Fluorescence Methods Applied to the Description of Urea-Dependent YME1L Protease Unfolding

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
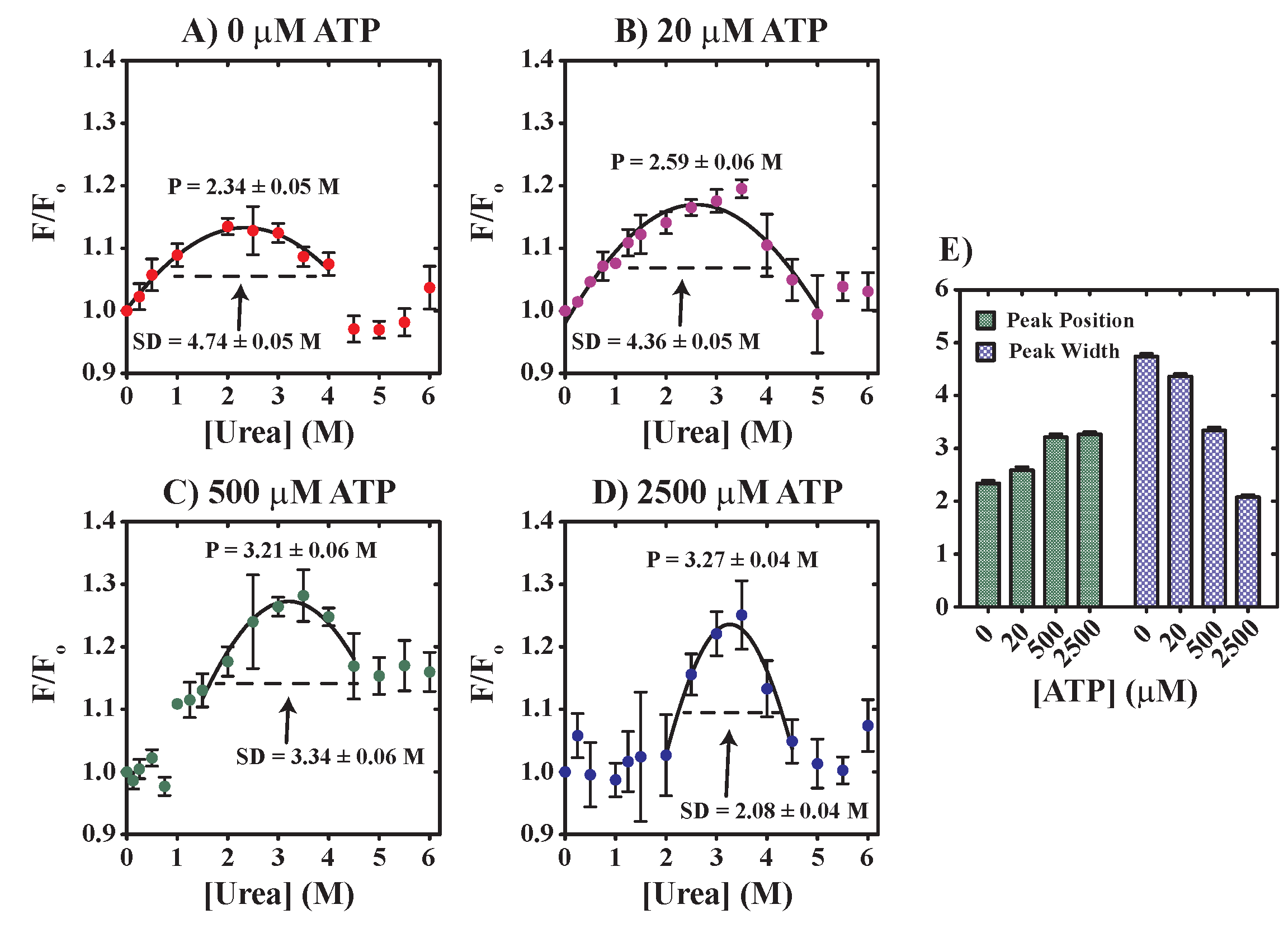
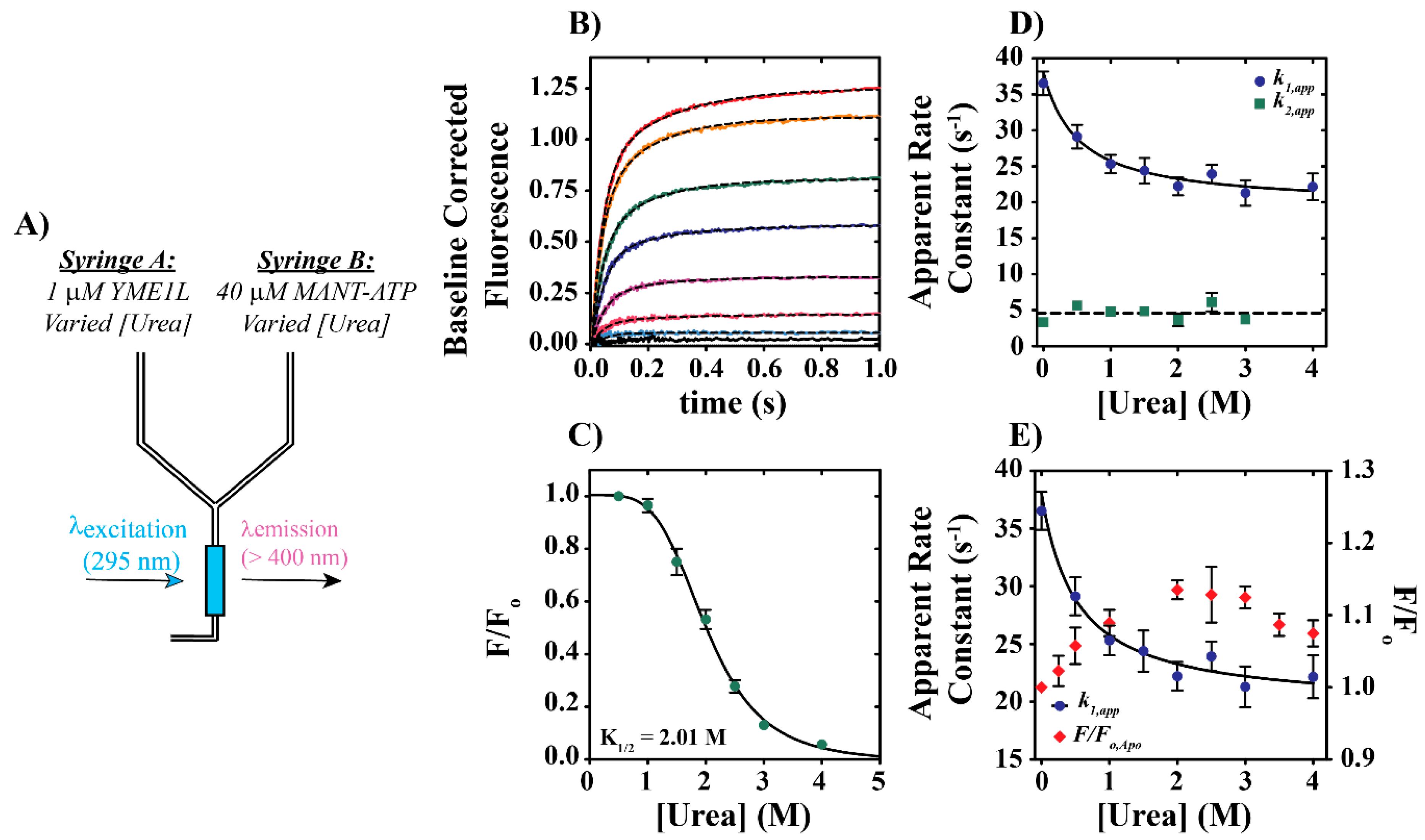
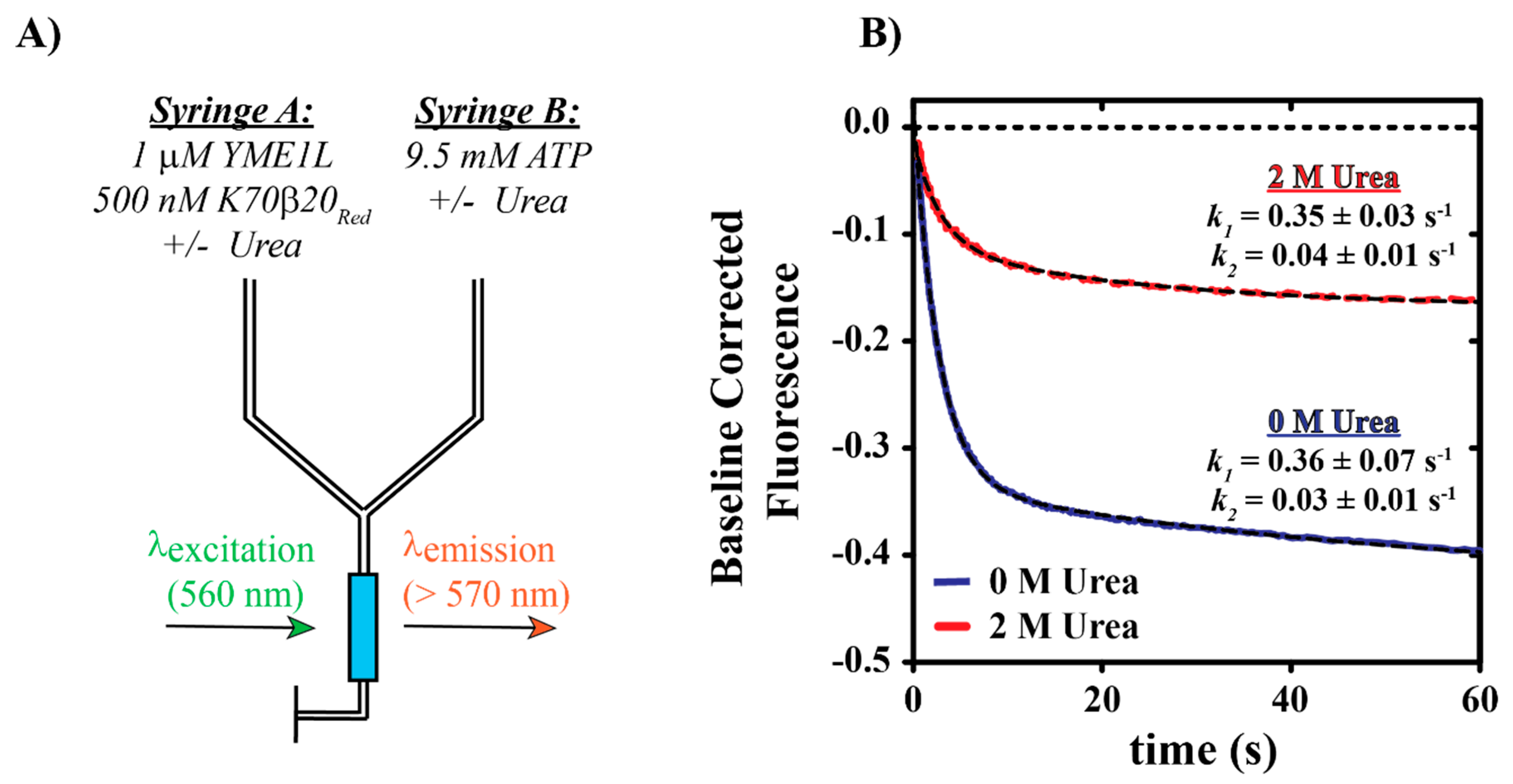
2.2.1. Stopped-Flow Fluorescence Assays
2.2.2. Equilibrium Unfolding Experiments
3. Results
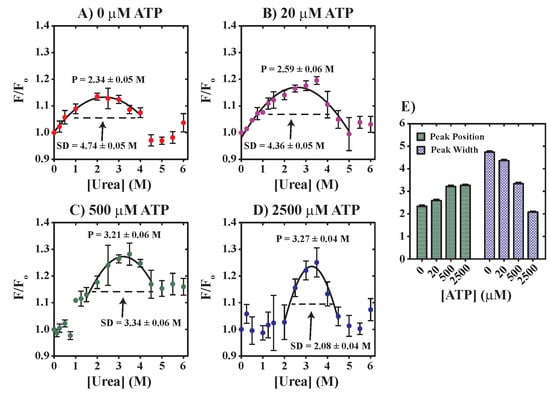
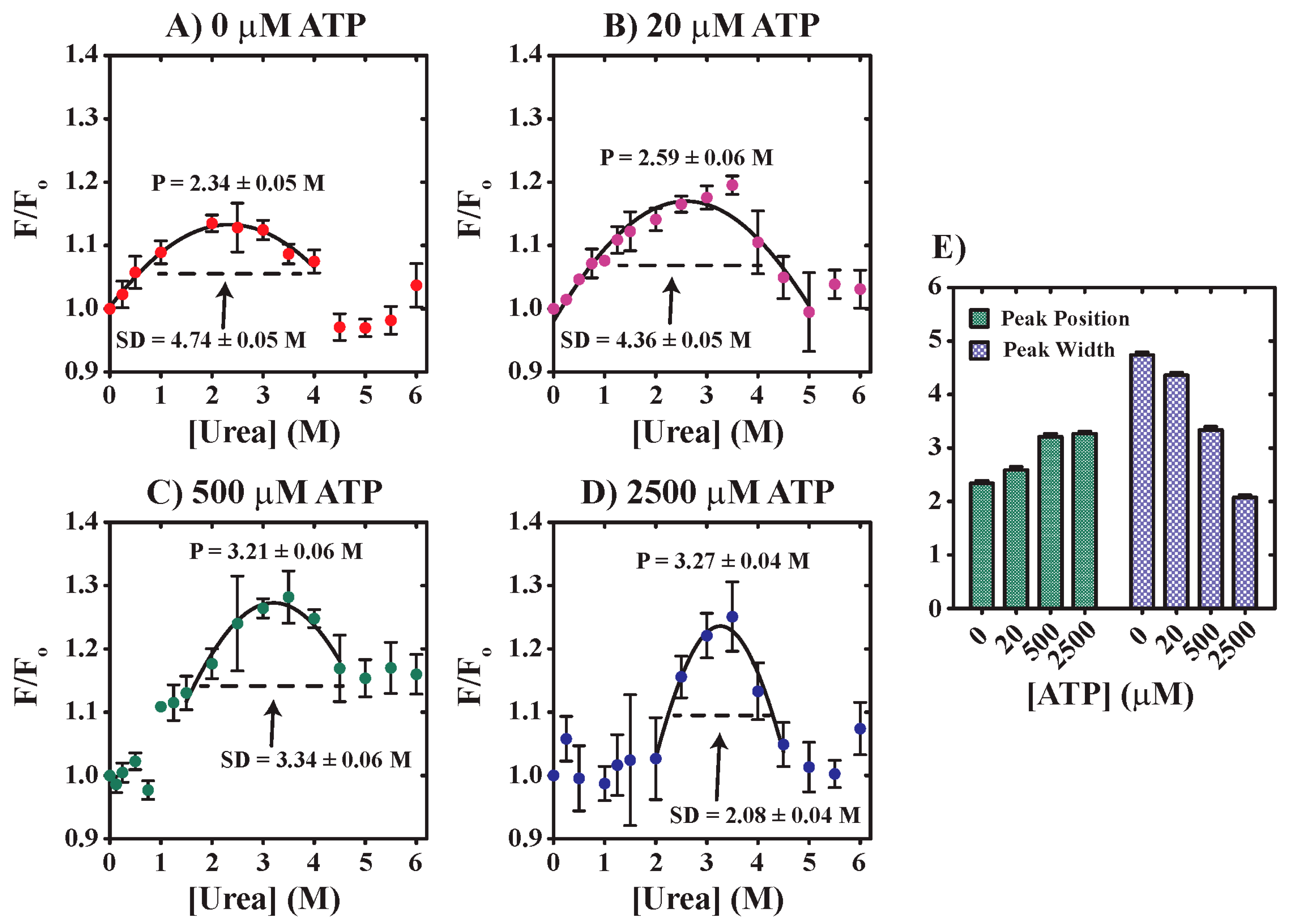
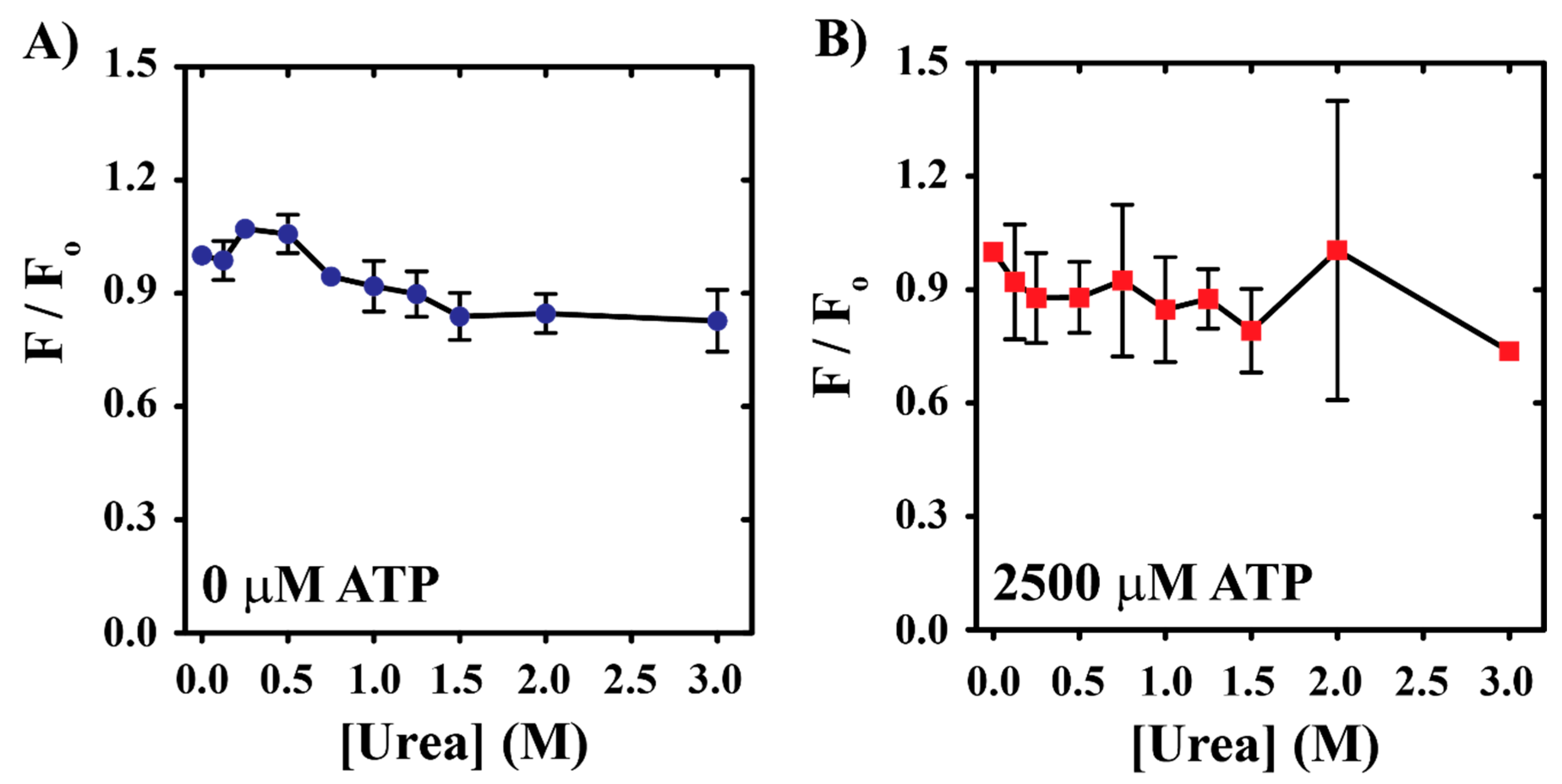
3.1. Acute Urea Stress Drives YME1L Denaturation
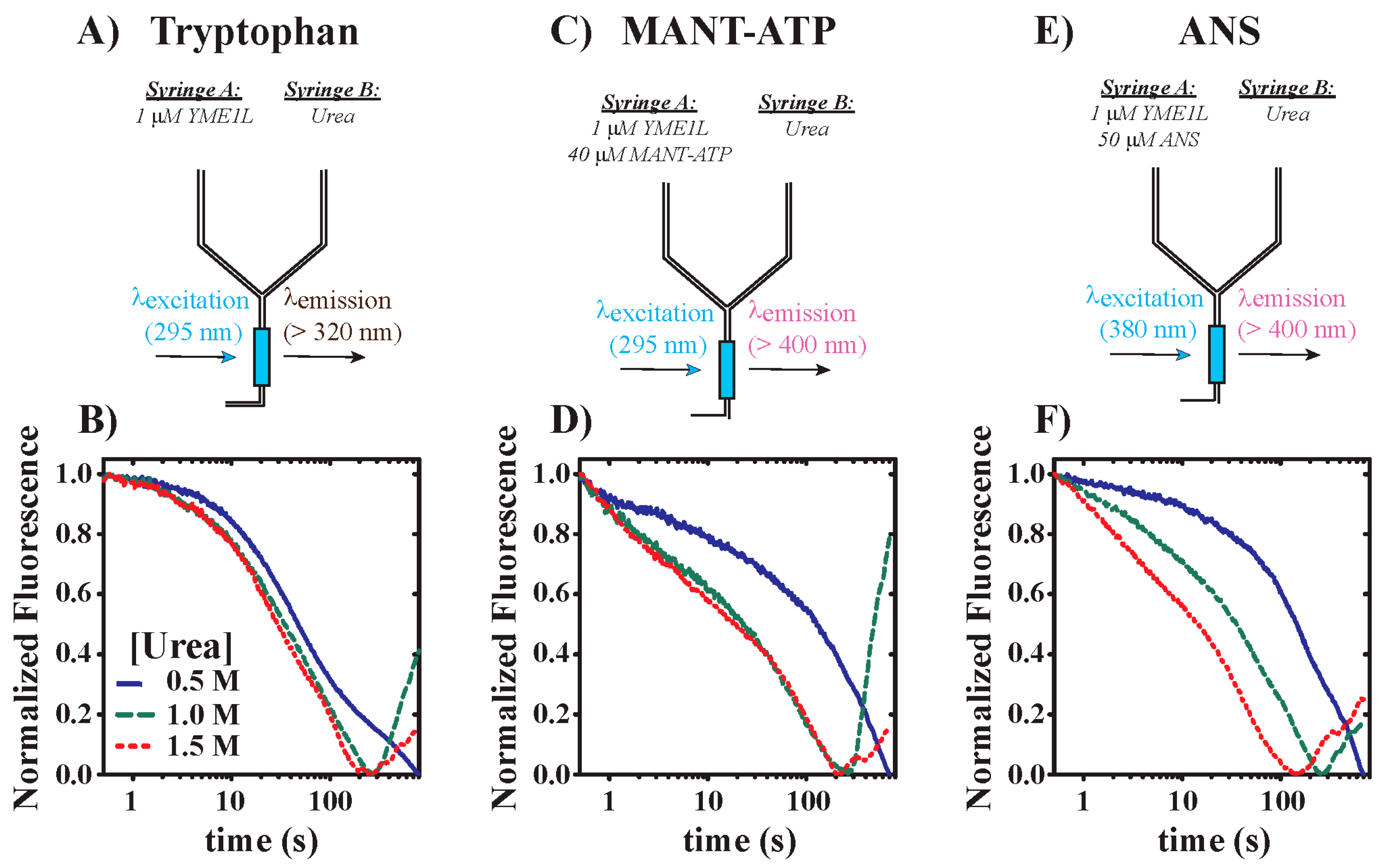
3.2. YME1L Incubation with Urea Drives Alterations in Tryptophan Solvent Environments
3.3. Evaluation of YME1L—The Functional Impact of Urea on Nucleotide Binding and Unfoldase Activities
4. Discussion
4.1. Model for YME1L Denaturation
4.2. Comparison with Other ATP-Dependent Proteases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NTP | Nucleoside Triphosphate |
| ATP | Adenosine Triphosphate |
| AAA+ | ATPases Associated with Various Cellular Activities |
| YME1 | Yeast Mitochondrial Escape Protein 1 |
| NLLS | Non-Linear Least Squares |
| LLS | Linear Least Squares |
| LEM | Linear Extrapolation Method |
References
- Gottesman, S.; Maurizi, M.R. Regulation by proteolysis: Energy-dependent proteases and their targets. Microbiol. Rev. 1992, 56, 592–621. [Google Scholar] [CrossRef] [PubMed]
- Levytskyy, R.M.; Bohovych, I.; Khalimonchuk, O. Metalloproteases of the Inner Mitochondrial Membrane. Biochemistry 2017, 56, 4737–4746. [Google Scholar] [CrossRef]
- Sauer, R.T.; A Baker, T. AAA+ Proteases: ATP-Fueled Machines of Protein Destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Alhuwaider, A.A.H.; Dougan, D.A. AAA+ Machines of Protein Destruction in Mycobacteria. Front. Mol. Biosci. 2017, 4, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Striebel, F.; Kress, W.; Weber-Ban, E. Controlled destruction: AAA+ ATPases in protein degradation from bacteria to eukaryotes. Curr. Opin. Struct. Boil. 2009, 19, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, S.; Maurizi, M.R.; Wickner, S. Regulatory subunits of energy-dependent proteases. Cell 1997, 91, 435–438. [Google Scholar] [CrossRef] [Green Version]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Genet. 2015, 14, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erzberger, J.P.; Berger, J.M. EVOLUTIONARY RELATIONSHIPS AND STRUCTURAL MECHANISMS OF AAA+ PROTEINS. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef]
- Miller, J.; Enemark, E.J. Fundamental Characteristics of AAA+ Protein Family Structure and Function. Archaea 2016, 2016, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar]
- A Baker, T.; Sauer, R.T. ATP-dependent proteases of bacteria: Recognition logic and operating principles. Trends Biochem. Sci. 2006, 31, 647–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puchades, C.; Ding, B.; Song, A.; Wiseman, R.L.; Lander, G.C.; Glynn, S.E. Unique Structural Features of the Mitochondrial AAA+ Protease AFG3L2 Reveal the Molecular Basis for Activity in Health and Disease. Mol. Cell 2019, 75, 1073–1085.e6. [Google Scholar] [CrossRef] [PubMed]
- Puchades, C.; Rampello, A.J.; Shin, M.; Giuliano, C.J.; Wiseman, R.L.; Glynn, S.E.; Lander, G.C. Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 2017, 358, eaao0464. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Beuron, F.; Kessel, M.; Wickner, S.; Maurizi, M.R.; Steven, A.C. Translocation pathway of protein substrates in ClpAP protease. Proc. Natl. Acad. Sci. USA 2001, 98, 4328–4333. [Google Scholar] [CrossRef] [Green Version]
- Gatsogiannis, C.; Balogh, D.; Merino, F.; Sieber, S.A.; Raunser, S. Cryo-EM structure of the ClpXP protein degradation machinery. Nat. Struct. Mol. Boil. 2019, 26, 946–954. [Google Scholar] [CrossRef]
- Gates, S.N.; Yokom, A.L.; Lin, J.; Jackrel, M.; Rizo, A.N.; Kendsersky, N.M.; Buell, C.E.; Sweeny, E.A.; Mack, K.L.; Chuang, E.; et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp1. Science 2017, 357, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieniossek, C.; Niederhauser, B.; Baumann, U. The crystal structure of apo-FtsH reveals domain movements necessary for substrate unfolding and translocation. Proc. Natl. Acad. Sci. USA 2009, 106, 21579–21584. [Google Scholar] [CrossRef] [Green Version]
- Ruer, M.; Krainer, G.; Gröger, P.; Schlierf, M. ATPase and Protease Domain Movements in the Bacterial AAA+ Protease FtsH Are Driven by Thermal Fluctuations. J. Mol. Boil. 2018, 430, 4592–4602. [Google Scholar] [CrossRef]
- Himmelfarb, J.; Stenvinkel, P.; Ikizler, T.A.; Hakim, R.M. The elephant in uremia: Oxidant stress as a unifying concept of cardiovascular disease in uremia. Kidney Int. 2002, 62, 1524–1538. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Mircescu, G. Oxidative Stress: An Accomplice to Uremic Toxicity? J. Ren. Nutr. 2006, 16, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Glassock, R.J. Uremic Toxins: What Are They? An Integrated Overview of Pathobiology and Classification. J. Ren. Nutr. 2008, 18, 2–6. [Google Scholar] [CrossRef]
- Vanholder, R.; for the European Uremic Toxin Work Group (EUTox); De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Glorieux, G.; De Smet, R.; Lameire, N. New insights in uremic toxins. Kidney Int. 2003, 63, S6–S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, A.; Rehan, M.; Subbarao, N.; Rabbani, G.; Khan, R.H. Elimination of Endogenous Toxin, Creatinine from Blood Plasma Depends on Albumin Conformation: Site Specific Uremic Toxicity & Impaired Drug Binding. PLOS ONE 2011, 6, e17230. [Google Scholar] [CrossRef] [Green Version]
- Baker, M.J.; Tatsuta, T.; Langer, T. Quality Control of Mitochondrial Proteostasis. Cold Spring Harb. Perspect. Boil. 2011, 3, a007559. [Google Scholar] [CrossRef] [Green Version]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Glynn, S.E. Multifunctional Mitochondrial AAA Proteases. Front. Mol. Biosci. 2017, 4, 34. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Rampello, A.J.; Glynn, S.E. Engineered AAA+ proteases reveal principles of proteolysis at the mitochondrial inner membrane. Nat. Commun. 2016, 7, 13301. [Google Scholar] [CrossRef] [Green Version]
- Brambley, C.A.; Marsee, J.D.; Halper, N.; Miller, J.M. Characterization of Mitochondrial YME1L Protease Oxidative Stress-Induced Conformational State. J. Mol. Boil. 2019, 431, 1250–1266. [Google Scholar] [CrossRef]
- Hiratsuka, T. New ribose-modified fluorescent analogs of adenine and guanine nucleotides available as subtrates for various enzymes. Biochim. et Biophys. Acta (BBA) - Protein Struct. Mol. Enzym. 1983, 742, 496–508. [Google Scholar] [CrossRef]
- Ptitsyn, O. Molten Globule and Protein Folding. Adv. Protein Chem. 1995, 47, 83–229. [Google Scholar] [CrossRef] [PubMed]
- Gasymov, O.K.; Glasgow, B.J. ANS fluorescence: Potential to augment the identification of the external binding sites of proteins. Biochim. et Biophys. Acta (BBA) - Bioenerg. 2007, 1774, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szász, C.; Alexa, A.; Tóth, K.; Rakacs, M.; Langowski, J.; Tompa, P. Protein Disorder Prevails under Crowded Conditions. Biochemistry 2011, 50, 5834–5844. [Google Scholar] [CrossRef]
- Holm, J.; Lawaetz, A.J.; Hansen, S.I. Ligand binding induces a sharp decrease in hydrophobicity of folate binding protein assessed by 1-anilinonaphthalene-8-sulphonate which suppresses self-association of the hydrophobic apo-protein. Biochem. Biophys. Res. Commun. 2012, 425, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 2nd ed.; Springer: New York, NY, USA, 1999; p. 954. [Google Scholar]
- Pace, C. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986, 131, 266–280. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Shaw, K.L. Linear extrapolation method of analyzing solvent denaturation curves. Proteins 2000, 41 (Suppl. 4), 1–7. [Google Scholar] [CrossRef]
- Bagnasco, S.M. How Renal Cells Handle Urea. Cell. Physiol. Biochem. 2000, 10, 379–384. [Google Scholar] [CrossRef]
- Zhang, Z.; Dmitrieva, N.; Park, J.-H.; Levine, R.L.; Burg, M.B. High urea and NaCl carbonylate proteins in renal cells in culture and in vivo, and high urea causes 8-oxoguanine lesions in their DNA. Proc. Natl. Acad. Sci. USA 2004, 101, 9491–9496. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Rozanov, D.; Cheltsov, A.; Nilsen, A.; Boniface, C.; Forquer, I.; Korkola, J.; Gray, J.; Tyner, J.; Tognon, C.E.; Mills, G.B.; et al. Targeting mitochondria in cancer therapy could provide a basis for the selective anti-cancer activity. PLoS ONE 2019, 14, e0205623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, D.R.; Cotter, T. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Suno, R.; Niwa, H.; Tsuchiya, D.; Zhang, X.; Yoshida, M.; Morikawa, K. Structure of the Whole Cytosolic Region of ATP-Dependent Protease FtsH. Mol. Cell 2006, 22, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Bieniossek, C.; Schalch, T.; Bumann, M.; Meister, M.; Meier, R.; Baumann, U. The molecular architecture of the metalloprotease FtsH. Proc. Natl. Acad. Sci. USA 2006, 103, 3066–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, C.; Prakash, S.; Lu, C.Z.; Matouschek, A.; Gross, C.A. Lack of a robust unfoldase activity confers a unique level of substrate specificity to the universal AAA protease FtsH. Mol. Cell 2011, 659–669. [Google Scholar] [CrossRef]
- Cooper, K.W.; Baneyx, F. Escherichia coli FtsH (HflB) Degrades a Membrane-Associated TolAI–II-β-Lactamase Fusion Protein under Highly Denaturing Conditions. Protein Expr. Purif. 2001, 21, 323–332. [Google Scholar] [CrossRef]
- Rudyak, S.G.; Brenowitz, M.; E Shrader, T. Mg2+-linked oligomerization modulates the catalytic activity of the Lon (La) protease from Mycobacterium smegmatis. Biochemistry 2001, 40, 9317–9323. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, S.; Pickens, A.; Rodriguez, J.L.; Marsee, J.D.; Miller, J.M. Fluorescence Methods Applied to the Description of Urea-Dependent YME1L Protease Unfolding. Biomolecules 2020, 10, 656. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040656
Moore S, Pickens A, Rodriguez JL, Marsee JD, Miller JM. Fluorescence Methods Applied to the Description of Urea-Dependent YME1L Protease Unfolding. Biomolecules. 2020; 10(4):656. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040656
Chicago/Turabian StyleMoore, Sydney, Alyssa Pickens, Jessica L. Rodriguez, Justin D. Marsee, and Justin M. Miller. 2020. "Fluorescence Methods Applied to the Description of Urea-Dependent YME1L Protease Unfolding" Biomolecules 10, no. 4: 656. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10040656