuzo, O.; Li, Y.; Han, R. Inhibition of farnesyl protein transferase, h-ras oncogene expression and p21ras membrane association by natural products in human solid tumor cell lines. J. Asian Nat. Prod. Res. 1998, 1, 29–51. [Google Scholar] [CrossRef]
- Singh, S.B.; Zink, D.L.; Liesch, J.M.; Ball, R.G.; Goetz, M.A.; Bolessa, E.A.; Giacobbe, R.A.; Silverman, K.C.; Bills, G.F.; Pelaez, F.; et al. Preussomerins and Deoxypreussomerins: Novel Inhibitors of Ras Farnesyl-Protein Transferase. J. Org. Chem. 1994, 59, 6296–6302. [Google Scholar] [CrossRef]
- Omura, S.; Van Der Pyl, D.; Inokoshi, J.; Takahashi, Y.; Takeshima, H. Pepticinnamins, new farnesyl-protein transferase inhibitors produced by an actinomycete i. producing strain, fermentation, isolation and biological activity. J. Antibiot. 1993, 46, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, J.B.; Pompliano, D.L.; Mosser, S.D.; Rands, E.; Lingham, R.B.; Singh, S.B.; Scolnick, E.M.; Kohl, N.E.; Oliff, A. Selective inhibition of farnesyl-protein transferase blocks Ras processing in vivo. J. Biol. Chem. 1993, 268, 7617–7620. [Google Scholar] [PubMed]
- Nogueira, A.; Vala, H.; Vasconcelos-Nóbrega, C.; Faustino-Rocha, A.I.; Pires, C.A.; Colaço, A.; Oliveira, P.A.; Pires, M.J. Long-term treatment with chaethomellic acid A reduces glomerulosclerosis and arteriolosclerosis in a rat model of chronic kidney disease. Biomed. Pharmacother. 2017, 96, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.H.; Pham, T.H.; Lee, J.; Lee, J.; Ryu, H.W.; Oh, S.R.; Oh, J.W.; Yoon, D.Y. Methyl linderone suppresses TPA-stimulated IL-8 and MMP-9 expression via the ERK/STAT3 pathway in MCF-7 breast cancer cells. J. Microbiol. Biotechnol. 2020, 30, 325–332. [Google Scholar] [CrossRef]
- Costa, S.M.O.; Lemos, T.L.G.; Pessoa, O.D.L.; Pessoa, C.; Montenegro, R.C.; Braz-Filho, R. Chemical constituents from Lippia sidoides and cytotoxic activity. J. Nat. Prod. 2001, 64, 792–795. [Google Scholar] [CrossRef]
- Ho, C.L.; Wang, J.L.; Lee, C.C.; Cheng, H.Y.; Wen, W.C.; Cheng, H.H.Y.; Chen, M.C.M. Antroquinonol blocks Ras and Rho signaling via the inhibition of protein isoprenyltransferase activity in cancer cells. Biomed. Pharmacother. 2014, 68, 1007–1014. [Google Scholar] [CrossRef]
- Lee, S.H.; Lee, M.Y.; Kang, H.M.; Han, D.C.; Son, K.H.; Yang, D.C.; Do Sung, N.; Lee, C.W.; Kim, H.M.; Kwon, B.M. Anti-tumor activity of the farnesyl-protein transferase inhibitors arteminolides, isolated from Artemisa. Bioorganic Med. Chem. 2003, 11, 4545–4549. [Google Scholar] [CrossRef]
- Gbelcová, H.; Rimpelová, S.; Knejzlík, Z.; Šáchová, J.; Kolář, M.; Strnad, H.; Repiská, V.; D’Acunto, W.C.; Ruml, T.; Vítek, L. Isoprenoids responsible for protein prenylation modulate the biological effects of statins on pancreatic cancer cells. Lipids Health Dis. 2017, 16, 250. [Google Scholar] [CrossRef] [Green Version]
- Gelb, M.H.; Tamanoi, F.; Yokoyama, K.; Ghomashchi, F.; Esson, K.; Gould, M.N. The inhibition of protein prenyltransferases by oxygenated metabolites of limonene and perillyl alcohol. Cancer Lett. 1995, 91, 169–175. [Google Scholar] [CrossRef]
- Chaudhary, S.C.; Siddiqui, M.S.; Athar, M.; Alam, M.S. D-Limonene modulates inflammation, oxidative stress and Ras-ERK pathway to inhibit murine skin tumorigenesis. Hum. Exp. Toxicol. 2012, 31, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Afshordel, S.; Kern, B.; Clasohm, J.; König, H.; Priester, M.; Weissenberger, J.; Kögel, D.; Eckert, G.P. Lovastatin and perillyl alcohol inhibit glioma cell invasion, migration, and proliferation—Impact of Ras-/Rho-prenylation. Pharmacol. Res. 2015, 91, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.A.; Baenziger, N.C.; Gloer, J.B. Structure of Preussomerin A: An Unusual New Antifungal Metabolite from the Coprophilous Fungus Preussia isómera. J. Am. Chem. Soc. 1990, 112, 6718–6719. [Google Scholar] [CrossRef]
- Weber, H.A.; Gloer, J.B. The Preussomerins: Novel Antifungal Metabolites from the Coprophilous Fungus Preussia isomera Cain. J. Org. Chem. 1991, 56, 4355–4360. [Google Scholar] [CrossRef]
- Polishook, J.D.; Dombrowski, A.W.; Tsou, N.N.; Salituro, G.M.; Curotto, J.E. Preussomerin D from the Endophyte Hormonema Dematioides. Mycologia 1993, 85, 62–64. [Google Scholar] [CrossRef]
- Weerapreeyakul, N.; Anorach, R.; Khuansawad, T.; Yenjai, C.; Isaka, M. Synthesis of bioreductive esters from fungal compounds. Chem. Pharm. Bull. 2007, 55, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Vigushin, D.M.; Brooke, G.; Willows, D.; Coombes, R.C.; Moody, C.J. Pyrazino[1,2-a]indole-1,4-diones, simple analogues of gliotoxin, as selective inhibitors of geranylgeranyltransferase I. Bioorganic Med. Chem. Lett. 2003, 13, 3661–3663. [Google Scholar] [CrossRef]
- Ōmura, S.; Tomoda, H. Microbial metabolites affecting lipid biosynthesis. Pure Appl. Chem. 1994, 66, 2267–2270. [Google Scholar] [CrossRef] [Green Version]
- Santa Maria, K.C.; Chan, A.N.; O’Neill, E.M.; Li, B. Targeted Rediscovery and Biosynthesis of the Farnesyl-Transferase Inhibitor Pepticinnamin E. ChemBioChem 2019, 20, 1387–1393. [Google Scholar] [CrossRef]
- Thutewohl, M.; Kissau, L.; Popkirova, B.; Karaguni, I.M.; Nowak, T.; Bate, M.; Kuhlmann, J.; Müller, O.; Waldmann, H. Identification of mono- and bisubstrate inhibitors of protein farnesyltransferase and inducers of apoptosis from a pepticinnamin E library. Bioorganic Med. Chem. 2003, 11, 2617–2626. [Google Scholar] [CrossRef]
- Singh, S.B.; Jayasuriya, H.; Silverman, K.C.; Bonfiglio, C.A.; Williamson, J.M.; Lingham, R.B. Efficient syntheses, human and yeast farnesyl-protein transferase inhibitory activities of chaetomellic acids and analogues. Bioorganic Med. Chem. 2000, 8, 571–580. [Google Scholar] [CrossRef]
- Oh, H.M.; Choi, S.K.; Lee, J.M.; Lee, S.K.; Kim, H.Y.; Han, D.C.; Kim, H.M.; Son, K.H.; Kwon, B.M. Cyclopentenediones, inhibitors of farnesyl protein transferase and anti-tumor compounds, isolated from the fruit of Lindera erythrocarpa Makino. Bioorganic Med. Chem. 2005, 13, 6182–6187. [Google Scholar] [CrossRef] [PubMed]
- Cadelis, M.M.; Bourguet-Kondracki, M.L.; Dubois, J.; Valentin, A.; Barker, D.; Copp, B.R. Discovery and preliminary structure-activity relationship studies on tecomaquinone i and tectol as novel farnesyltransferase and plasmodial inhibitors. Bioorganic Med. Chem. 2016, 24, 3102–3107. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.J.; Wang, W.; Huang, S.Y.; Hong, Y.; Li, G.; Lin, S.; Tian, J.; Cai, Z.; Wang, H.M.D.; Ma, D.L.; et al. Inhibition of the Ras/Raf interaction and repression of renal cancer xenografts in vivo by an enantiomeric iridium(III) metal-based compound. Chem. Sci. 2017, 8, 4756–4763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirn, M.J.; Bok, S.H.; Kwon, B.M.; Shin, J.; Seo, Y. Arteminolide, an inhibitor of farnesyl transferase from Artemisia sylvatica. J. Org. Chem. 1998, 63, 7111–7113. [Google Scholar] [CrossRef]
- Lee, S.H.; Seo, Y.; Kim, H.K.; Kang, H.M.; Kim, J.H.; Son, K.H.; Lee, H.; Kwon, B.M.; Shin, J.; Seo, J.M. Arteminolides B, C, and D, new inhibitors of farnesyl protein transferase from Artemisia argyi. J. Org. Chem. 2002, 67, 7670–7675. [Google Scholar] [CrossRef]
- Wen, J.; Shi, H.; Xu, Z.; Chang, H.; Jia, C.; Zan, K.; Jiang, Y.; Tu, P. Dimeric guaianolides and sesquiterpenoids from Artemisia anomala. J. Nat. Prod. 2010, 73, 67–70. [Google Scholar] [CrossRef]
- Lin, H.C.; Lin, M.H.; Liao, J.H.; Wu, T.H.; Lee, T.H.; Mi, F.L.; Wu, C.H.; Chen, K.C.; Cheng, C.H.; Lin, C.W. Antroquinonol, a ubiquinone derivative from the mushroom antrodia camphorata, inhibits colon cancer stem cell-like properties: Insights into the molecular mechanism & inhibitory targets. J. Agric. Food Chem. 2017, 65, 51–59. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Sreekala, C.; Zhang, Z.; Budhraja, A.; Ding, S.; Son, Y.; Wang, X.; Hitron, A.; Hyun-jung, K.; Wang, L.; et al. Cancer Prevention with Promising Natural Products: Mechanisms of Action and Molecular Targets. Anticancer Agents Med Chem. 2012, 12, 1159–1184. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Agnihotri, N. Piperlongumine, a piper alkaloid targets Ras/PI3K/Akt/mTOR signaling axis to inhibit tumor cell growth and proliferation in DMH/DSS induced experimental colon cancer. Biomed. Pharmacother. 2019, 109, 1462–1477. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, L.X.; Ouyang, L.; Cheng, Y.; Liu, B. Plant natural compounds: Targeting pathways of autophagy as anti-cancer therapeutic agents. Cell Prolif. 2012, 45, 466–476. [Google Scholar] [CrossRef]
- Alqathama, A.; Prieto, J.M. Natural products with therapeutic potential in melanoma metastasis. Nat. Prod. Rep. 2015, 32, 1170–1182. [Google Scholar] [CrossRef]
- Ganaie, A.A.; Siddique, H.R.; Sheikh, I.A.; Parray, A.; Wang, L.; Panyam, J.; Villalta, P.W.; Deng, Y.; Konety, B.R.; Saleem, M. A novel terpenoid class for prevention and treatment of KRAS-driven cancers: Comprehensive analysis using in situ, in vitro, and in vivo model systems. Mol. Carcinog. 2020, 59. [Google Scholar] [CrossRef]
- Kim, C.K.; Wang, D.; Bokesch, H.R.; Fuller, R.W.; Smith, E.; Henrich, C.J.; Durrant, D.E.; Morrison, D.K.; Bewley, C.A.; Gustafson, K.R. Swinhopeptolides A and B: Cyclic Depsipeptides from the Sponge Theonella swinhoei That Inhibit Ras/Raf Interaction. J. Nat. Prod. 2020, 83, 1288–1294. [Google Scholar] [CrossRef]
- Sturm, S.; Gil, R.R.; Chai, H.B.; Ngassapa, O.D.; Santisuk, T.; Reutrakul, V.; Howe, A.; Moss, M.; Besterman, J.M.; Yang, S.L.; et al. Lupane derivatives from Lophopetalum wallichii with farnesyl protein transferase inhibitory activity. J. Nat. Prod. 1996, 59, 658–663. [Google Scholar] [CrossRef]
- Thornburg, C.C.; Britt, J.R.; Evans, J.R.; Akee, R.K.; Whitt, J.A.; Trinh, S.K.; Harris, M.J.; Thompson, J.R.; Ewing, T.L.; Shipley, S.M.; et al. NCI Program for Natural Product Discovery: A Publicly-Accessible Library of Natural Product Fractions for High-Throughput Screening. ACS Chem. Biol. 2018, 13, 2484–2497. [Google Scholar] [CrossRef]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.Z.; Xu, X. Use of Natural Products as Chemical Library for Drug Discovery and Network Pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Mechanism of Action | Source | Models | Ref. |
|---|---|---|---|---|---|
Quercetin  | K-, N-, and HRAS | indirect inhibition of expression | red grapes and red wine | colon cancer cells | Zhang et al., 2015 [155] |
Prostratin  | KRas | Inhibition of CaM interaction | mamala tree of Samoa, Homalanthus nutans | pancreatic cancer murine models | Wang et al., 2015 [59] |
| Avicin G | K- and HRas | indirect delocalization | Acacia victoriae | cells expressing mGFP-KRasG12V | Garrido et al., 2020 [157] |
 | |||||
Bryostatin-1  | K-Ras4B | direct delocalization | marine organism Bugula neritina | Bivona et al., 2006 [26] | |
| Compound | Source | Models | Ref. |
|---|---|---|---|
| Manumycin A | Streptomyces parvulus | castration-resistant prostate cancer (CRPC) C4-2B | Datta et al., 2017 [169] |
 | |||
D-Limonene  | orange peels; other plants essential oils | W 1-38. CACW, A549 and PaCa cells | Chen et al., 1998 [171] |
Preussomerin G  | Preussia isomera and Harmonema dematioides | BC-1 and NCI-H187 cells | Singh et al., 1994 [172] |
Gliotoxin  | Aspergillus, Trichoderma, and Penicillium | human colon carcinoma (LoVo) cells | Nagase et al., 1997 and Saha et al., 2009 [164,165] |
Pepticinnamin E  | Actinobacteria bacterium | Kidney Vero cells | Omura et al., 1993 [173] |
Chaetomellic acid A  | Chaetomella acutiseta | murine model of renal fibrosis | Gibbs et al. 1993; Nogueira et al. 2017 [174,175] |
Methyl linderone  | fruits of Lindera erythrocarpa | human breast cancer cells MCF-7 | Yoon et al., 2020 [176] |
Tectol  | Brazilian Lippia sidoides | HL60 (human promyelocytic leukemia) and CEM (human acute lymphoblastic leukemia) | Costa et al., 2001 [177] |
Antroquinonol  | Antrodia camphorata | Human lung cancer (A549 and H838), liver cancer (HepG2 and Hep3B), and leukemia (K562 and THP-1) cells | Ho et al., 2014 [178] |
Artemidolide C  | Artemisia spp. | SW620 (colon), MDA-MB-231 (breast), HCT116 (colon), and MCF7 (breast) | Lee et al., 2003 [179] |
Statins (lovastatin, simvastatin)  | Aspergillus terreus | pancreatic cancer cells | Gbelcová et al., 2017 [180] |
| Compound | Target | Mechanism of Action | Source | Models | Ref. |
|---|---|---|---|---|---|
5CQA, 5-O-caffeoylquinic acid  | HRas | direct inhibition of nucleotide exchange and Raf1 binding | coffee | KRasG13D breast cancer cells | Palmioli et al., 2017 [150] |
Lupeol  | KRas | direct inhibition of nucleotide exchange | many edible fruits and vegetables | human and murine KRAS-driven cancer models | Ganaie et al., 2020 [207] |
Swinhopeptolides  | unknown | direct inhibition of Raf1 binding | Papua New Guinea marine sponge Theonella swinhoei cf. verrucosa | unknown | Kim et al., 2020 [208] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tisi, R.; Gaponenko, V.; Vanoni, M.; Sacco, E. Natural Products Attenuating Biosynthesis, Processing, and Activity of Ras Oncoproteins: State of the Art and Future Perspectives. Biomolecules 2020, 10, 1535. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10111535
Tisi R, Gaponenko V, Vanoni M, Sacco E. Natural Products Attenuating Biosynthesis, Processing, and Activity of Ras Oncoproteins: State of the Art and Future Perspectives. Biomolecules. 2020; 10(11):1535. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10111535
Chicago/Turabian StyleTisi, Renata, Vadim Gaponenko, Marco Vanoni, and Elena Sacco. 2020. "Natural Products Attenuating Biosynthesis, Processing, and Activity of Ras Oncoproteins: State of the Art and Future Perspectives" Biomolecules 10, no. 11: 1535. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10111535