Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation and Cellular Bioenergetics in Human Down Syndrome Fibroblasts


Abstract
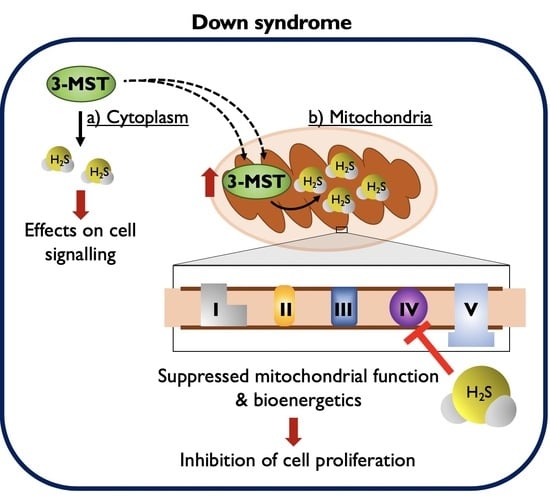
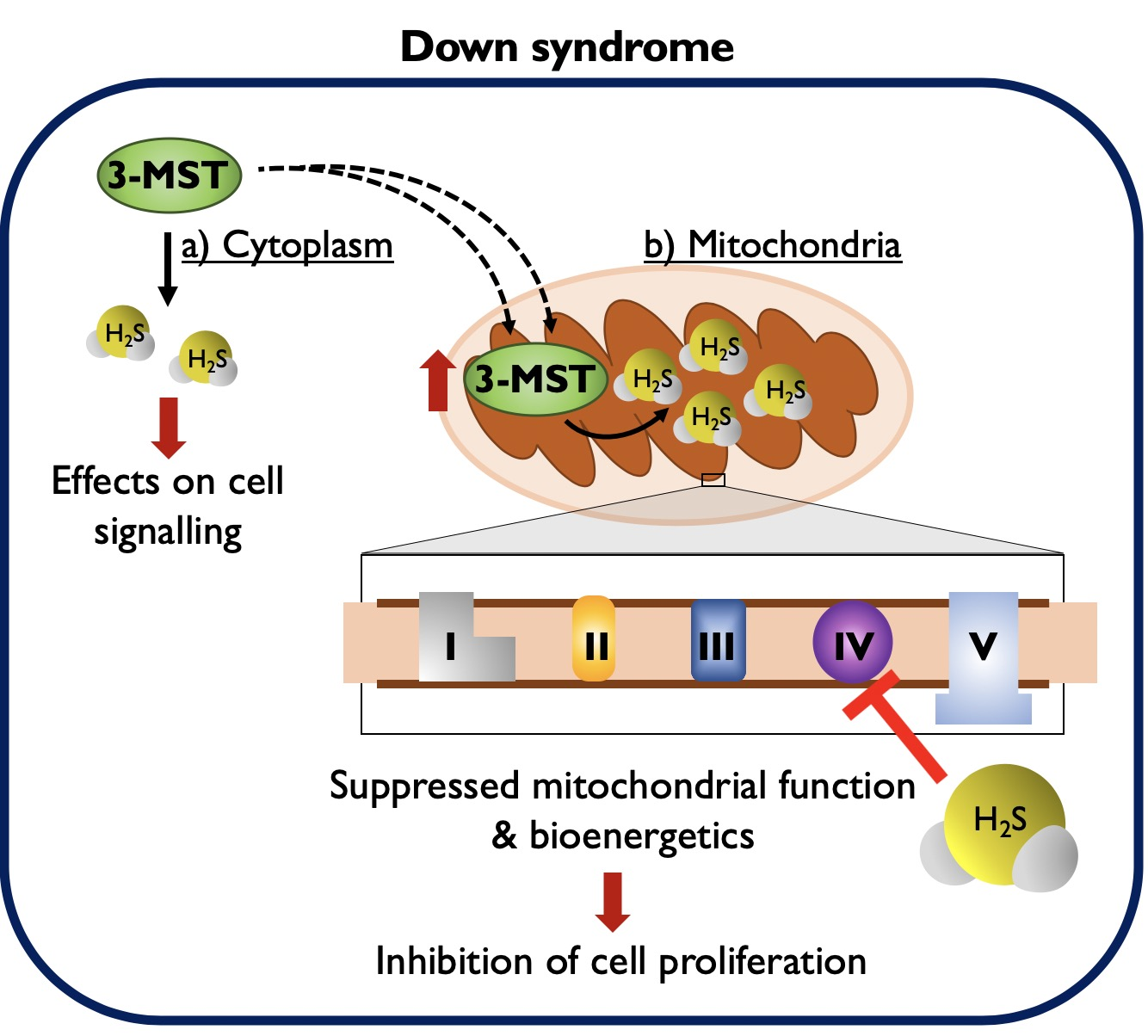
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Cell Treatments
2.4. Cell Proliferation Assay
2.5. Quantification of H2S Levels in Live Cells
2.6. Measurement of Mitochondrial Respiration
2.7. Sample Preparation for Whole-Cell Protein Extraction
2.8. Sample Preparation for Mitochondrial and Cytosolic Protein Extractions
2.9. Western Blotting
2.10. Statistics
3. Results
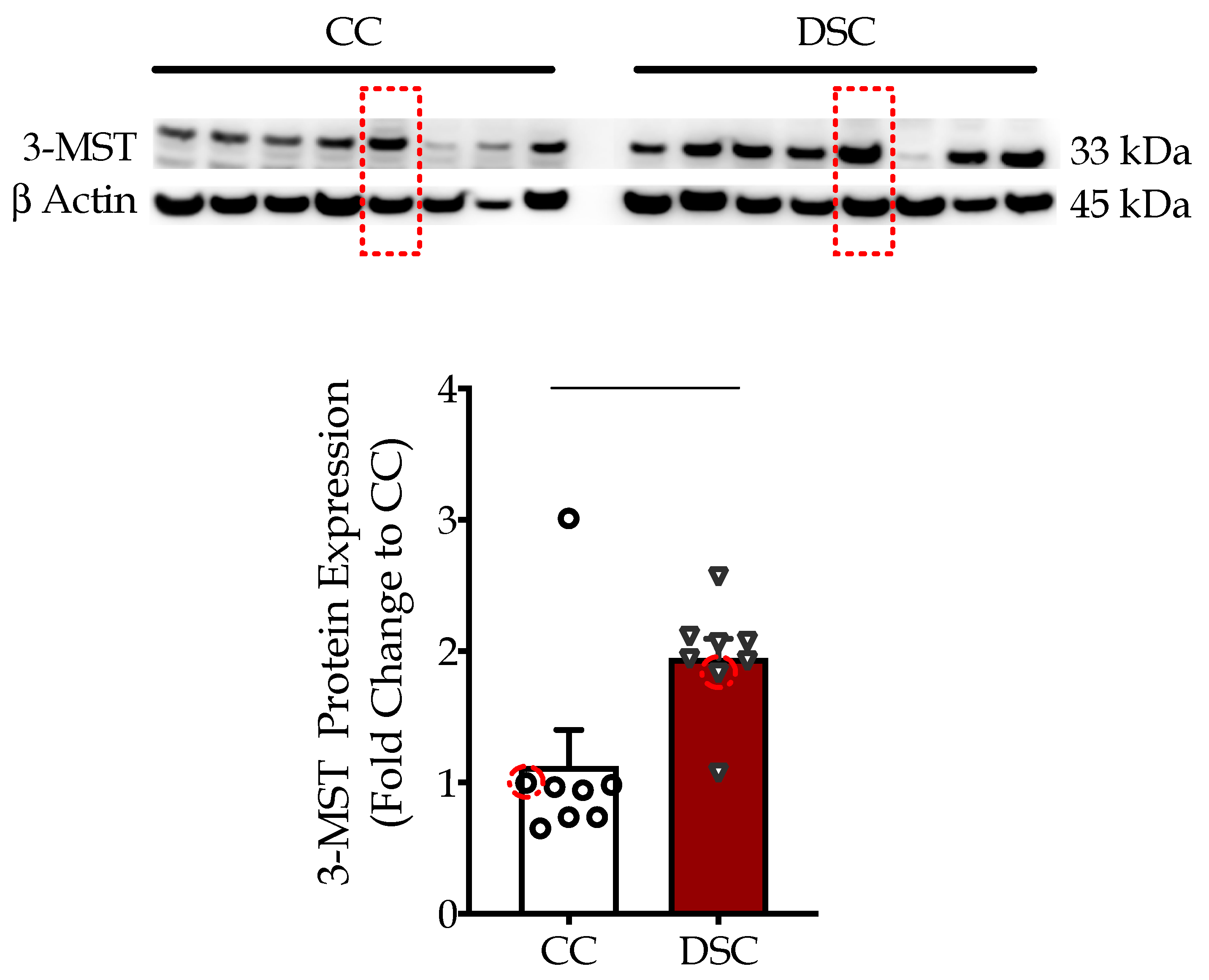
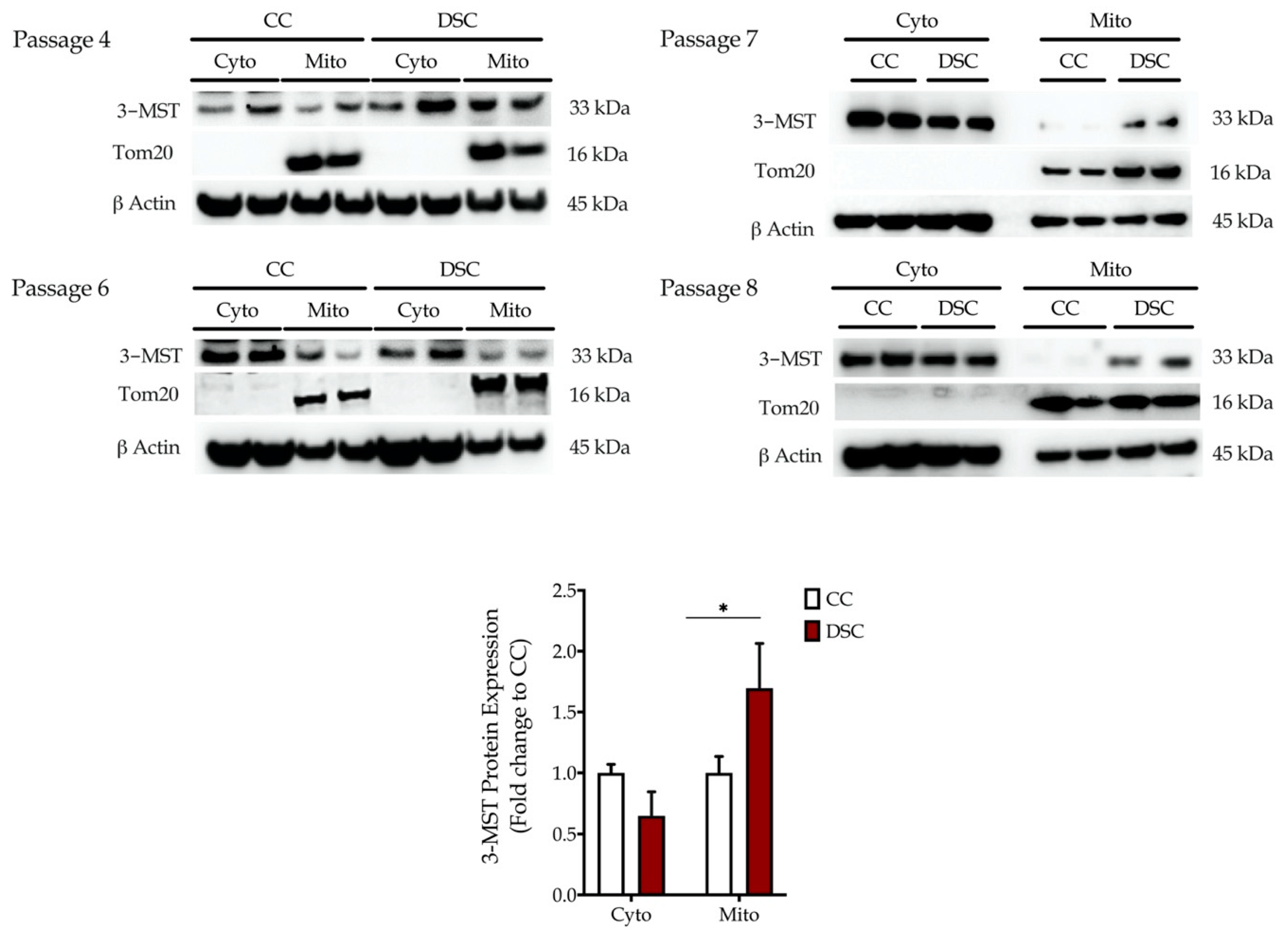
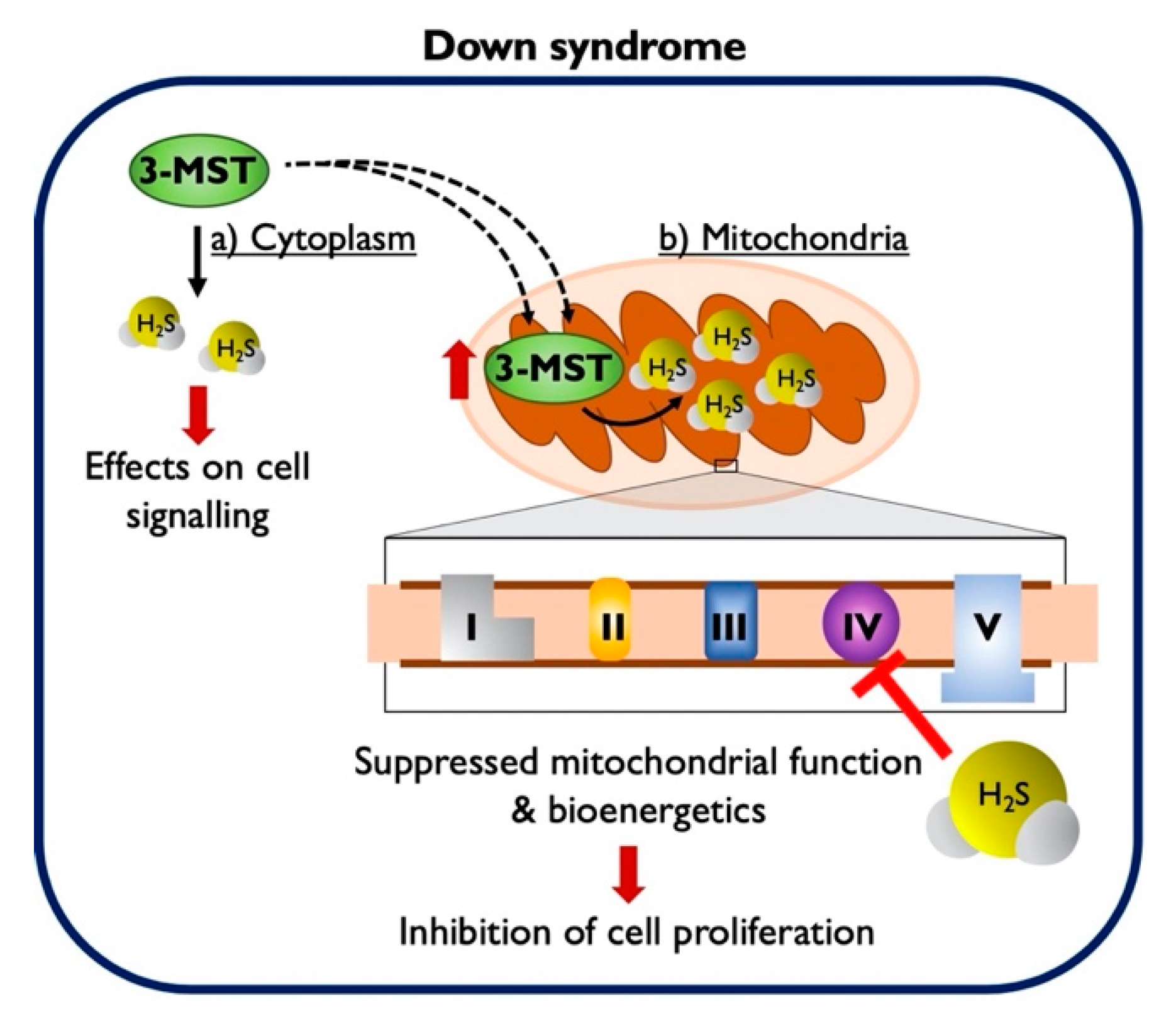
3.1. Down Syndrome Fibroblasts Overexpress 3-MST, which Accumulates in the Mitochondria
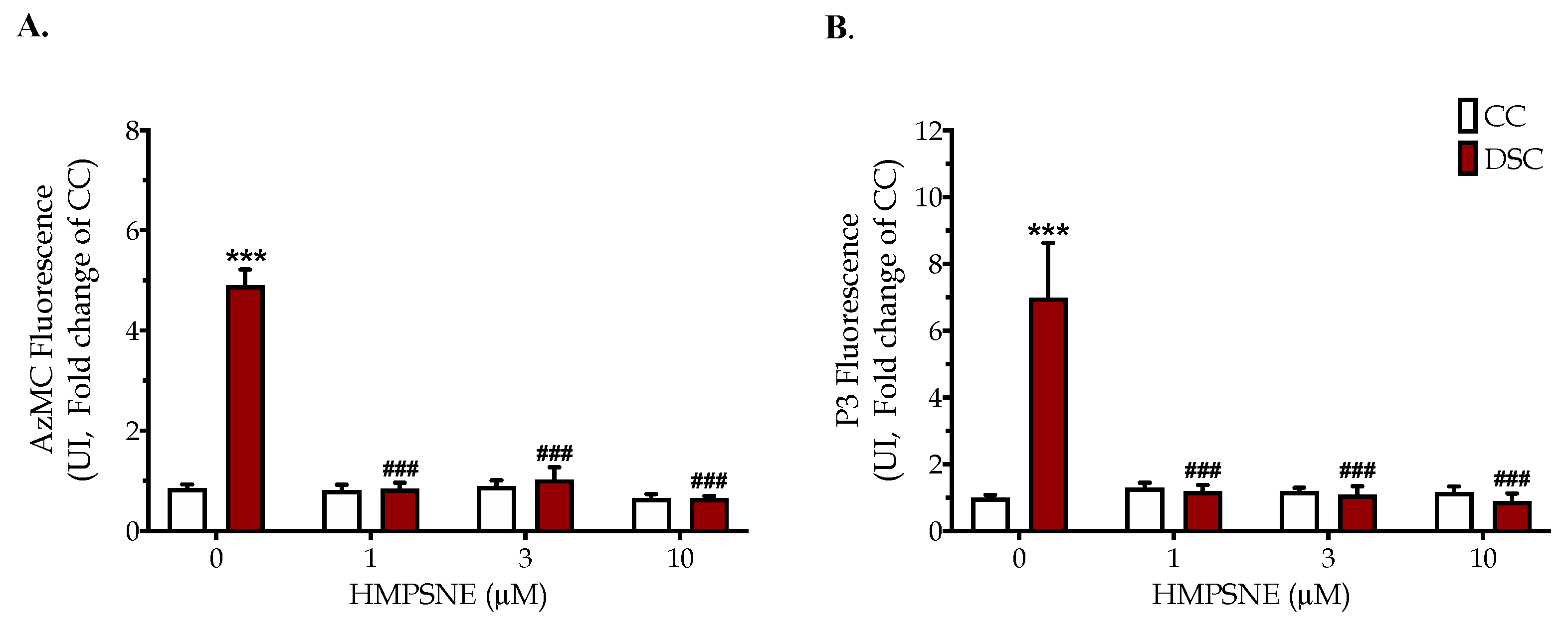
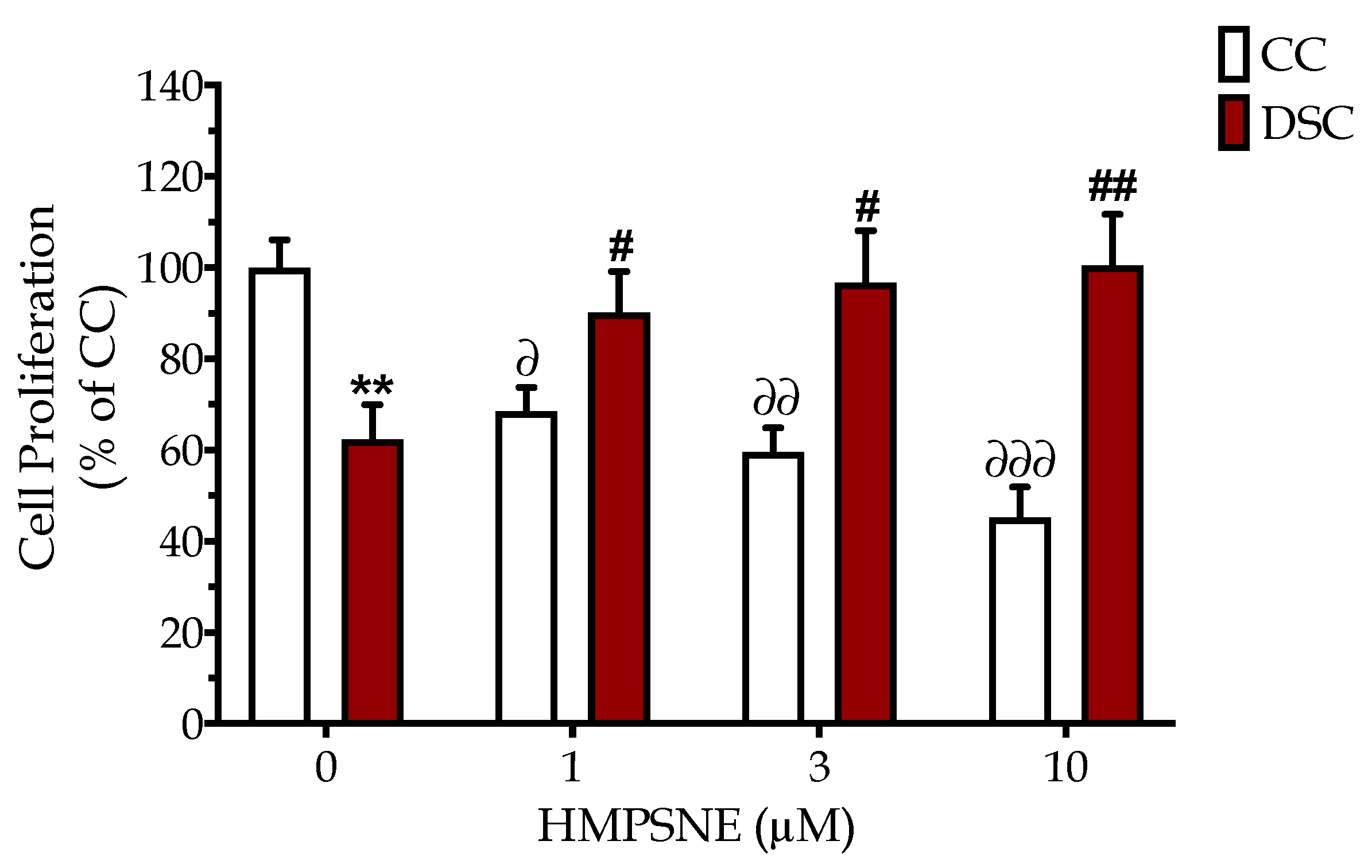
3.2. HMPSNE Inhibits H2S Production and Restores the Cellular Proliferation Rate in Down Syndrome Cells
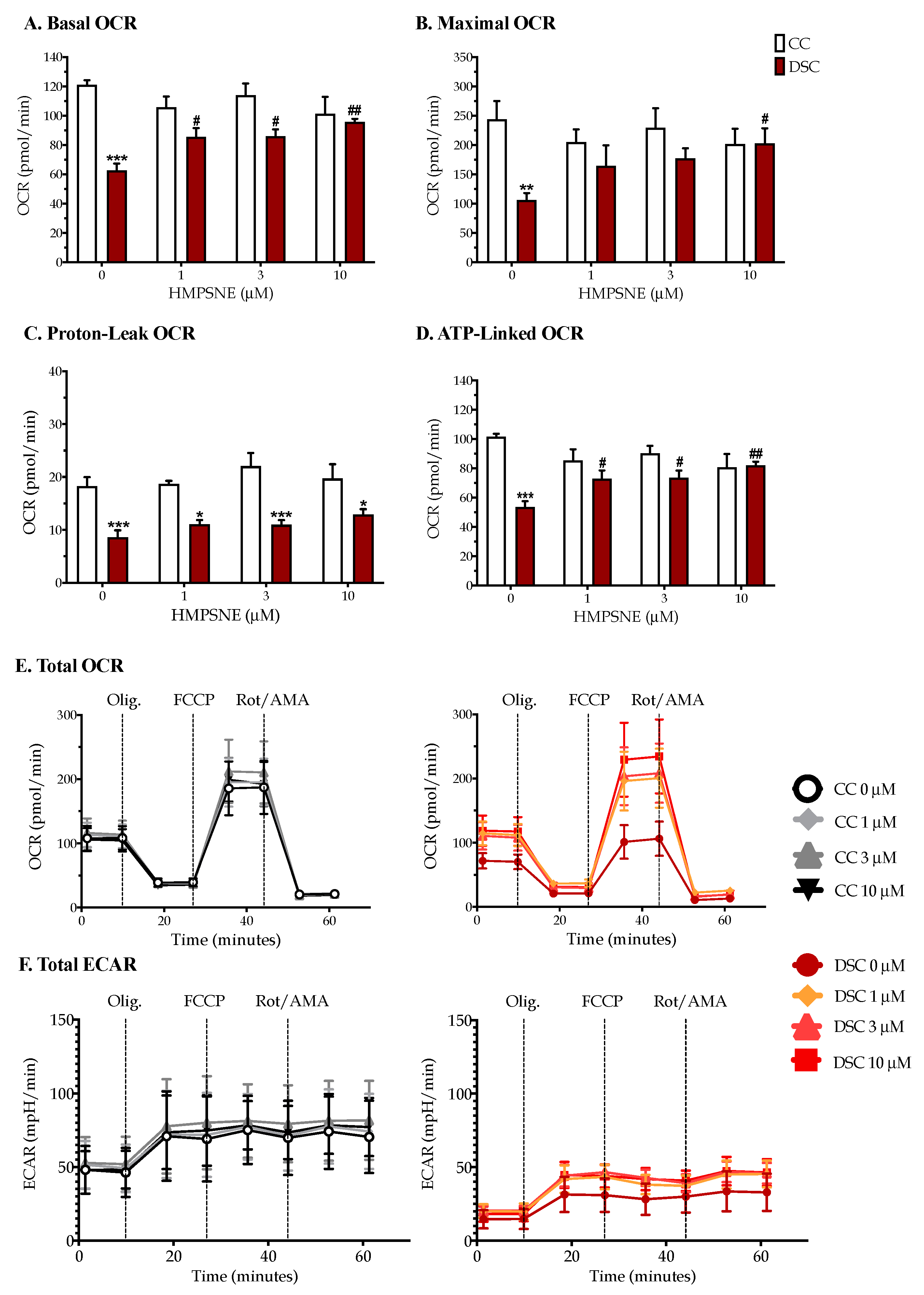
3.3. HMPSNE Normalizes Cellular Bioenergetics in Down Syndrome Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Calì, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in Down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Braidy, N.; De Rasmo, D.; Signorile, A.; Rossi, L.; Atanasov, A.G.; Volpicella, M.; Henrion-Caude, A.; Nabavi, S.M.; Vacca, R.A. Mitochondria as pharmacological targets in Down syndrome. Free Radic. Biol. Med. 2018, 114, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. The re-emerging pathophysiological role of the cystathionine-β-synthase—Hydrogen sulfide system in Down syndrome. FEBS J. 2020, in press. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.W.; Moore, P.K. H2S synthesizing enzymes: Biochemistry and molecular aspects. Handb. Exp. Pharmacol. 2015, 230, 3–25. [Google Scholar]
- Kimura, H. Physiological roles of hydrogen sulfide and polysulfides. Handb. Exp. Pharmacol. 2015, 230, 61–81. [Google Scholar]
- Szabo, C.; Papapetropoulos, A. International union of basic and clinical pharmacology. CII: Pharmacological modulation of H2S levels: H2S donors and H2S biosynthesis inhibitors. Pharmacol. Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef] [Green Version]
- Panagaki, T.; Randi, E.B.; Augsburger, F.; Szabo, C. Overproduction of H2S, generated by CBS, inhibits mitochondrial Complex IV and suppresses oxidative phosphorylation in Down syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 18769–18771. [Google Scholar] [CrossRef] [Green Version]
- Szczesny, B.; Módis, K.; Yanagi, K.; Coletta, C.; Le Trionnaire, S.; Perry, A.; Wood, M.E.; Whiteman, M.; Szabo, C. AP39, a novel mitochondria-targeted hydrogen sulfide donor, stimulates cellular bioenergetics, exerts cytoprotective effects and protects against the loss of mitochondrial DNA integrity in oxidatively stressed endothelial cells in vitro. Nitric Oxide 2014, 41, 120–130. [Google Scholar] [CrossRef]
- Singha, S.; Kim, D.; Moon, H.; Wang, T.; Kim, K.H.; Shin, Y.H.; Jung, J.; Seo, E.; Lee, S.J.; Ahn, K.H. Toward a selective, sensitive, fast-responsive, and biocompatible two-photon probe for hydrogen sulfide in live cells. Anal. Chem. 2015, 87, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Sasakura, K.; Suwanai, Y.; Toma-Fukai, S.; Shimamoto, K.; Takano, Y.; Shibuya, N.; Terai, T.; Komatsu, T.; Ueno, T.; et al. Discovery and mechanistic characterization of selective inhibitors of H2S-producing enzyme: 3-mercaptopyruvate sulfurtransferase (3MST) targeting active-site cysteine persulfide. Sci. Rep. 2017, 7, 40227. [Google Scholar] [CrossRef] [PubMed]
- Módis, K.; Coletta, C.; Erdélyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Augsburger, F.; Randi, E.B.; Jendly, M.; Ascencao, K.; Dilek, N.; Szabo, C. Role of 3-mercaptopyruvate sulfurtransferase in the regulation of proliferation, migration, and bioenergetics in murine colon cancer cells. Biomolecules 2020, 10, 447. [Google Scholar] [CrossRef] [Green Version]
- Panagaki, T.; Michael, M.; Hölscher, C. Liraglutide restores chronic ER stress, autophagy impairments and apoptotic signalling in SH-SY5Y cells. Sci. Rep. 2017, 7, 16158. [Google Scholar] [CrossRef]
- Kamoun, P. Mental retardation in Down syndrome: A hydrogen sulfide hypothesis. Med. Hypotheses 2001, 57, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Kamoun, P.P. Mental retardation in Down syndrome: Two ways to treat. Med. Hypotheses 2019, 131, 109289. [Google Scholar] [CrossRef]
- Belardinelli, M.C.; Chabli, A.; Chadefaux-Vekemans, B.; Kamoun, P. Urinary sulfur compounds in Down syndrome. Clin. Chem. 2001, 47, 1500–1501. [Google Scholar] [CrossRef]
- Kamoun, P.; Belardinelli, M.C.; Chabli, A.; Lallouchi, K.; Chadefaux-Vekemans, B. Endogenous hydrogen sulfide overproduction in Down syndrome. Am. J. Med. Genet. A 2003, 116A, 310–311. [Google Scholar] [CrossRef]
- Chadefaux, B.; Rethoré, M.O.; Raoul, O.; Ceballos, I.; Poissonnier, M.; Gilgenkranz, S.; Allard, D. Cystathionine beta synthase: Gene dosage effect in trisomy 21. Biochem. Biophys. Res. Commun. 1985, 128, 40–44. [Google Scholar] [CrossRef]
- Taub, J.W.; Huang, X.; Matherly, L.H.; Stout, M.L.; Buck, S.A.; Massey, G.V.; Becton, D.L.; Chang, M.N.; Weinstein, H.J.; Ravindranath, Y. Expression of chromosome 21-localized genes in acute myeloid leukemia: Differences between Down syndrome and non-Down syndrome blast cells and relationship to in vitro sensitivity to cytosine arabinoside and daunorubicin. Blood 1999, 94, 1393–1400. [Google Scholar] [PubMed]
- Ge, Y.; Jensen, T.L.; Matherly, L.H.; Taub, J.W. Transcriptional regulation of the cystathionine-beta -synthase gene in Down syndrome and non-Down syndrome megakaryocytic leukemia cell lines. Blood 2003, 101, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, A.; Kanaumi, T.; Takashima, S.; Enokido, Y.; Nagai, Y.; Kimura, H. Cystathionine beta-synthase is enriched in the brains of Down’s patients. Biochem. Biophys. Res. Commun. 2005, 338, 1547–1550. [Google Scholar] [CrossRef] [PubMed]
- Marechal, D.; Brault, V.; Leon, A.; Martin, D.; Lopes Pereira, P.; Loaëc, N.; Birling, M.C.; Friocourt, G.; Blondel, M.; Herault, Y. CBS overdosage is necessary and sufficient to induce cognitive phenotypes in mouse models of Down syndrome and interacts genetically with Dyrk1a. Hum. Mol. Genet. 2019, 28, 1561–1577. [Google Scholar] [PubMed] [Green Version]
- Sullivan, K.D.; Lewis, H.C.; Hill, A.A.; Pandey, A.; Jackson, L.P.; Cabral, J.M.; Smith, K.P.; Liggett, L.A.; Gomez, E.B.; Galbraith, M.D.; et al. Trisomy 21 consistently activates the interferon response. Elife 2016, 5, e16220. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Pennings, J.L.; Massingham, L.J.; Wick, H.C.; Siegel, A.E.; Tantravahi, U.; Bianchi, D.W. An integrated human/murine transcriptome and pathway approach to identify prenatal treatments for Down syndrome. Sci. Rep. 2016, 6, 32353. [Google Scholar] [CrossRef] [Green Version]
- Pelleri, M.C.; Cattani, C.; Vitale, L.; Antonaros, F.; Strippoli, P.; Locatelli, C.; Cocchi, G.; Piovesan, A.; Caracausi, M. Integrated quantitative transcriptome maps of human trisomy 21 tissues and cells. Front. Genet. 2018, 9, 125. [Google Scholar] [CrossRef] [Green Version]
- Sriroopreddy, R.; Sajeed, R. Differentially expressed gene (DEG) based protein-protein interaction (PPI) network identifies a spectrum of gene interactome, transcriptome and correlated miRNA in nondisjunction Down syndrome. Int. J. Biol. Macromol. 2019, 122, 1080–1089. [Google Scholar] [CrossRef]
- Sobol, M.; Klar, J.; Laan, L.; Shahsavani, M.; Schuster, J.; Annerén, G.; Konzer, A.; Mi, J.; Bergquist, J.; Nordlund, J.; et al. Transcriptome and proteome profiling of neural induced pluripotent stem cells from individuals with Down syndrome disclose dynamic dysregulations of key pathways and cellular functions. Mol. Neurobiol. 2019, 56, 7113–7127. [Google Scholar] [CrossRef] [Green Version]
- Moreira-Filho, C.A.; Bando, S.Y.; Bertonha, F.B.; Silva, F.N.; Costa Lda, F.; Ferreira, L.R.; Furlanetto, G.; Chacur, P.; Zerbini, M.C.; Carneiro-Sampaio, M. Modular transcriptional repertoire and MicroRNA target analyses characterize genomic dysregulation in the thymus of Down syndrome infants. Oncotarget 2016, 7, 7497–7533. [Google Scholar] [CrossRef]
- Ruike, Y.; Ichimura, A.; Tsuchiya, S.; Shimizu, K.; Kunimoto, R.; Okuno, Y.; Tsujimoto, G. Global correlation analysis for micro-RNA and mRNA expression profiles in human cell lines. J. Hum. Genet. 2008, 53, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.D.; Liu, N.; Xu, X.J. Bioinformatics analysis of biomarkers and transcriptional factor motifs in Down syndrome. Braz. J. Med. Biol. Res. 2014, 47, 834–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helguera, P.; Seiglie, J.; Rodriguez, J.; Hanna, M.; Helguera, G.; Busciglio, J. Adaptive downregulation of mitochondrial function in Down syndrome. Cell Metab. 2013, 17, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Borel, C.; Li, L.; Müller, T.; Williams, E.G.; Germain, P.L.; Buljan, M.; Sajic, T.; Boersema, P.J.; Shao, W.; et al. Systematic proteome and proteostasis profiling in human Trisomy 21 fibroblast cells. Nat. Commun. 2017, 8, 1212. [Google Scholar] [CrossRef] [PubMed]
- Yim, A.; Koti, P.; Bonnard, A.; Marchiano, F.; Dürrbaum, M.; Garcia-Perez, C.; Villaveces, J.; Gamal, S.; Cardone, G.; Perocchi, F.; et al. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Nucleic Acids Res. 2020, 48, 605–632. [Google Scholar] [CrossRef] [Green Version]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)-hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2020, 154, 104083. [Google Scholar] [CrossRef]
- Oláh, G.; Módis, K.; Törö, G.; Hellmich, M.R.; Szczesny, B.; Szabo, C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem. Pharmacol. 2018, 149, 186–204. [Google Scholar] [CrossRef]
- Toliver-Kinsky, T.; Cui, W.; Törö, G.; Lee, S.J.; Shatalin, K.; Nudler, E.; Szabo, C. H2S, a bacterial defense mechanism against the host immune response. Infect. Immun. 2018, 87, e00272-18. [Google Scholar] [CrossRef] [Green Version]
- Abdollahi Govar, A.; Törő, G.; Szaniszlo, P.; Pavlidou, A.; Bibli, S.I.; Thanki, K.; Resto, V.A.; Chao, C.; Hellmich, M.R.; Szabo, C.; et al. 3-Mercaptopyruvate sulfurtransferase supports endothelial cell angiogenesis and bioenergetics. Br. J. Pharmacol. 2020, 177, 866–883. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.J.; Wang, M.J.; Moore, P.K.; Jin, H.M.; Yao, T.; Zhu, Y.C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef]
- Huang, Y.; Li, F.; Tong, W.; Zhang, A.; He, Y.; Fu, T.; Liu, B. Hydrogen sulfide, a gaseous transmitter, stimulates proliferation of interstitial cells of Cajal via phosphorylation of AKT protein kinase. Tohoku J. Exp. Med. 2010, 221, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: Mechanisms and implications. Am. J. Physiol. Cell Physiol. 2017, 312, C3–C15. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.W.; Teo, X.Y.; Tay, E.Y.; Tan, C.H.; Hagen, T.; Moore, P.K.; Deng, L.W. Utilizing hydrogen sulfide as a novel anti-cancer agent by targeting cancer glycolysis and pH imbalance. Br. J. Pharmacol. 2014, 171, 4322–4336. [Google Scholar] [CrossRef] [Green Version]
- Paul, B.D.; Snyder, S.H. H2S: A Novel gasotransmitter that signals by sulfhydration. Trends Biochem. Sci. 2015, 40, 687–700. [Google Scholar] [CrossRef] [Green Version]
- Ostrakhovitch, E.A.; Akakura, S.; Sanokawa-Akakura, R.; Tabibzadeh, S. 3-Mercaptopyruvate sulfurtransferase disruption in dermal fibroblasts facilitates adipogenic trans-differentiation. Exp. Cell Res. 2019, 385, 111683. [Google Scholar] [CrossRef]
- Zhang, D.; Du, J.; Tang, C.; Huang, Y.; Jin, H. H2S-induced sulfhydration: Biological function and detection methodology. Front. Pharmacol. 2017, 8, 608. [Google Scholar] [CrossRef] [Green Version]
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H2S and polysulfide production and possible SOx production. Br. J. Pharmacol. 2018, 175, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Annerén, G.; Edman, B. Down syndrome—A gene dosage disease caused by trisomy of genes within a small segment of the long arm of chromosome 21, exemplified by the study of effects from the superoxide-dismutase type 1 (SOD-1) gene. APMIS Suppl. 1993, 40, 71–79. [Google Scholar]
- Muchová, J.; Žitňanová, I.; Ďuračková, Z. Oxidative stress and Down syndrome. Do antioxidants play a role in therapy? Physiol. Res. 2014, 63, 535–542. [Google Scholar]
- London, J.; Ndiaye, F.K.; Bui, L.C.; Souchet, B.; Daubigney, F.; Magnan, C.; Luquet, S.; Dairou, J.; Janel, N.; Rouch, C. Alterations in the serotonin and dopamine pathways by cystathionine beta synthase overexpression in murine brain. Mol. Neurobiol. 2019, 56, 3958–3971. [Google Scholar] [CrossRef] [PubMed]
- Régnier, V.; Billard, J.M.; Gupta, S.; Potier, B.; Woerner, S.; Paly, E.; Ledru, A.; David, S.; Luilier, S.; Bizot, J.C.; et al. Brain phenotype of transgenic mice overexpressing cystathionine β-synthase. PLoS ONE 2012, 7, e29056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, N.; Nagano, M.; Ito, T.; Shimamura, K.; Akimoto, T.; Suzuki, H. Antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice exhibit increased anxiety-like behaviors: A model for human mercaptolactate-cysteine disulfiduria. Sci. Rep. 2013, 3, 1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ide, M.; Ohnishi, T.; Toyoshima, M.; Balan, S.; Maekawa, M.; Shimamoto-Mitsuyama, C.; Iwayama, Y.; Ohba, H.; Watanabe, A.; Ishii, T.; et al. Excess hydrogen sulfide and polysulfides production underlies a schizophrenia pathophysiology. EMBO Mol. Med. 2019, 11, e10695. [Google Scholar] [CrossRef]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference ID | Group ID | Origin | Description | Gender | Age at Sampling |
|---|---|---|---|---|---|
| DETROIT 551 | CC | LGC STANDARDS | DIPLOID | FEMALE | FETUS |
| GM08447 | CC | CORIELL INSTITUTE | DIPLOID | FEMALE | NEWBORN |
| CCD1064SK | CC | LGC STANDARDS | DIPLOID | MALE | NEWBORN |
| GM05756 | CC | CORIELL INSTITUTE | DIPLOID | MALE | 2 MONTHS |
| GM00041 | CC | CORIELL INSTITUTE | DIPLOID | FEMALE | 5 MONTHS |
| GM05659 | CC | CORIELL INSTITUTE | DIPLOID | MALE | 12 MONTHS |
| 3-FCYPR10000286 | CC | JéRôME LEJEUNE INSTITUTE | DIPLOID | MALE | 5 YEARS |
| 1-FCYPR10000368 | CC | JéRôME LEJEUNE INSTITUTE | DIPLOID | FEMALE | 12 YEARS |
| GM04616 | DSC | CORIELL INSTITUTE | TRISOMY 21 | FEMALE | NEWBORN |
| DETROIT 532 | DSC | LGC STANDARDS | TRISOMY 21 | MALE | 2 MONTHS |
| GM02571 | DSC | CORIELL INSTITUTE | TRISOMY 21 | FEMALE | 3 MONTHS |
| AG07096 | DSC | CORIELL INSTITUTE | TRISOMY 21 | MALE | 5 MONTHS |
| AG05397 | DSC | CORIELL INSTITUTE | TRISOMY 21 | MALE | 1 YEAR |
| DETROIT 539 | DSC | LGC STANDARDS | TRISOMY 21 | FEMALE | 2 YEARS |
| 3-FCYPR10000285 | DSC | JéRôME LEJEUNE INSTITUTE | TRISOMY 21 | MALE | 5 YEARS |
| 3-FCYPR10000369 | DSC | JéRôME LEJEUNE INSTITUTE | TRISOMY 21 | FEMALE | 9 YEARS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panagaki, T.; Randi, E.B.; Szabo, C. Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation and Cellular Bioenergetics in Human Down Syndrome Fibroblasts. Biomolecules 2020, 10, 653. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040653
Panagaki T, Randi EB, Szabo C. Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation and Cellular Bioenergetics in Human Down Syndrome Fibroblasts. Biomolecules. 2020; 10(4):653. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040653
Chicago/Turabian StylePanagaki, Theodora, Elisa B. Randi, and Csaba Szabo. 2020. "Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation and Cellular Bioenergetics in Human Down Syndrome Fibroblasts" Biomolecules 10, no. 4: 653. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040653