New Lipidyl-Cyclodextrins Obtained by Ring Opening of Methyl Oleate Epoxide Using Ball Milling

 , , , , , and
, , , , , and
Abstract
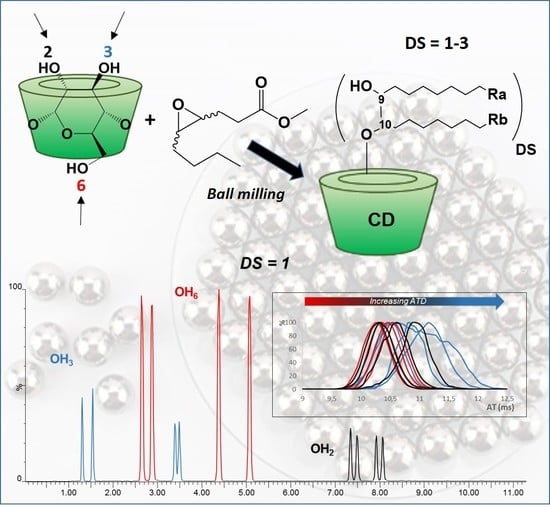
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Flash Chromatography
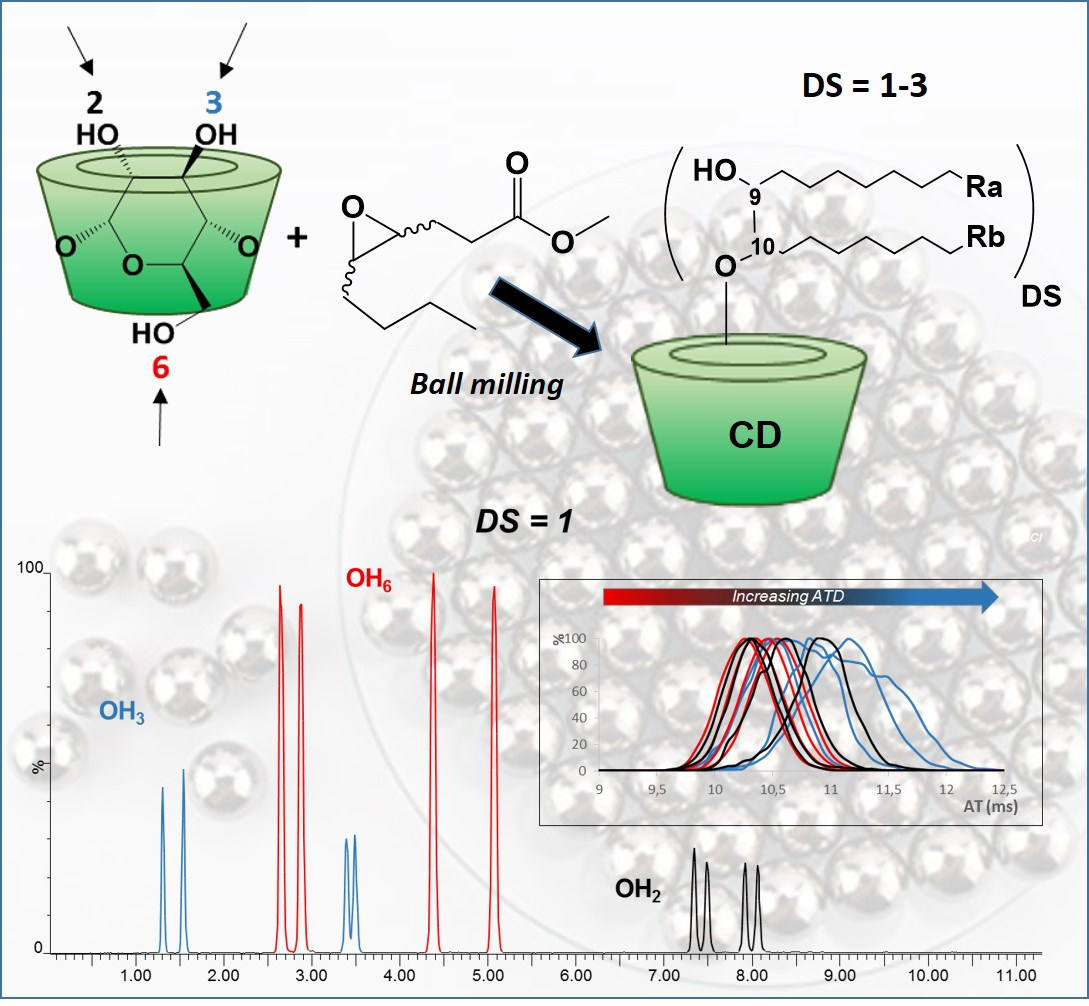
2.3. Ball Milling
2.4. Mass Spectrometry Analysis
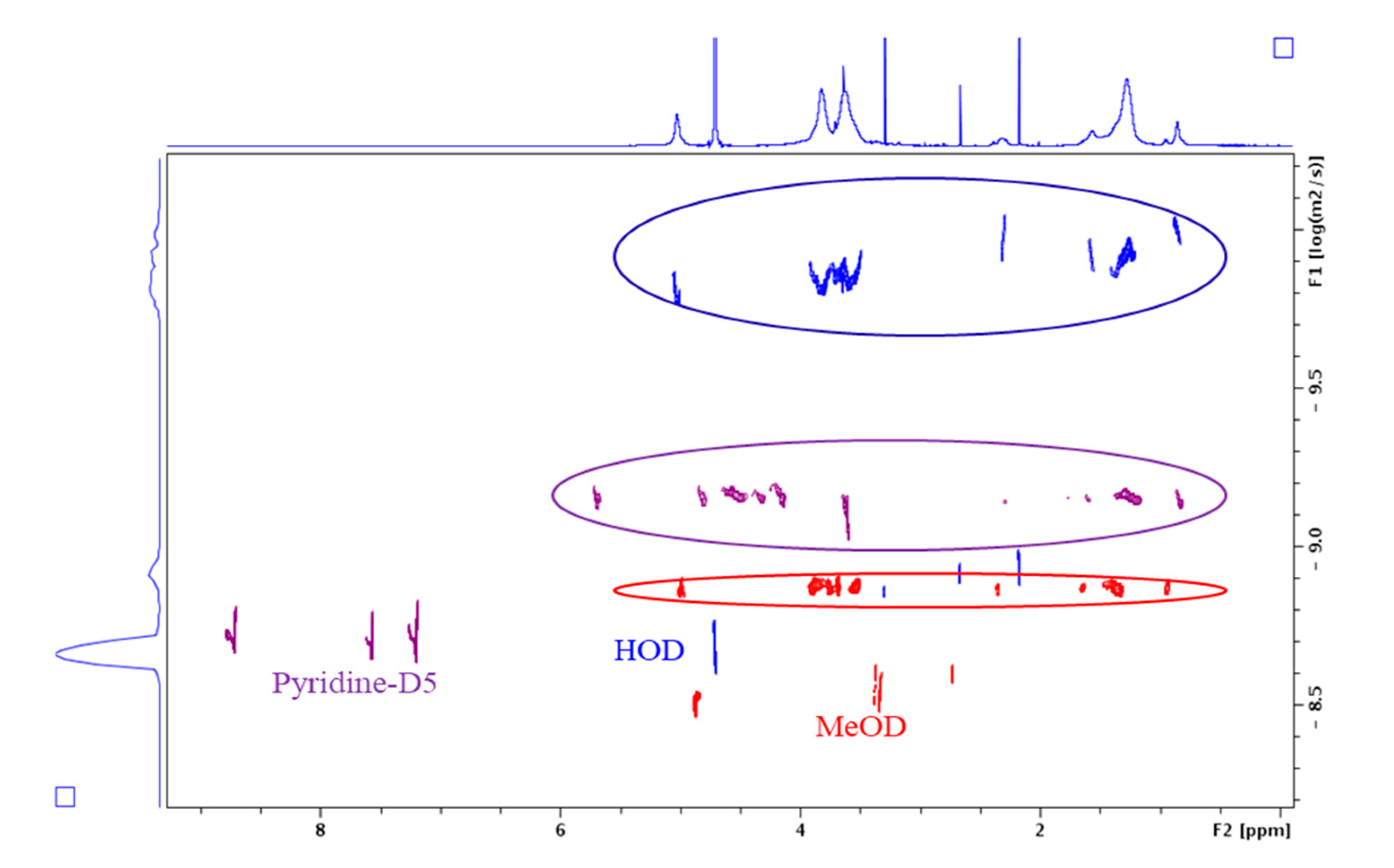
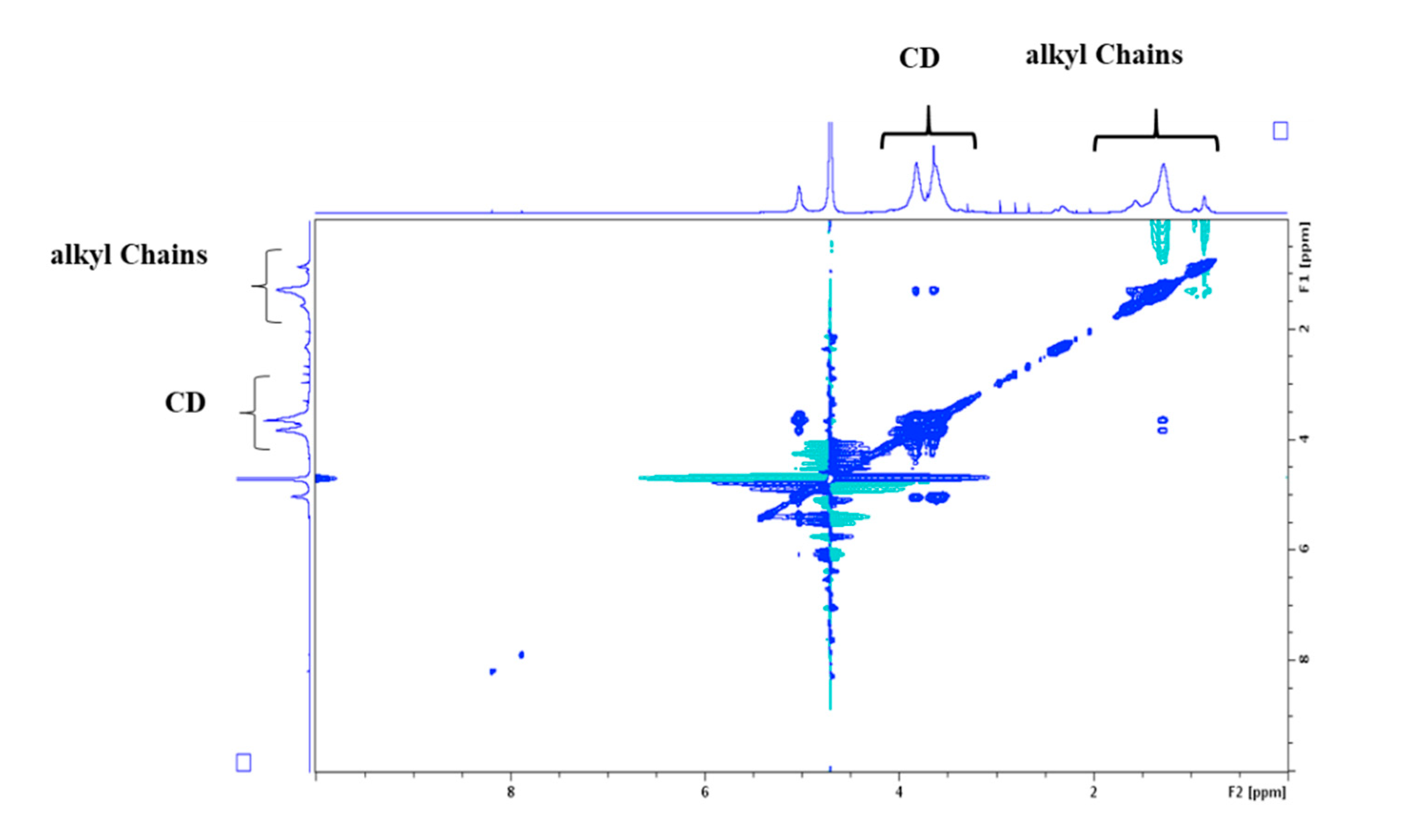
2.5. NMR Analysis
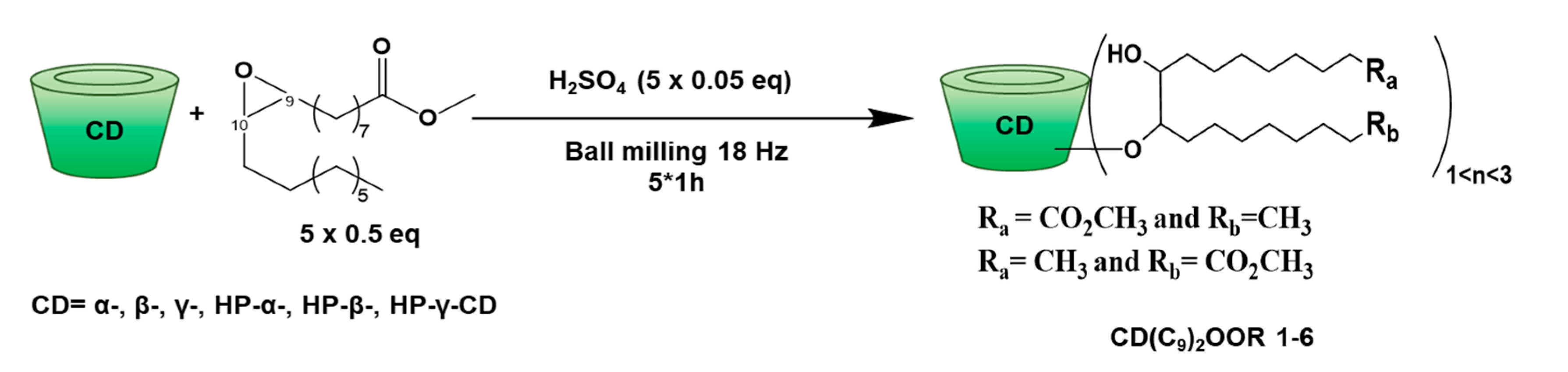
2.6. Synthesis and Product Characterization
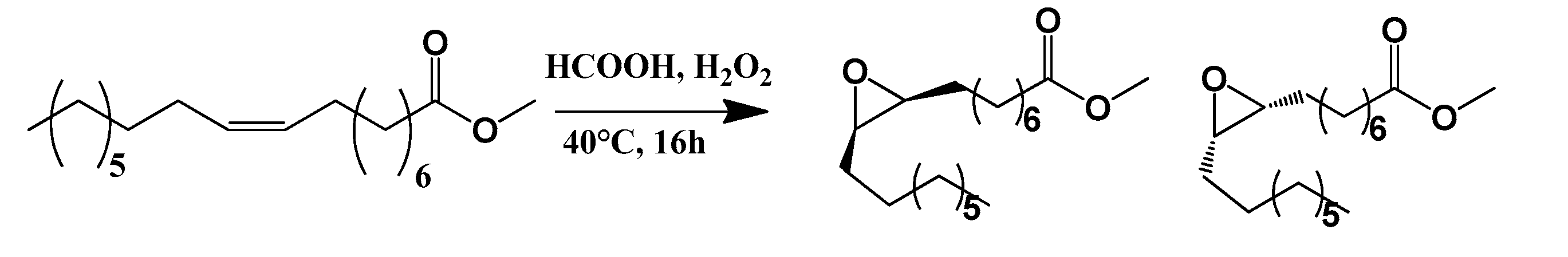
2.6.1. Methyl Oleate Epoxide
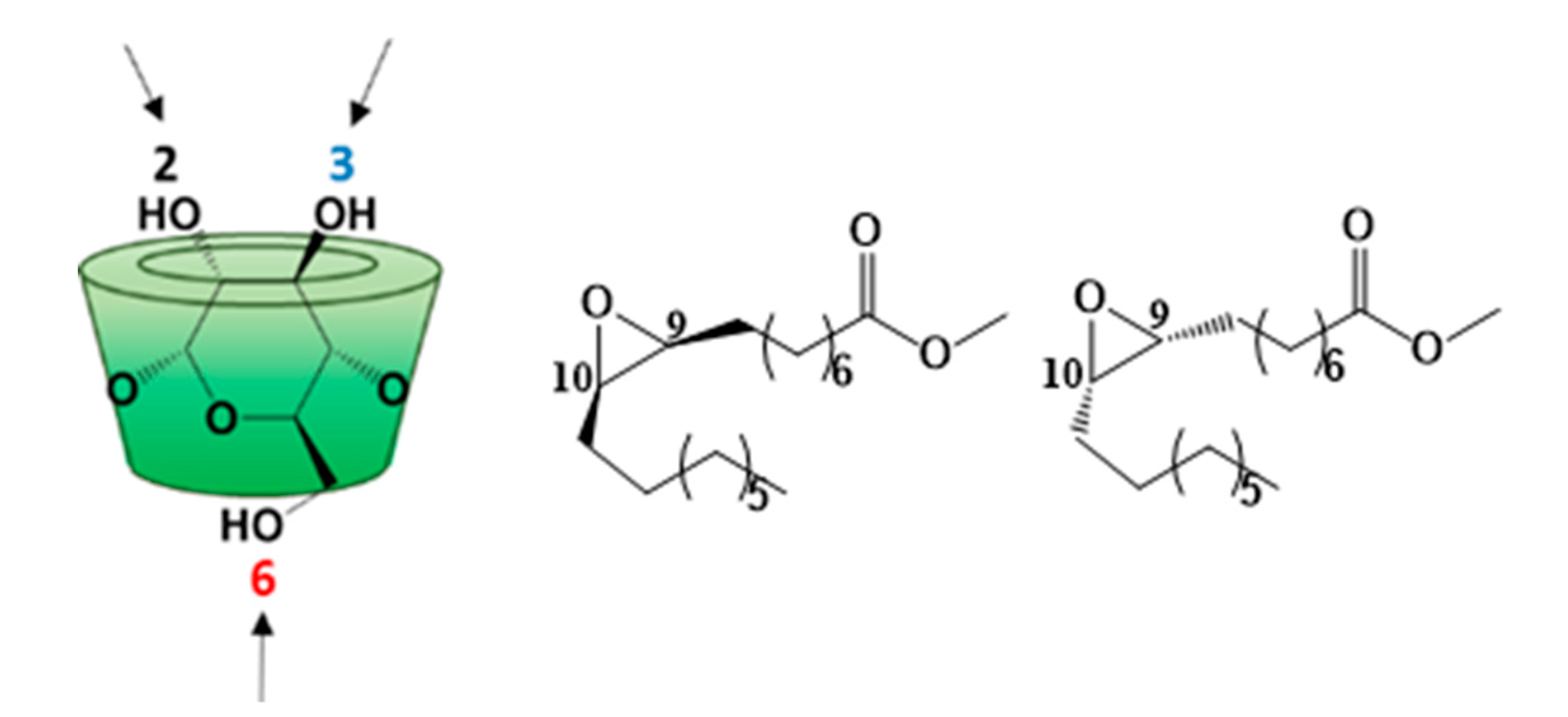
2.6.2. CD(C9)2OOMe
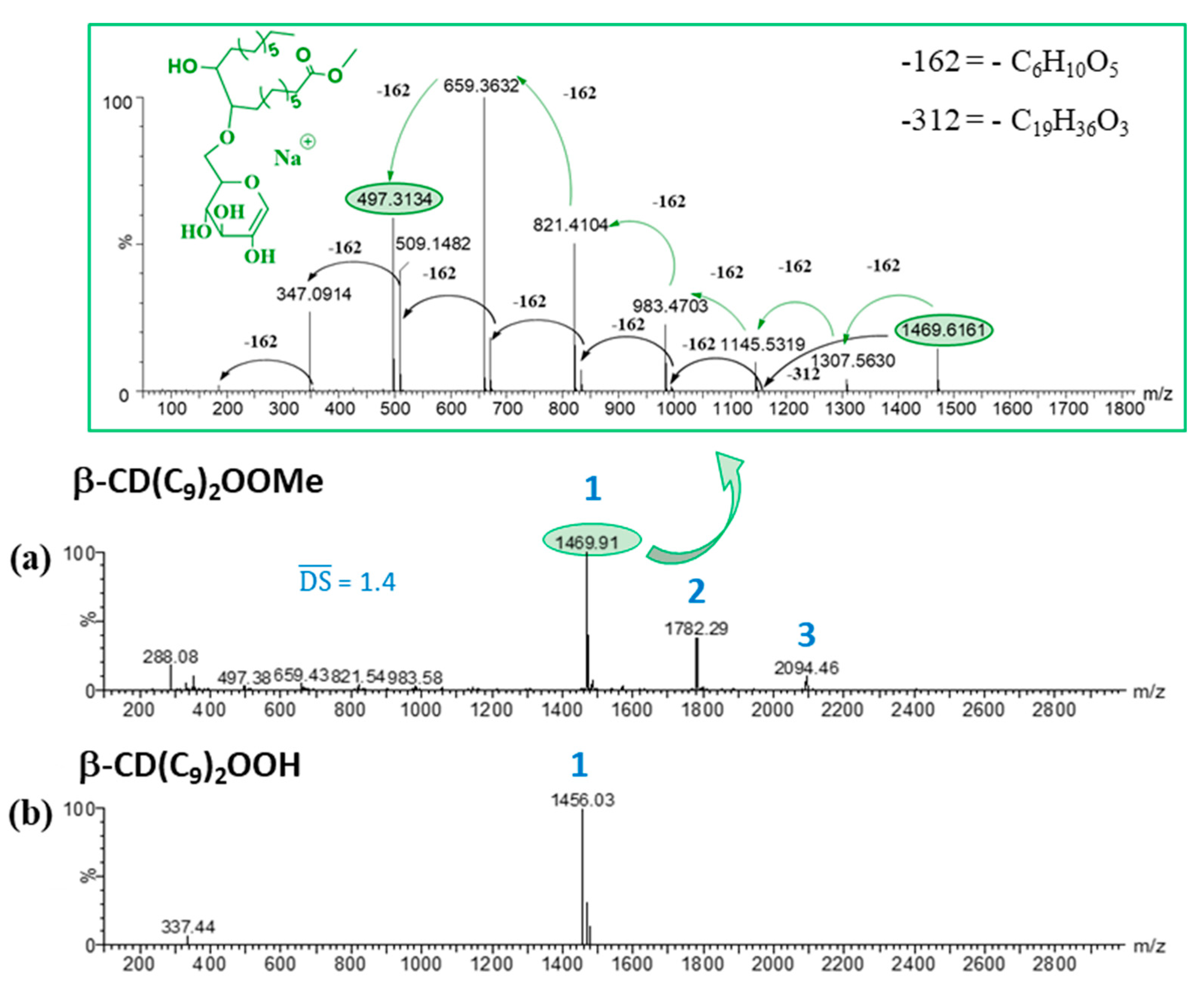
- β-CD(C9)2OOMe, 1: Yield: 55%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 5.69–5.60 (H1), 4.85–3.55 (H2, H3, H4, H5, H6), 3.63 (-OCH3), 2.30–1.01 (CH2 (alkyl chains)), 0.84 (CH3). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 174.4 (C=O), 104.3 (C1), 85.2–83.0 (C4), 75.3–72.7 (C2, C3, C5), 62.4–61.6 (C6), 51.7 (-OCH3), 34.5–23.0 (CH2 (alkyl chains)), 14.6 (CH3). HRMS (ESI): m/z DS1: 1469.6233 ([M + Na]+ (C61H106O38Na requires 1469.6260)), m/z DS2: 1781.8888 ([M + Na]+ (C80H142O41Na requires 1781.8924)) SI Figure S1.
- α-CD(C9)2OOMe, 2: Yield: 28%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 5.77–5.38 (H1), 5.01–3.49 (H2, H3, H4, H5, H6), 2.66–1.06 (CH2 (alkyl chains)), 0.84 (CH3). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 174.3 (C=O), 104.5–103.5 (C1), 86.1–82.4 (C4), 76.4–70.0 (C2, C3, C5), 62.4–60.8 (C6), 51.6 (-OCH3), 35.1–22.7 (CH2 (alkyl chains)), 14.6 (CH3). HRMS (ESI): m/z DS1: 1307.5717 ([M + Na]+ (C55H96O33Na requires 1307.5732)), m/z DS2: 1619.8401 ([M + Na]+ (C74H132O36Na requires 1619.8396)) SI Figure S2.
- γ-CD(C9)2OOMe, 3: Yield: 33%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 5.98–5.45 (H1), 4.79–3.52 (H2, H3, H4, H5, H6), 3.63 (-OCH3), 2.50–1.06 (CH2 (alkyl chains)), 0.81 (CH3). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 174.2 (C=O), 103.6 (C1), 85.5–81.0 (C4), 75.6–71.3 (C2, C3, C5), 63.2–60.9 (C6), 51.6 (-OCH3), 35.0–22.6 (CH2 (alkyl chains)), 14.6 (CH3). HRMS (ESI): m/z DS1: 1631.6769 ([M + Na]+ (C67H116O43Na requires 1631.6788)), m/z DS2: 1943.9395 ([M + Na]+ (C86H152O46Na requires 1943.9452)) SI Figure S3.
- HP-α-CD(C9)2OOMe, 4: Yield: 33%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 6.42–5.24 (H1), 4.92–3.55 (H2, H3, H4, H5, H6), 3.63 (-OCH3), 2.43–1.46 (CH2 (alkyl chains)), 1.50–1.03 (CH3 (hydroxypropyl groups)), 0.84 (CH3 (alkyl chains)). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 174.3 (C=O), 105.0–100.5 (C1), 86.1–77.4 (C4), 76.0–66.0 (C2, C3, C5), 63.0–60.7 (C6), 51.6 (-OCH3), 34.8–22.9 (CH2 (alkyl chains)), 21.0–19.9 (CH3 (hydroxypropyl groups)), 14.6 (CH3). HRMS (ESI): m/z DS1: 1597.7808 ([M + Na]+ (C70H126O38Na requires 1597.7825)), m/z DS2: 1852.0050 ([M + Na]+ (C86H156O40Na requires 1852.0071)) SI Figure S4.
- HP-β-CD(C9)2OOMe, 5: Yield: 35%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 6.22–5.35 (H1), 4.80–3.55 (H2, H3, H4, H5, H6), 3.62 (-OCH3), 2.50–1.46 (CH2 (alkyl chains)), 1.50–1.10 (CH3 (hydroxypropyl groups)), 0.83 (CH3 (alkyl chains)). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 174.3 (C=O), 105.0–100.0 (C1), 85.4–78.0 (C4), 75.7–66.2 (C2, C3, C5), 62.4–61.3 (C6), 51.6 (-OCH3), 35.0–23.1 (CH2 (alkyl chains)), 21.1–19.9 (CH3 (hydroxypropyl groups)), 14.6 (CH3). HRMS (ESI): m/z DS1: 1875.9177 ([M + Na]+ (C82H148O45Na requires 1875.9190)), m/z DS2: 2188.1821 ([M + Na]+ (C101H184O48Na requires 2188.1855)), m/z DS3: 2500.4465 ([M + Na]+ (C120H220O51Na requires 2500.4519)). SI Figure S5.
- HP-γ-CD(C9)2OOMe, 6: Yield: 44%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 6.49–5.31 (H1), 4.70–3.49 (H2, H3, H4, H5, H6), 3.63 (-OCH3), 2.50–1.46 (CH2 (alkyl chains)), 1.44–1.01 (CH3 (hydroxypropyl groups)), 0.82 (CH3 (alkyl chains)). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 174.3 (C=O), 104.6–98.9 (C1), 86.0–78.4 (C4), 75.8–65.6 (C2, C3, C5), 63.0–60.2 (C6), 51.7 (-OCH3), 34.9–22.7 (CH2 (alkyl chains)), 20.9–19.6 (CH3 (hydroxypropyl groups)), 14.6 (CH3). HRMS (ESI): m/z DS1: 1979.9336 ([M + Na]+ (C85H152O49Na requires 1979.9300)), m/z DS2: 2292.1956 ([M + Na]+ (C104H188O52Na requires 2292.1964)), m/z DS3: 2604.4609 ([M + Na]+ (C123H224O55Na requires 2604.4629)) SI Figure S6.
2.6.3. CD(C9)2OOH
- α-CD(C9)2OOH, 7: Yield: 78%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 5.77–5.38 (H1), 5.01–3.49 (H2, H3, H4, H5, H6), 2.66–1.06 (CH2 (alkyl chains)), 0.84 (CH3). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 176.7 (C=O), 104.7–103.1 (C1), 85.7–81.9 (C4), 76.3–69.4 (C2, C3, C5), 62.7–60.8 (C6), 35.9–22.9 (CH2 (alkyl chains)), 14.6 (CH3). HRMS (ESI): m/z: 1293.5549 ([M + Na]+ (C54H94O33Na requires 1293.5575)) SI Figure S7.
- β-CD(C9)2OOH, 8: Yield: 69%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 5.67(H1), 4.97–3.49 (H2, H3, H4, H5, H6), 2.56–1.01 (CH2 (alkyl chains)), 0.81 (CH3).13C NMR (151 MHz, Pyridine-d5) δ (ppm): 176.5 (C=O), 104.3 (C1), 85.4–82.7 (C4), 75.5–72.2 (C2, C3, C5), 62.5–61.2 (C6), 35.4–22.9 (CH2 (alkyl chains)), 14.6 (CH3). HRMS (ESI): m/z: 1455.6115 ([M + Na]+ (C60H104O38Na requires 1455.6103)) SI Figure S8.
- γ-CD(C9)2OOH, 9: Yield: 89%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 6.03–5.20 (H1), 5.10–3.47 (H2, H3, H4, H5, H6), 2.67–1.00 (CH2 (alkyl chains)), 0.84 (CH3). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 176.6 (C=O), 105.2–94.6 (C1), 85.2–78.2 (C4), 76.7–68.6 (C2, C3, C5), 64.9–60.2 (C6), 36.0–22.4 (CH2 (alkyl chains)), 14.6 (CH3). HRMS (ESI): m/z: 1617.6648 ([M + Na]+ (C66H114O43Na requires 1617.6632)) SI Figure S9.
- HP-α-CD(C9)2OOH, 10: Yield: 82%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 5.72–5.31 (H1), 4.89–3.31 (H2, H3, H4, H5, H6), 2.78–1.47 (CH2 (alkyl chains)), 1.50–1.00 (CH3 (hydroxypropyl groups)), 0.84 (CH3 (alkyl chains)). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 176.7 (C=O), 105.2–100.0 (C1), 87.3–76.8 (C4), 76.6–65.1 (C2, C3, C5), 63.8–60.4 (C6), 36.6–22.4 (CH2 (alkyl chains)), 21.5–18.8 (CH3 (hydroxypropyl groups)), 14.6 (CH3). HRMS (ESI): m/z: 1525.7257 ([M + Na]+ (C66H118O37Na requires 1525.7250)) SI Figure S10.
- HP-β-CD(C9)2OOH, 11: Yield: 84%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 6.00–5.35 (H1), 5.09–3.09 (H2, H3, H4, H5, H6), 2.96–1.57 (CH2 (alkyl chains)), 1.56–1.00 (CH3 (hydroxypropyl groups)), 0.84 (CH3 (alkyl chains)). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 176.6 (C=O), 105.2–100.0 (C1), 85.6–77.5 (C4), 75.7–65.7 (C2, C3, C5), 63.1–60.2 (C6), 36.2–22.6 (CH2 (alkyl chains)), 21.3–19.8 (CH3 (hydroxypropyl groups)), 14.6 (CH3). HRMS (ESI): m/z: 1861.9044 ([M + Na]+ (C81H146O45Na requires 1861.9034)) SI Figure S11.
- HP-γ-CD(C9)2OOH, 12: Yield: 89%. ¹H NMR (600 MHz, Pyridine-d5) δ (ppm): 5.99–5.27 (H1), 4.80–3.24 (H2, H3, H4, H5, H6), 2.60–1.47 (CH2 (alkyl chains)), 1.47–1.02 (CH3 (hydroxypropyl groups)), 0.83 (CH3 (alkyl chains)). 13C NMR (151 MHz, Pyridine-d5) δ (ppm): 176.5 (C=O), 104.4–98.8 (C1), 85.9–77.6 (C4), 76.2–65.7 (C2, C3, C5), 62.9–60.4 (C6), 35.7–22.8 (CH2 (alkyl chains)), 20.9–19.6 (CH3 (hydroxypropyl groups)), 14.6 (CH3). HRMS (ESI): m/z: 1965.9083 ([M + Na]+ (C84H150O49Na requires 1965.9143)) SI Figure S12.
2.7. In Vitro Models of Intestinal and Blood-Brain Barriers
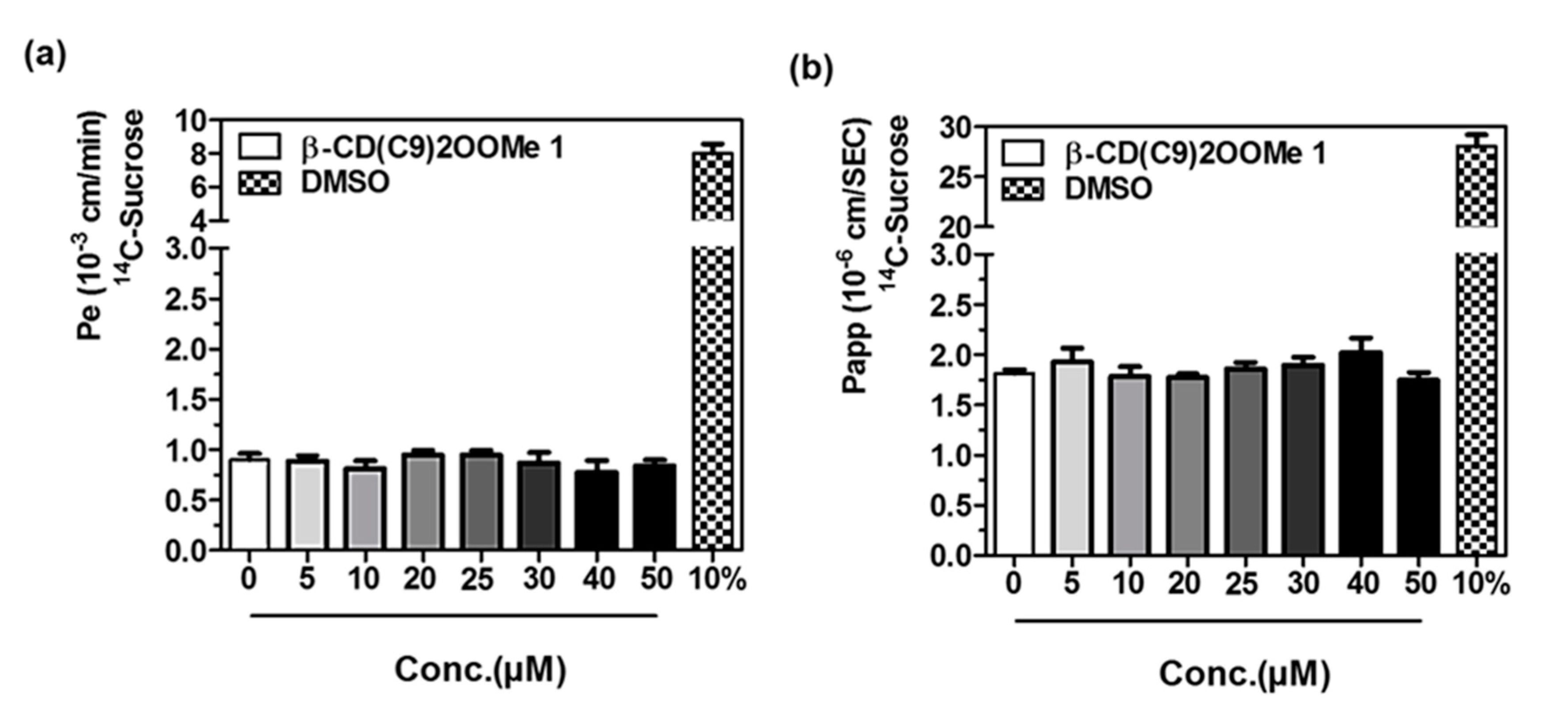
2.8. Permeability Assessment
3. Results and Discussion
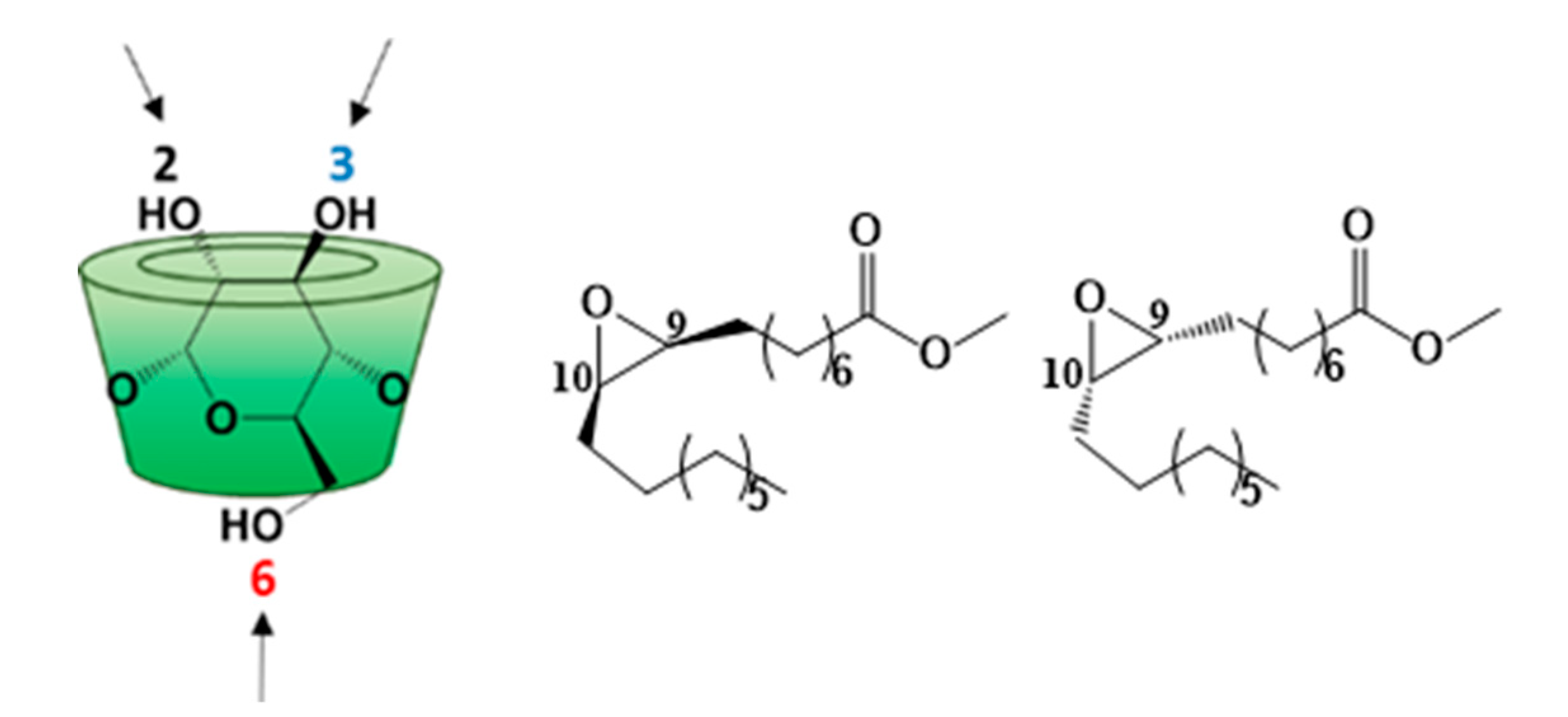
3.1. Synthesis
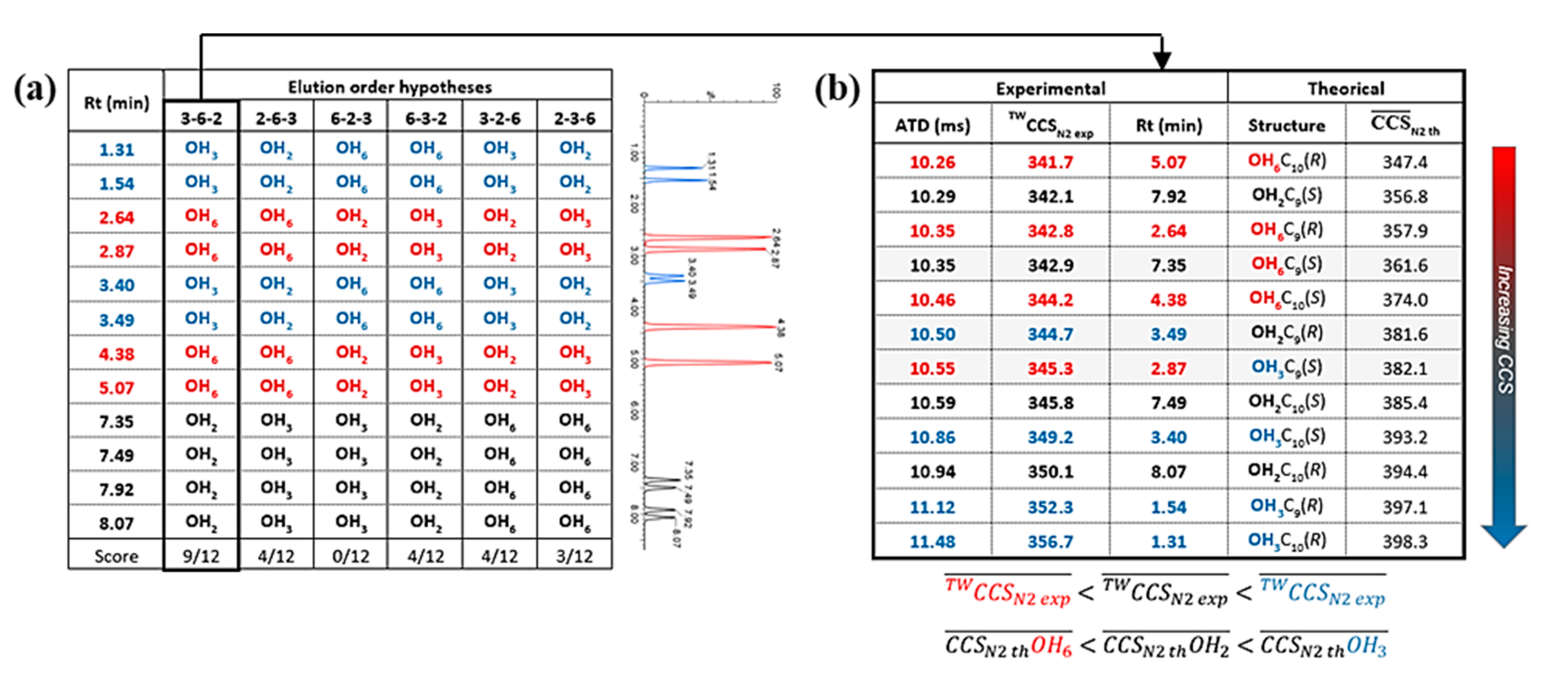
3.2. Characterization
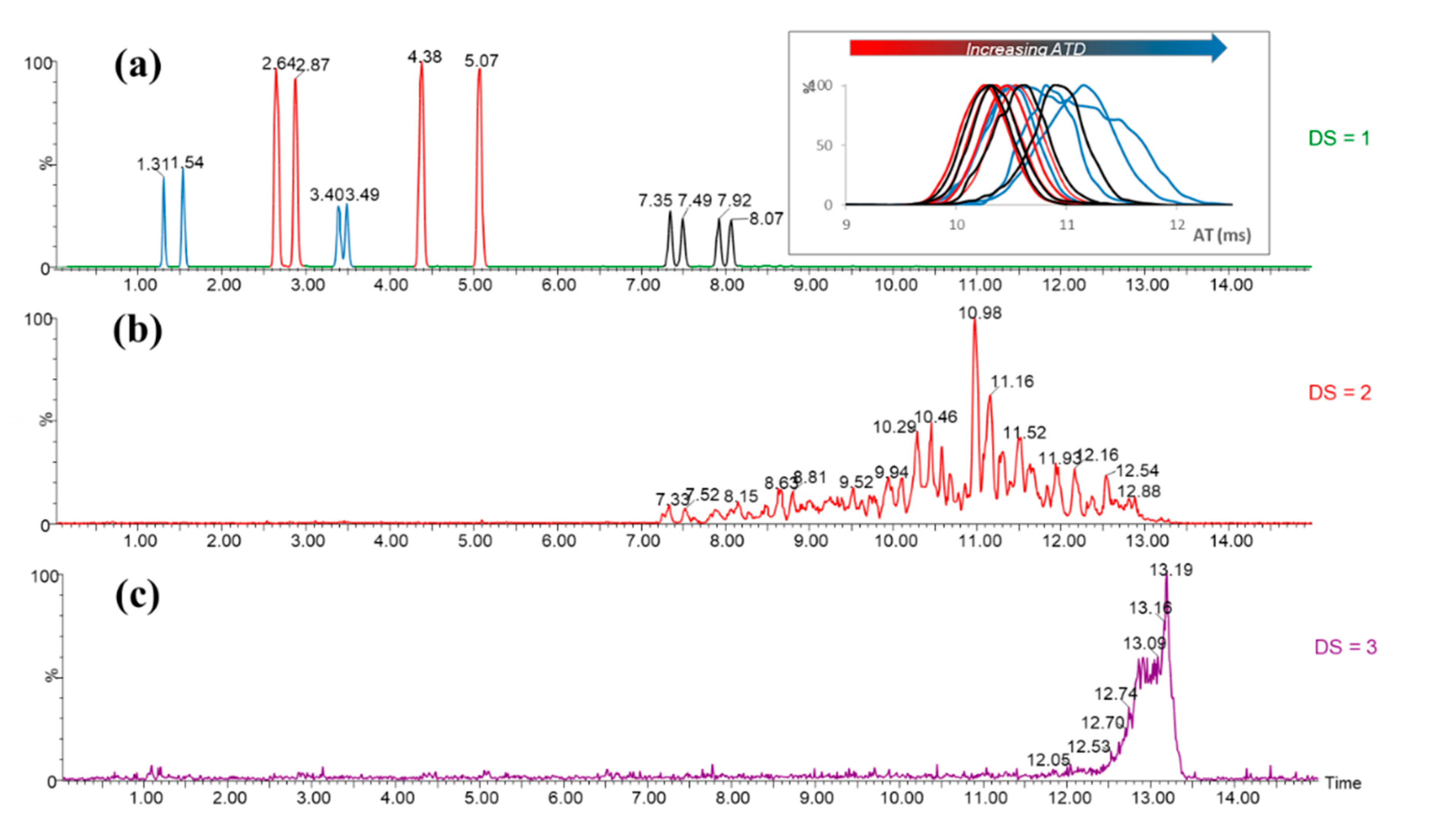
3.3. Self-Organization Properties
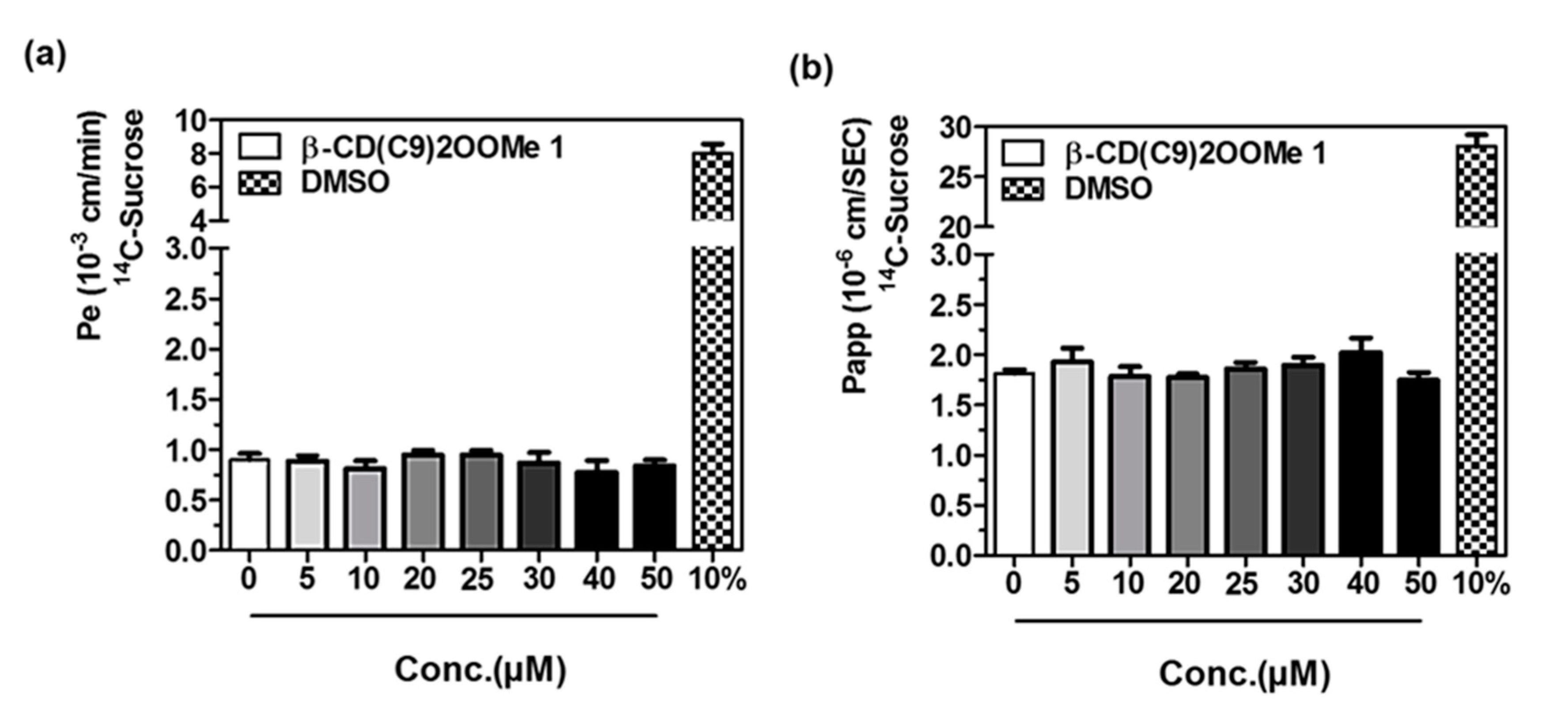
3.4. Preliminary Studies for L-CD Vectorization Potential Use of In Vitro Blood-Brain and Intestinal Barriers Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crini, G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975. [Google Scholar]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Deliv. Rev. 2007, 59, 645–666. [Google Scholar]
- Choisnard, L.; Gèze, A.; Putaux, J.L.; Wong, Y.S.; Wouessidjewe, D. Nanoparticles of β-Cyclodextrin Esters Obtained by Self-Assembling of Biotransesterifiedβ-Cyclodextrins. Biomacromolecules 2006, 7, 515–520. [Google Scholar]
- Bonnet, V.; Gervaise, C.; Djedaïni-Pilard, F.; Furlan, A.; Sarazin, C. Cyclodextrin nanoassemblies: A promising tool for drug delivery. Drug Discov. Today 2015, 20, 1120–1126. [Google Scholar] [CrossRef]
- Moutard, S.; Perly, B.; Godé, P.; Demailly, G.; Djedaïni-Pilard, F. Novel glycolipids based on cyclodextrins. J. Incl. Phenom. 2002, 44, 317–322. [Google Scholar]
- Gervaise, C.; Bonnet, V.; Wattraint, O.; Aubry, F.; Sarazin, C.; Jaffrès, P.A.; Djedaïni-Pilard, F. Synthesis of lipophosphoramidyl-cyclodextrins and their supramolecular properties. Biochimie 2012, 94, 66–74. [Google Scholar]
- Roux, M.; Bonnet, V.; Djedaïni-Pilard, F. Ordering of Saturated and Unsaturated Lipid Membranes near Their Phase Transitions Induced by an Amphiphilic Cyclodextrin and Cholesterol. Langmuir 2019, 35, 14376–14387. [Google Scholar] [CrossRef] [PubMed]
- Cravotto, G.; Nano, G.M.; Palmisano, G. A Sonochemical Protocol for the Synthesis of a Sonochemical Protocol for the Synthesis of Permodified. Synthesis 2001, 20, 37–41. [Google Scholar]
- Trotta, F.; Martina, K.; Robaldo, B.; Barge, A.; Cravotto, G. Recent advances in the synthesis of cyclodextrin derivatives under microwaves and power ultrasound. J. Incl. Phenom. Macrocycl. Chem. 2007, 57, 3–7. [Google Scholar]
- Martina, K.; Cravotto, G.; Caporaso, M.; Rinaldi, L.; Villalonga-Barber, C.; Ermondi, G. Efficient microwave-assisted synthetic protocols and in silico behaviour prediction of per-substituted β-cyclodextrins. Org. Biomol. Chem. 2013, 11, 5521–5527. [Google Scholar]
- Kotková, Z.; Kotek, J.; Jirák, D.; Jendelová, P.; Herynek, V.; Berková, Z.; Hermann, P.; Lukeš, I. Cyclodextrin-Based Bimodal Fluorescence/MRI Contrast Agents: An Efficient Approach to Cellular Imaging. Chemistry 2010, 16, 10094–10102. [Google Scholar]
- Cravotto, G.; Caporaso, M.; Jicsinszky, L.; Martina, K. Enabling technologies and green processes in cyclodextrin chemistry. Beilstein J. Org. Chem. 2016, 12, 278–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jicsinszky, L.; Caporaso, M.; Martina, K.; Gaudino, E.C.; Cravotto, G. Efficient mechanochemical synthesis of regioselective persubstituted cyclodextrins. Beilstein J. Org. Chem. 2016, 12, 2364–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jicsinszky, L.; Calsolaro, F.; Martina, K.; Bucciol, F.; Manzoli, M.; Cravotto, G. Reaction of oxiranes with cyclodextrins under high-energy ball-milling conditions. Beilstein J. Org. Chem. 2019, 15, 1448–1459. [Google Scholar] [CrossRef]
- Menuel, S.; Doumert, B.; Saitzek, S.; Ponchel, A.; Delevoye, L.; Monflier, E.; Hapiot, F. Selective Secondary Face Modification of Cyclodextrins by Mechanosynthesis. J. Org. Chem. 2015, 80, 6259–6266. [Google Scholar] [CrossRef]
- Peptu, C.; Balan-Porcarasu, M.; Šišková, A.; Škultéty, Ľ.; Mosnáček, J. Cyclodextrins tethered with oligolactides—Green synthesis and structural assessment. Beilstein J. Org. Chem. 2017, 13, 779–792. [Google Scholar] [CrossRef] [Green Version]
- Moutard, S.; Djedaïni-Pilard, F.; Meudal, S.; Luijten, W.; Perly, B.; Pilard, S. Structural identification of new glycolipids based on cyclodextrin using high-resolution positive and negative electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2535–2540. [Google Scholar] [CrossRef]
- Kieken, F.; West, C.; Keddadouche, K.; Elfakir, C.; Choisnard, L.; Gèze, A.; Wouessidjewe, D. Characterisation of complex amphiphilic cyclodextrin mixtures by high-performance liquid chromatography and mass spectrometry. J. Chromatogr. A 2008, 1189, 385–391. [Google Scholar] [CrossRef]
- Chizhov, A.O.; Tsvetkov, Y.E.; Nifantiev, N.E. Gas-Phase Fragmentation of Cyclic Oligosaccharides in Tandem Mass Spectrometry. Molecules 2019, 24, 2226. [Google Scholar] [CrossRef] [Green Version]
- Mu, Y.; Schulz, B.L.; Ferro, V. Applications of Ion Mobility-Mass Spectrometry in Carbohydrate Chemistry and Glycobiology. Molecules 2018, 23, 2557. [Google Scholar] [CrossRef] [Green Version]
- Epoune Lingome, C.; Gadenne, B.; Alfos, C.; Queneau, Y.; Moebs-Sanchez, S. Ring opening of epoxidized methyl or ethyl oleate by alkyl glycosides. Eur. J. Lipid Sci. Technol. 2017, 1600413, 1600413. [Google Scholar] [CrossRef]
- Pedroso, D.C.S.; Tellechea, A.; Moura, L.; Fidalgo-Carvalho, I.; Duarte, J.; Carvalho, E.; Ferreira, L. Improved Survival, Vascular Differentiation and Wound Healing Potential of Stem Cells Co-Cultured with Endothelial Cells. PLoS ONE 2011, 6, e16114. [Google Scholar] [CrossRef] [Green Version]
- Cecchelli, R.; Aday, S.; Sevin, E.; Almeida, C.; Culot, M.; Dehouck, L.; Coisne, C.; Engelhardt, B.; Dehouck, M.P.; Ferreira, L. A Stable and Reproducible Human Blood-Brain Barrier Model Derived from Hematopoietic Stem Cells. PLoS ONE 2014, 9, e99733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevin, E.; Dehouck, L.; Fabulas-da Costa, A.; Cecchelli, R.; Dehouck, M.P.; Lundquist, S.; Culot, M. Accelerated Caco-2 cell permeability model for drug discovery. J. Pharmacol. Toxicol. Methods 2013, 68, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, M.P.; Jolliet-Riant, P.; Brée, F.; Fruchart, J.C.; Cecchelli, R.; Tillement, J.P. Drug transfer across the blood-brain barrier: Correlation between in vitro and in vivo models. J. Neurochem. 1992, 58, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Pitha, J.; Trinadha Rao, C.; Lindberg, B.; Seffers, P. Distribution of substituents in 2-hydroxypropyl ethers of cyclomaltoheptaose. Carbohydr. Res. 1990, 200, 429–435. [Google Scholar] [CrossRef]
- Topchieva, I.N.; Mischnick, P.; Ku, G.; Polyakov, V.A.; Elezkaya, S.V.; Bystryzky, G.I.; Karezin, K.I. Novel Derivatives of Cyclodextrins, Modified with Poly (ethylene Oxide) and Their Complexation Properties. Bioconjugate Chem. 1998, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Renard, E.; Deratani, A.; Volet, G.; Sebille, B. Preparation and characterization of water soluble high molecular weight β-cyclodextrin-epichlorohydrin polymers. Eur. Polym. J. 1997, 33, 49–57. [Google Scholar] [CrossRef]
- Manta, C.; Peralta-Altier, G.; Gioia, L.; Méndez, M.F.; Seoane, G.; Ovsejevi, K. Synthesis of a thiol-β-cyclodextrin, a potential agent for controlling enzymatic browning in fruits and vegetables. J. Agric. Food Chem. 2013, 61, 11603–11609. [Google Scholar] [CrossRef]
- Martina, K.; Caporaso, M.; Tagliapietra, S.; Heropoulos, G.; Rosati, O.; Cravotto, G. Synthesis of water-soluble multidentate aminoalcohol β-cyclodextrin derivatives via epoxide opening. Carbohydr. Res. 2011, 346, 2677–2682. [Google Scholar] [CrossRef]
- Pierre, R.; Adam, I.; Fitremann, J.; Jérôme, F.; Bouchu, A.; Courtois, G.; Barrault, J.; Queneau, Y. Catalytic etherification of sucrose with 1,2-epoxydodecane: Investigation of homogeneous and heterogeneous catalysts. C. R. Chim. 2004, 7, 151–160. [Google Scholar] [CrossRef]
- Villandier, N.; Adam, I.; Jérôme, F.; Barrault, J.; Pierre, R.; Bouchu, A.; Fitremann, J.; Queneau, Y. Selective synthesis of amphiphilic hydroxyalkylethers of disaccharides over solid basic catalysts. Influence of the superficial hydrophilic-lipophilic balance of the catalyst. J. Mol. Catal. A Chem. 2006, 259, 67–77. [Google Scholar] [CrossRef]
- Albarrán-Preza, E.; Corona-Becerril, D.; Vigueras-Santiago, E.; Hernández-López, S. Sweet polymers: Synthesis and characterization of xylitol-based epoxidized linseed oil resins. Eur. Polym. J. 2016, 75, 539–551. [Google Scholar]
- Ranu, B.; Stolle, A. Ball Milling Towards Green Synthesis: Applications, Projects, Challenges. RSC Green Chem. Ser. 2014. [CrossRef] [Green Version]
- Rodríguez, B.; Bruckmann, A.; Bolm, C. A highly efficient asymmetric organocatalytic aldol reaction in a ball mill. Chem. A Eur. J. 2007, 13, 4710–4722. [Google Scholar]
- Gérard, E.M.C.; Sahin, H.; Encinas, A.; Bräse, S. Systematic study of a solvent-free mechanochemically induced domino oxa-Michael-aldol reaction in a ball mill. Synlett 2008, 17, 2702–2704. [Google Scholar]
- Lingome, C.E.; Pourceau, G.; Gobert-Deveaux, V.; Wadouachi, A. Efficient synthesis of glycosylamines in solventless conditions promoted by mechanical milling. RSC Adv. 2014, 4, 36350–36356. [Google Scholar] [CrossRef]
- Lanucara, F.; Holman, S.W.; Gray, C.J.; Eyers, C.E. The power of ion mobility-mass spectrometry for structural characterization and the study of conformational dynamics. Nat. Chem. 2014, 6, 281–294. [Google Scholar] [CrossRef]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef] [Green Version]
- Reading, E.; Munoz-Muriedas, J.; Roberts, A.D.; Dear, G.J.; Robinson, C.V.; Beaumont, C. Elucidation of Drug Metabolite Structural Isomers Using Molecular Modeling Coupled with Ion Mobility Mass Spectrometry. Anal. Chem. 2016, 88, 2273–2280. [Google Scholar] [CrossRef]
- Mathiron, D.; Lori, R.; Pilard, S.; Soundara Rajan, T.; Landy, D.; Mazzon, E.; Rollin, P.; Djedaïni-Pilard, F. A Combined Approach of NMR and Mass Spectrometry Techniques Applied to the α-Cyclodextrin/Moringin Complex for a Novel Bioactive Formulation. Molecules 2018, 23, 1714. [Google Scholar] [CrossRef] [Green Version]
- Oliva, E.; Mathiron, D.; Bertaut, E.; Landy, D.; Cailleu, D.; Pilard, S.; Clément, C.; Courot, E.; Bonnet, V.; Djedaïni-Pilard, F. Physico-chemical studies of resveratrol, methyl-jasmonate and cyclodextrin interactions: An approach to resveratrol bioproduction optimization. RSC Adv. 2018, 8, 1528–1538. [Google Scholar] [CrossRef] [Green Version]
- Furlan, A.L.; Buchoux, S.; Miao, Y.; Banchet, V.; Létévé, M.; Lambertyn, V.; Michel, J.; Sarazin, C.; Bonnet, V. Nanoparticles based on lipidyl-β-cyclodextrins: Synthesis, characterization, and experimental and computational biophysical studies for encapsulation of atazanavir. New J. Chem. 2018, 42, 20171–20179. [Google Scholar] [CrossRef]
- Ravid, O.; Elhaik Goldman, S.; Macheto, D.; Bresler, Y.; De Oliveira, R.I.; Liraz-Zaltsman, S.; Gosselet, F.; Dehouck, L.; Beeri, M.S.; Cooper, I. Blood-Brain Barrier Cellular Responses Toward Organophosphates: Natural Compensatory Processes and Exogenous Interventions to Rescue Barrier Properties. Front. Cell Neurosci. 2018, 12, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jähne, E.A.; Eigenmann, D.E.; Sampath, C.; Butterweck, V.; Culot, M.; Cecchelli, R.; Gosselet, F.; Walter, F.R.; Deli, M.A.; Smieško, M.; et al. Pharmacokinetics and In Vitro Blood-Brain Barrier Screening of the Plant-Derived Alkaloid Tryptanthrin. Planta Med. 2016, 82, 1021–1030. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD(C9)2OOMe | (ESI)+ | (ESI)− | MWa g.mol−1 | Yield % |
|---|---|---|---|---|
| β-CD(C9)2OOMe 1 | 1.4 | 1.3 | 1577.3 | 55% |
| α-CD(C9)2OOMe 2 | 1.3 | 1.2 | 1378.2 | 28% |
| γ-CD(C9)2OOMe 3 | 1.3 | 1.2 | 1712.5 | 33% |
| HP-α-CD(C9)2OOMe 4 | 1.2 | 1.1 | 1571.7 | 33% |
| HP-β-CD(C9)2OOMe 5 | 1.3 | 1.3 | 1892.5 | 35% |
| HP-γ-CD(C9)2OOMe 6 | 1.2 | 1.2 | 2031.9 | 44% |
| CD(C9)2OOH | Time (h) | MWa (g.mol−1) | Yield |
|---|---|---|---|
| α-CD(C9)2OOH 7 | 4.5 | 1271.0 | 78% |
| β-CD(C9)2OOH 8 | 8 | 1433.0 | 69% |
| γ-CD(C9)2OOH 9 | 1.5 | 1595.0 | 89% |
| HP-α-CD(C9)2OOH 10 | 2 | 1499.5 | 82% |
| HP-β-CD(C9)2OOH 11 | 2 | 1736.3 | 84% |
| HP-γ-CD(C9)2OOH 12 | 1 | 1887.3 | 89% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, E.; Mathiron, D.; Rigaud, S.; Monflier, E.; Sevin, E.; Bricout, H.; Tilloy, S.; Gosselet, F.; Fenart, L.; Bonnet, V.; et al. New Lipidyl-Cyclodextrins Obtained by Ring Opening of Methyl Oleate Epoxide Using Ball Milling. Biomolecules 2020, 10, 339. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10020339
Oliva E, Mathiron D, Rigaud S, Monflier E, Sevin E, Bricout H, Tilloy S, Gosselet F, Fenart L, Bonnet V, et al. New Lipidyl-Cyclodextrins Obtained by Ring Opening of Methyl Oleate Epoxide Using Ball Milling. Biomolecules. 2020; 10(2):339. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10020339
Chicago/Turabian StyleOliva, Estefania, David Mathiron, Sébastien Rigaud, Eric Monflier, Emmanuel Sevin, Hervé Bricout, Sébastien Tilloy, Fabien Gosselet, Laurence Fenart, Véronique Bonnet, and et al. 2020. "New Lipidyl-Cyclodextrins Obtained by Ring Opening of Methyl Oleate Epoxide Using Ball Milling" Biomolecules 10, no. 2: 339. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10020339