Differentiation of Murine C2C12 Myoblasts Strongly Reduces the Effects of Myostatin on Intracellular Signaling


Abstract
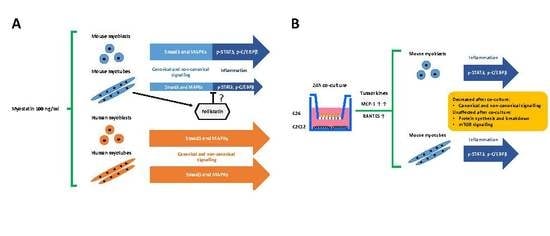
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Production and Administration of Myostatin
2.3. Transwell® Method, Coculture of C2C12 and C26 Cells
2.4. SUnSET Method for the Analysis of Protein Synthesis, Protein Extraction, and Western Blotting
2.5. RNA Extraction, cDNA Synthesis, and Quantitative Real-Time PCR
2.6. Multiplex Cytokine Assay
2.7. Statistical Analyses
3. Results
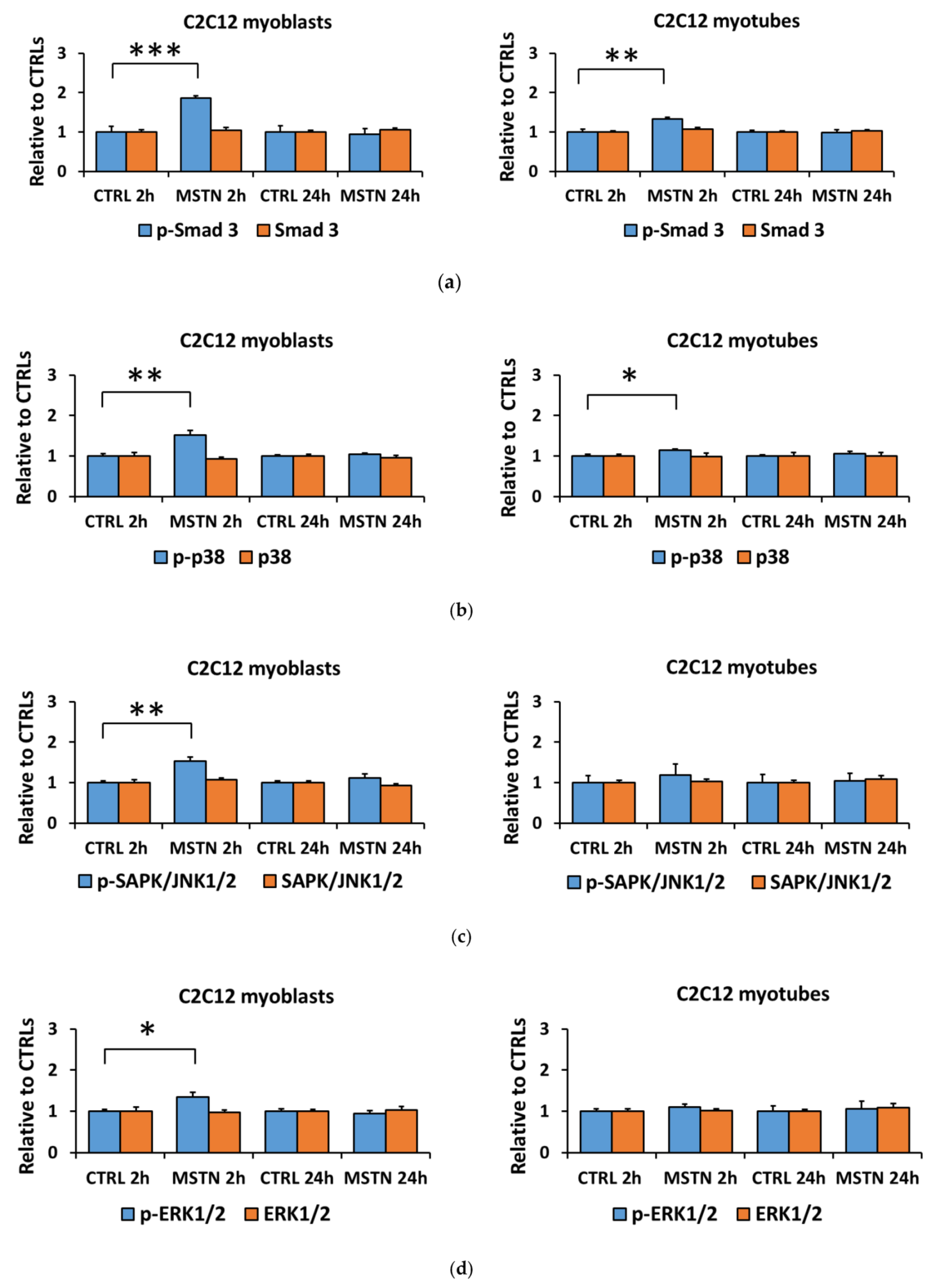
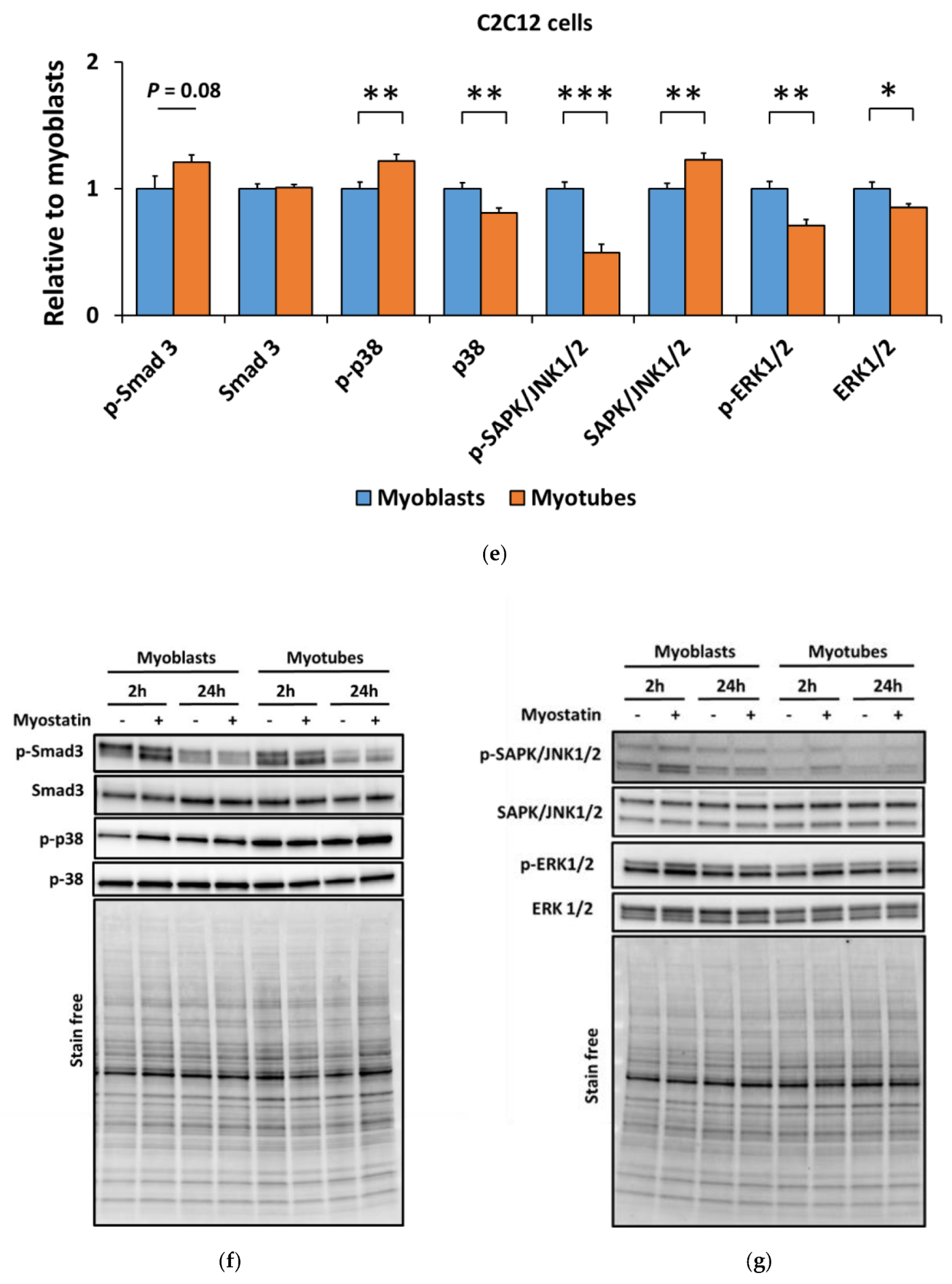
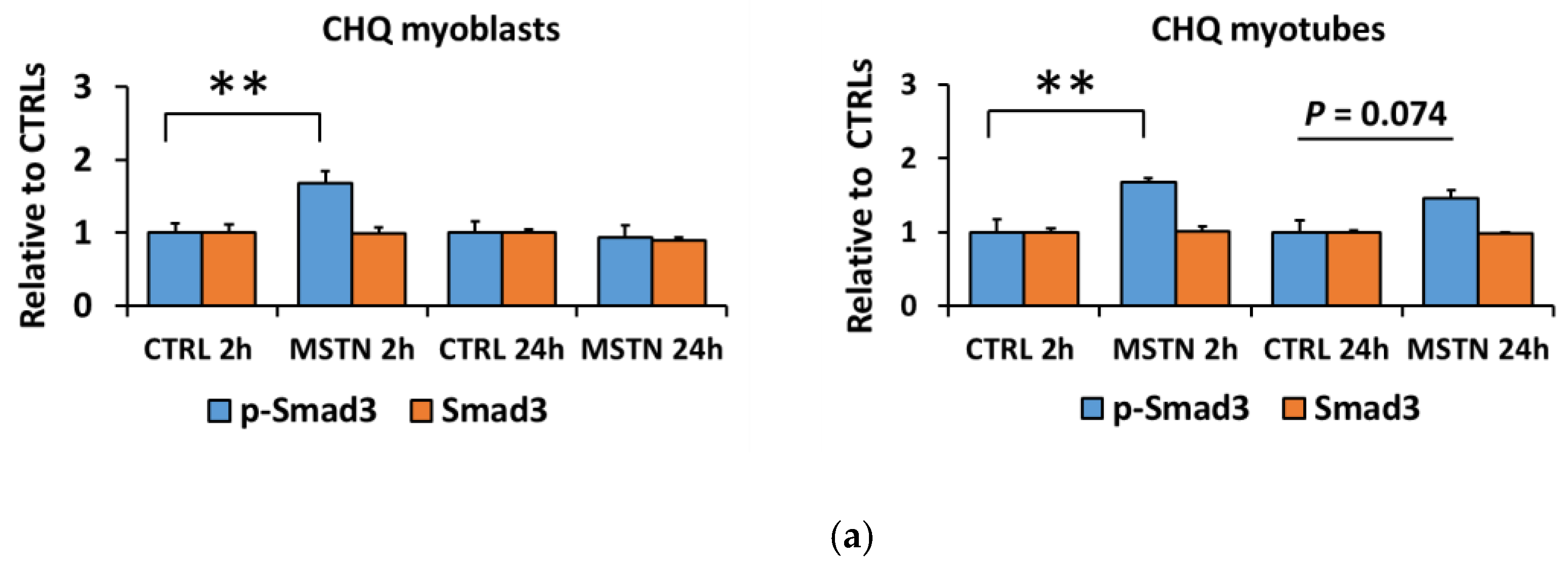
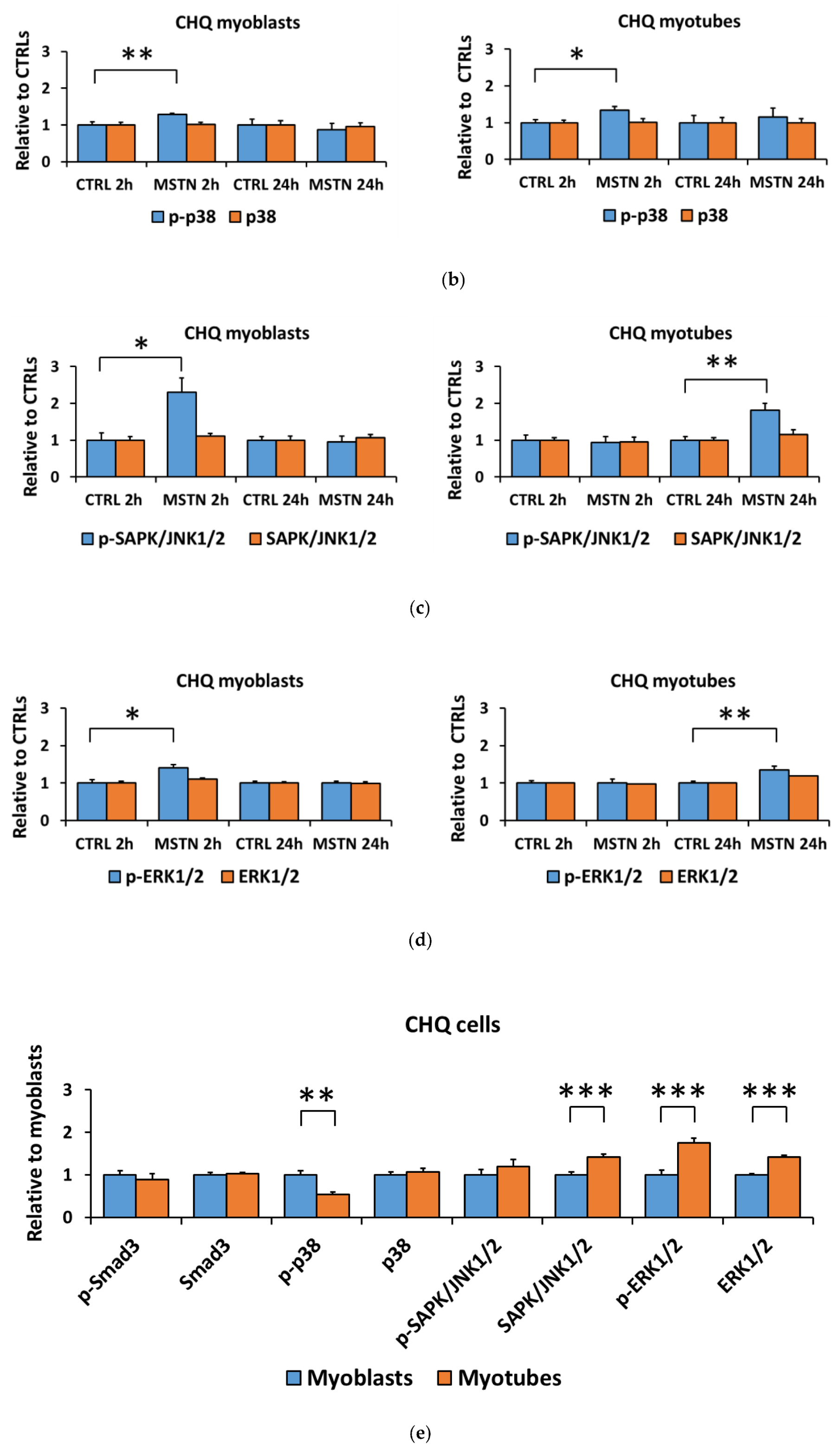
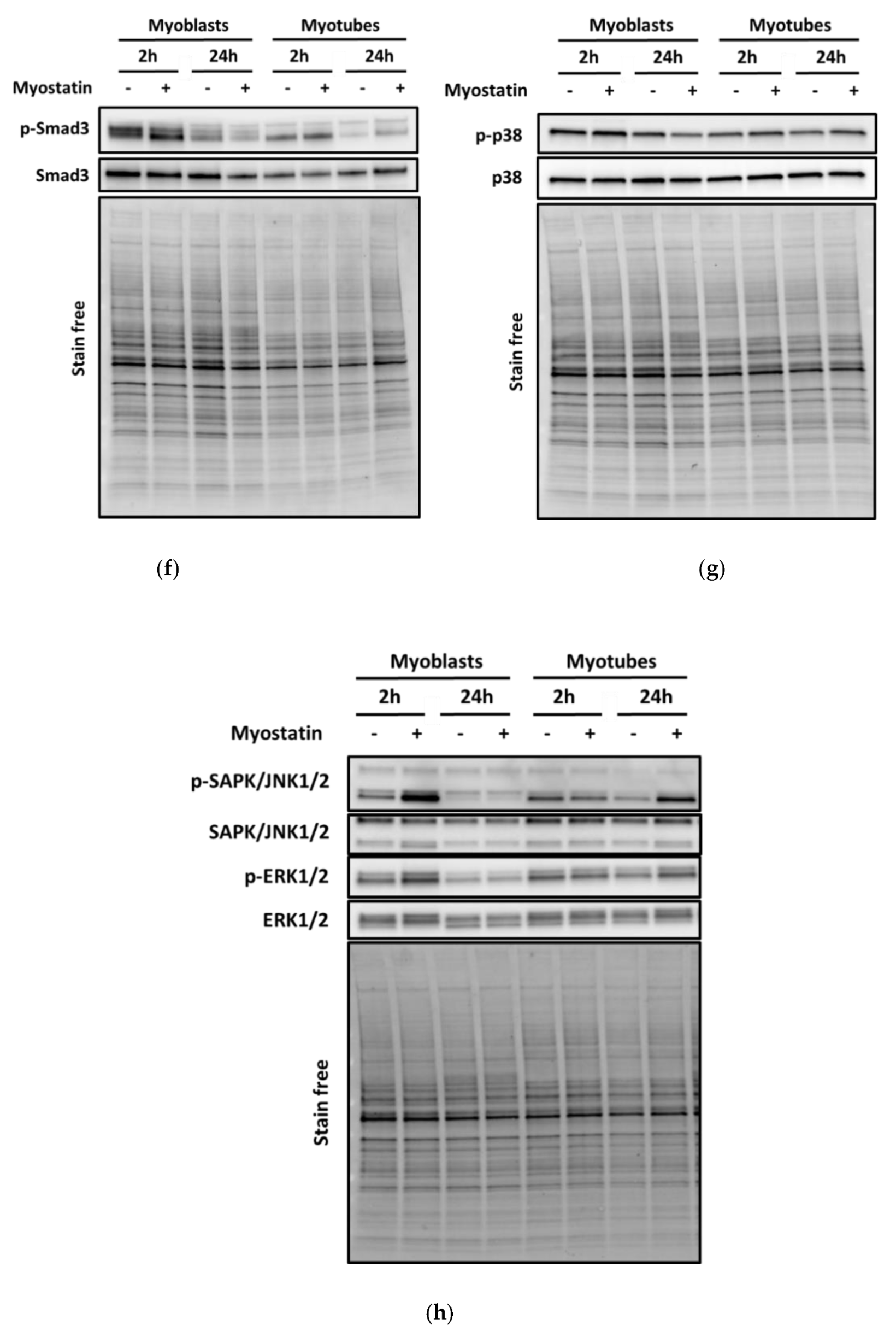
3.1. Myostatin Had a Larger Effect on the Canonical, Noncanonical, and Inflammatory Signaling in C2C12 Myoblasts than in Myotubes
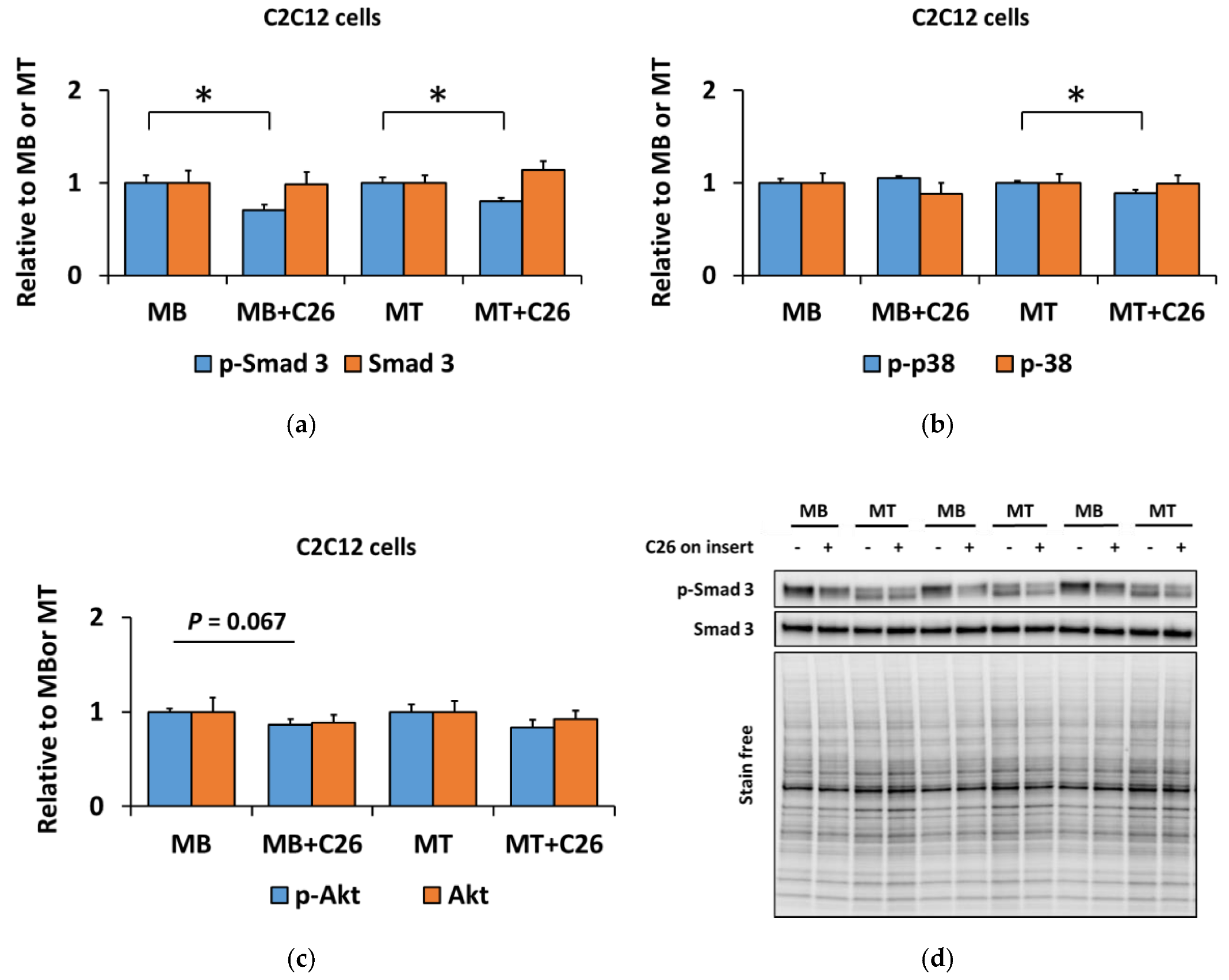
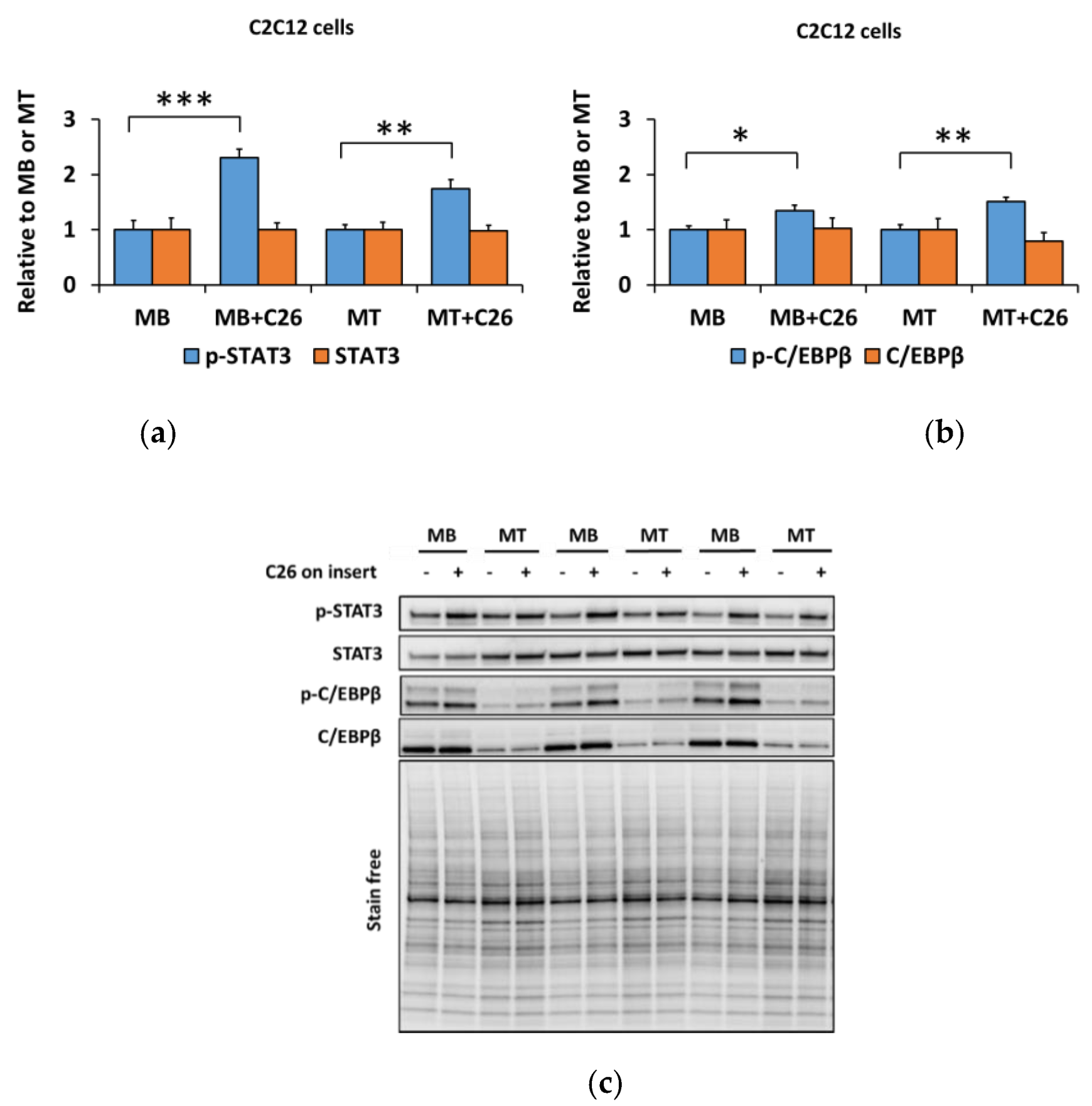
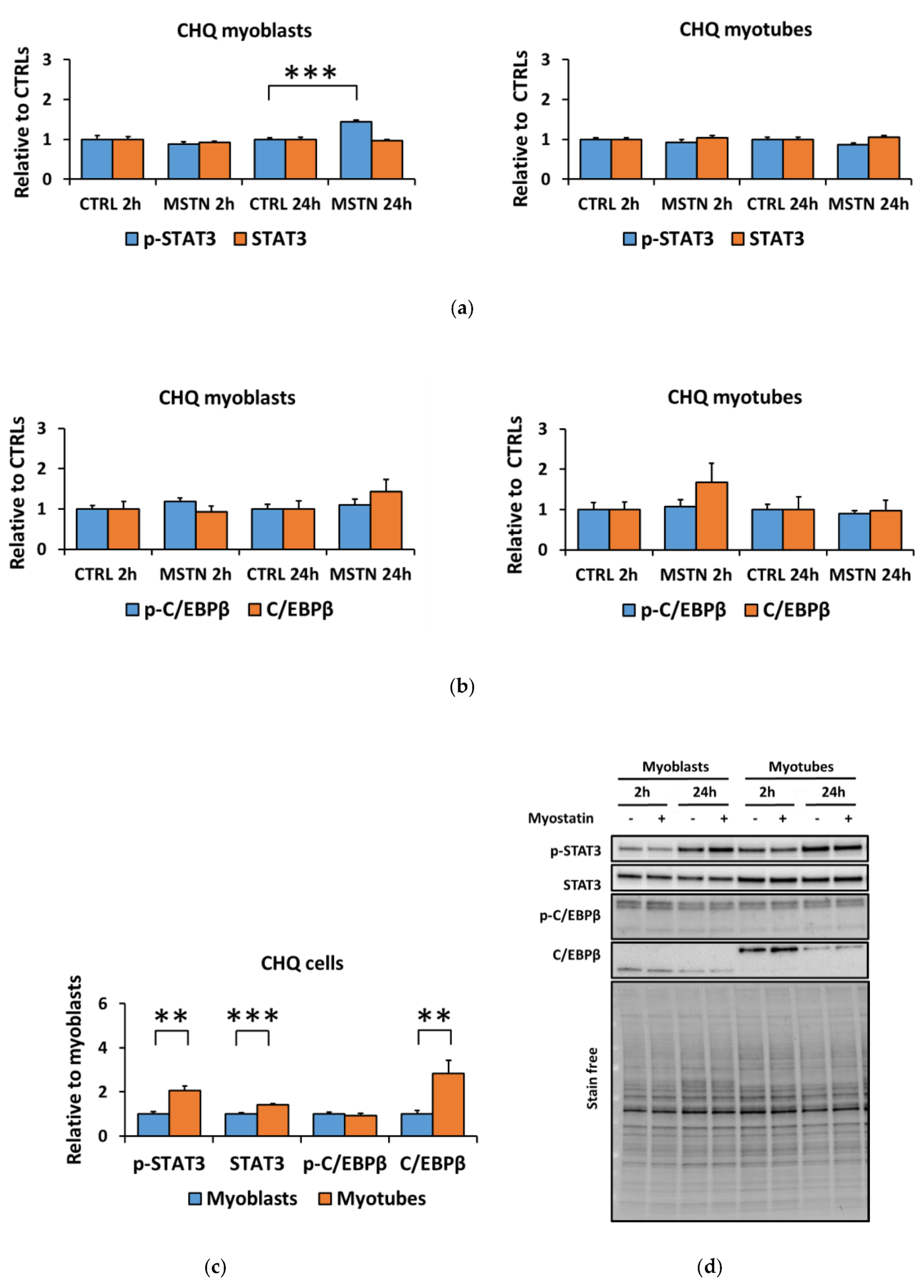
3.2. The Coculture with C26 Cells Had Minor Effects on Canonical and Noncanonical Signaling in C2C12 Cells, but Increased Inflammatory Signaling Similar to the Effect of Myostatin
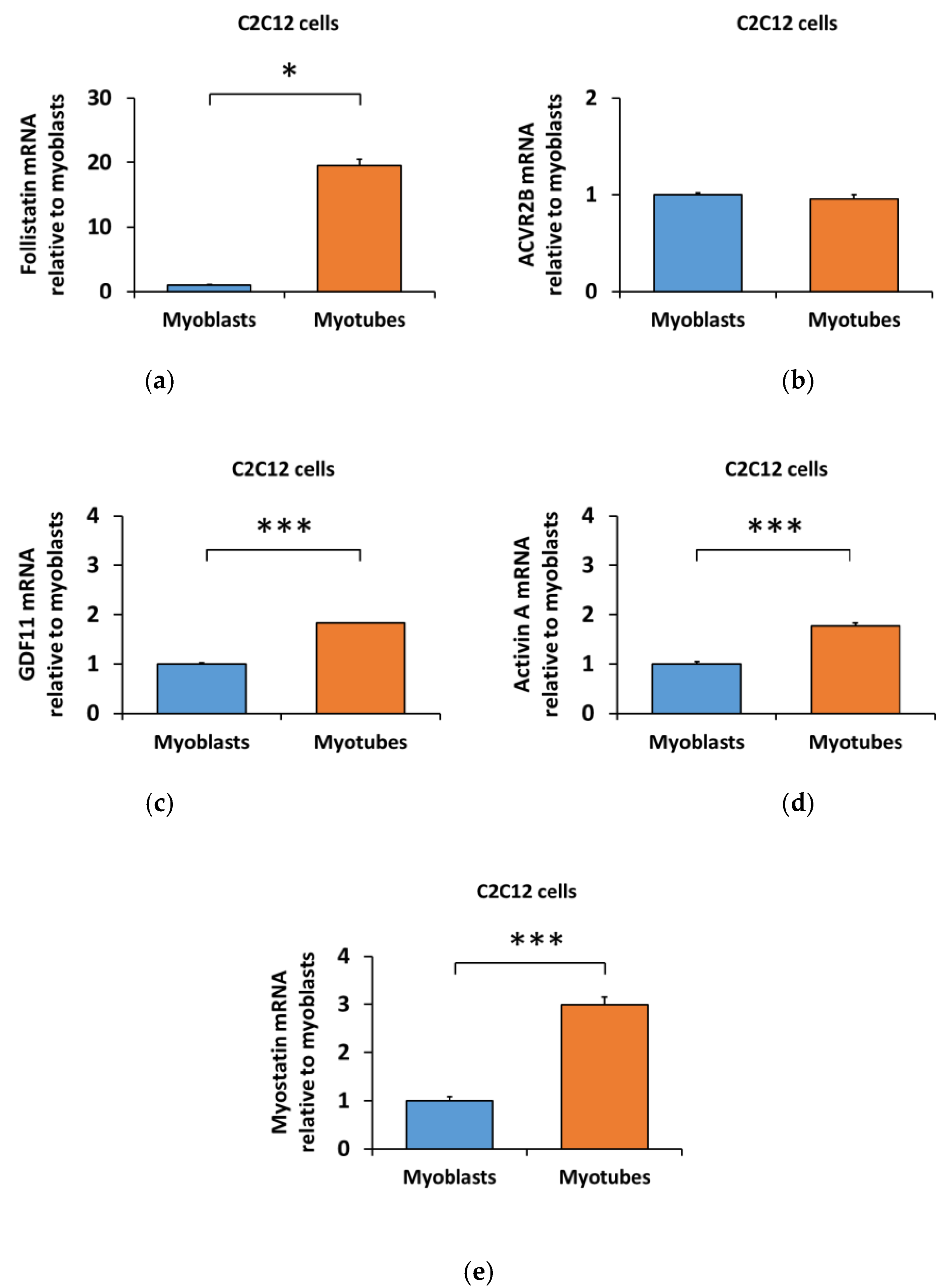
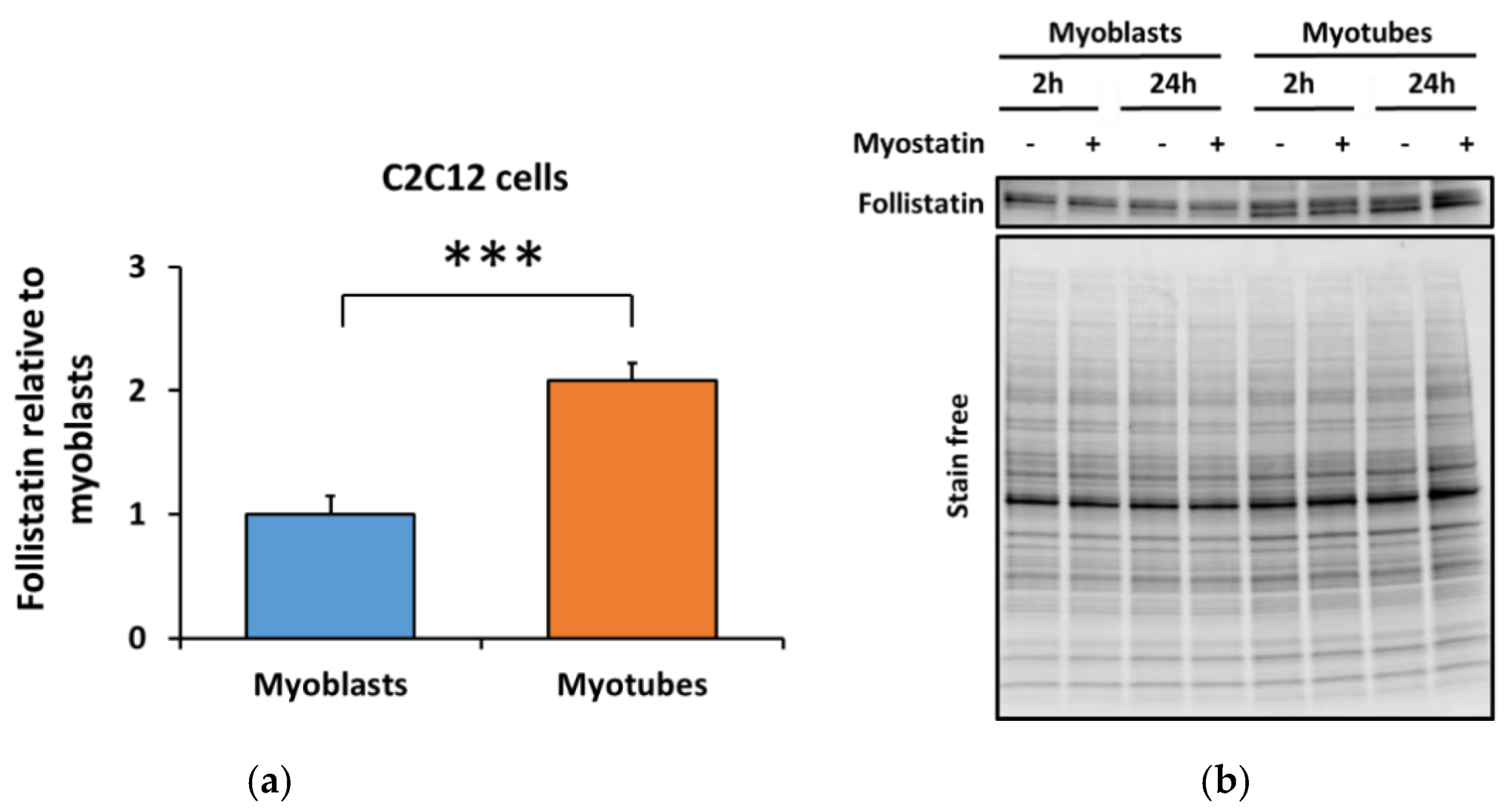
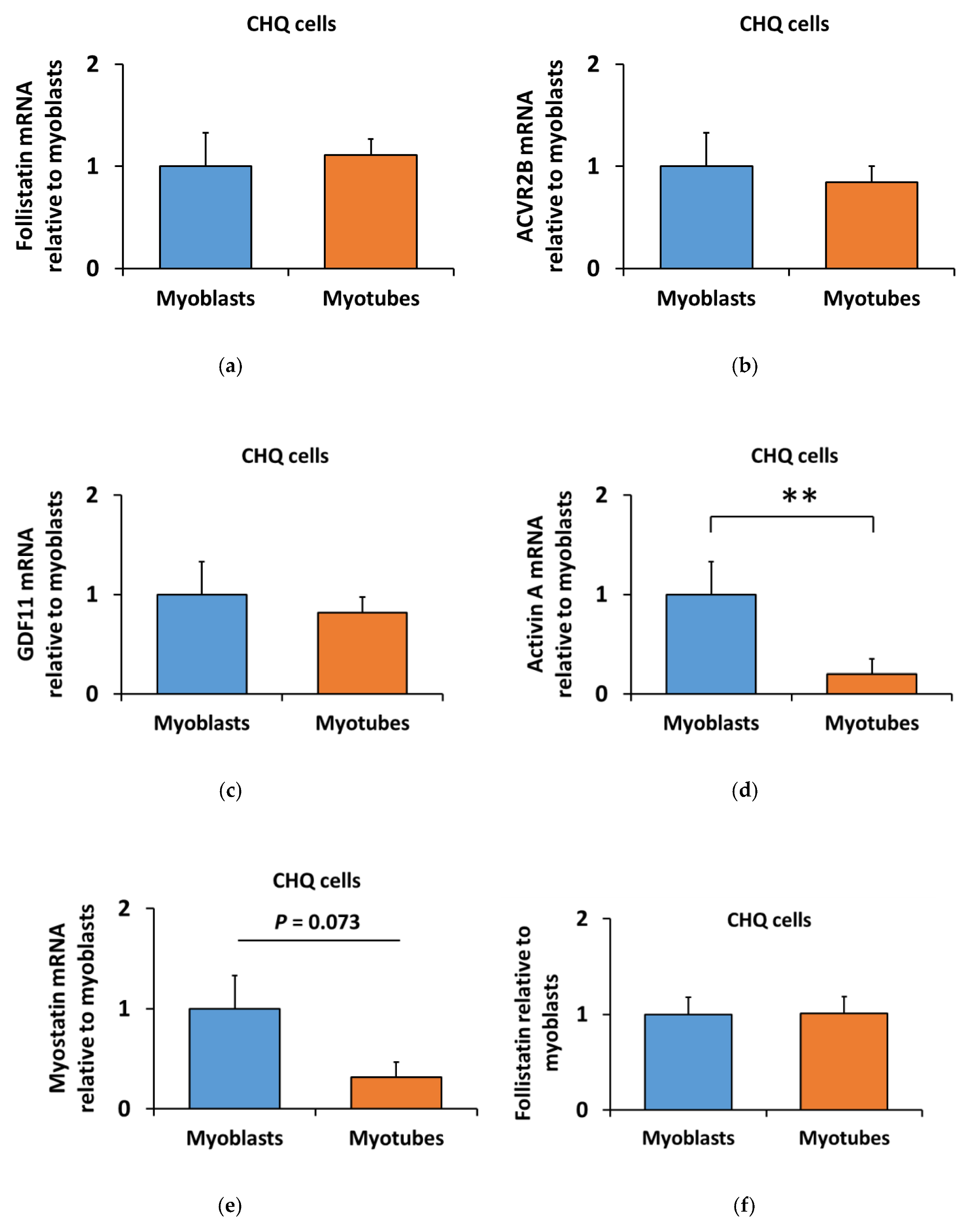
3.3. Lower Myostatin Responsiveness in C2C12 Myotubes Was Associated with Altered Gene Expression of Myostatin Regulators
3.4. Human Skeletal Myotubes Are Responsive to Myostatin
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACVR2 | Activin receptor type 2 |
| ACVR2B | Activin receptor type 2B |
| ALK4 or 5 | Activin receptor-like kinase 4 or 5 |
| BCA | Bicinchoninic Acid |
| C26-CM | Colon 26 carcinoma cell conditioned medium |
| C/EBPβ | CCAAT-enhancer-binding protein β |
| DM | Differentiation medium |
| DMEM | Dulbecco´s modified Eagle´s medium |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FBS | Fetal bovine serum |
| GDF | Growth differentiation factor |
| GM | Growth medium |
| HS | Horse serum |
| IL-6 | Interleukin-6 |
| MAPK(s) | Mitogen-activated protein kinase(s) |
| MCP-1 | Monocyte chemoattractant protein-1 or CCL2 |
| PBS | Phosphate buffered saline |
| P/S | Penicillin-streptomycin |
| RANTES | Regulated on activation, normal T cell expressed and secreted or CCL5 |
| SAPK/JNK1/2 | Stress-activated protein kinase/c-Jun N-terminal kinase 1/2 |
| STAT3 | Signal transducer and activator of transcription 3 |
| Smad3 | Small mothers against decapentaplegic 3 |
| TGF-β | Transforming growth factor-β |
References
- Lee, S.; Reed, L.A.; Davies, M.V.; Girgenrath, S.; Goad, M.E.; Tomkinson, K.N.; Wright, J.F.; Barker, C.; Ehrmantraut, G.; Holmstrom, J. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 18117–18122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, A.; Patel, K. Signalling and the control of skeletal muscle size. Exp. Cell Res. 2010, 316, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kambadur, R.; Matthews, K.G.; Somers, W.G.; Devlin, G.P.; Conaglen, J.V.; Fowke, P.J.; Bass, J.J. Myostatin, a transforming growth factor-β superfamily member, is expressed in heart muscle and is upregulated in cardiomyocytes after infarct. J. Cell. Physiol. 1999, 180, 1–9. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S. Regulation of skeletal muscle mass in mice by a new TGF-p superfamily member. Nature 1997, 387, 83. [Google Scholar] [CrossRef]
- Rodriguez, J.; Vernus, B.; Chelh, I.; Cassar-Malek, I.; Gabillard, J.; Sassi, A.H.; Seiliez, I.; Picard, B.; Bonnieu, A. Myostatin and the skeletal muscle atrophy and hypertrophy signaling pathways. Cell. Mol. Life Sci. 2014, 71, 4361–4371. [Google Scholar] [CrossRef]
- Dschietzig, T.B. Myostatin—From the mighty mouse to cardiovascular disease and cachexia. Clin. Chim. Acta 2014, 433, 216–224. [Google Scholar] [CrossRef]
- Egerman, M.A.; Glass, D.J. Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Elkina, Y.; von Haehling, S.; Anker, S.D.; Springer, J. The role of myostatin in muscle wasting: An overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143. [Google Scholar] [CrossRef] [Green Version]
- Kingsley, D.M. The TGF-beta superfamily: New members, new receptors, and new genetic tests of function in different organisms. Genes Dev. 1994, 8, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Goebel, E.J.; Hart, K.N.; McCoy, J.C.; Thompson, T.B. Structural biology of the TGFβ family. Exp. Biol. Med. 2019, 244, 1530–1546. [Google Scholar] [CrossRef] [PubMed]
- Nissinen, T.A.; Hentilä, J.; Penna, F.; Lampinen, A.; Lautaoja, J.H.; Fachada, V.; Holopainen, T.; Ritvos, O.; Kivelä, R.; Hulmi, J.J. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J. Cachexia Sarcopenia Muscle 2018, 9, 514–529. [Google Scholar] [CrossRef] [PubMed]
- Lach-Trifilieff, E.; Minetti, G.C.; Sheppard, K.; Ibebunjo, C.; Feige, J.N.; Hartmann, S.; Brachat, S.; Rivet, H.; Koelbing, C.; Morvan, F. An antibody blocking activin type II receptors induces strong skeletal muscle hypertrophy and protects from atrophy. Mol. Cell. Biol. 2014, 34, 606–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iskenderian, A.; Liu, N.; Deng, Q.; Huang, Y.; Shen, C.; Palmieri, K.; Crooker, R.; Lundberg, D.; Kastrapeli, N.; Pescatore, B. Myostatin and activin blockade by engineered follistatin results in hypertrophy and improves dystrophic pathology in mdx mouse more than myostatin blockade alone. Skelet. Muscle 2018, 8, 34. [Google Scholar] [CrossRef]
- Lee, S.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [Green Version]
- Abdelmoez, A.M.; Sardón Puig, L.; Smith, J.A.; Gabriel, B.M.; Savikj, M.; Dollet, L.; Chibalin, A.V.; Krook, A.; Zierath, J.R.; Pillon, N.J. Comparative profiling of skeletal muscle models reveals heterogeneity of transcriptome and metabolism. Am. J. Physiol. Cell Physiol. 2019, 318, C615–C626. [Google Scholar] [CrossRef] [Green Version]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, B.D.; Wiedeback, B.D.; Hoversten, K.E.; Jackson, M.F.; Walker, R.G.; Thompson, T.B. Myostatin stimulates, not inihibits, C2C12 myoblast proliferation. Endocrinology 2014, 155, 670–675. [Google Scholar] [CrossRef] [Green Version]
- Pèrié, L.; Parenté, A.; Brun, C.; Magnol, L.; Pélissier, P.; Blanquet, V. Enhancement of C2C12 myoblast proliferation and differentiation by GASP-2, a myostatin inhibitor. Biochem. Biophys. Rep. 2016, 6, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Uemura, K.; Hayashi, M.; Itsubo, T.; Oishi, A.; Iwakawa, H.; Komatsu, M.; Uchiyama, S.; Kato, H. Myostatin promotes tenogenic differentiation of C2C12 myoblast cells through Smad. FEBS Open Bio 2017, 7, 522–532. [Google Scholar] [CrossRef]
- Graham, Z.A.; De Gasperi, R.; Bauman, W.A.; Cardozo, C.P. Recombinant myostatin reduces highly expressed microRNAs in differentiating C2C12 cells. Biochem. Biophys. Rep. 2017, 9, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Burks, T.N.; Cohn, R.D. Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 2011, 1, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloise, E.; Ciarmela, P.; Dela Cruz, C.; Luisi, S.; Petraglia, F.; Reis, F.M. Activin a in mammalian physiology. Physiol Rev. 2018, 99, 739–780. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577. [Google Scholar] [CrossRef]
- Rebbapragada, A.; Benchabane, H.; Wrana, J.L.; Celeste, A.J.; Attisano, L. Myostatin signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Mol. Cell. Biol. 2003, 23, 7230–7242. [Google Scholar] [CrossRef] [Green Version]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef]
- Tisdale, M.J. Reversing cachexia. Cell 2010, 142, 511–512. [Google Scholar] [CrossRef] [Green Version]
- Philip, B.; Lu, Z.; Gao, Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell. Signal. 2005, 17, 365–375. [Google Scholar] [CrossRef]
- Yang, W.; Chen, Y.; Zhang, Y.; Wang, X.; Yang, N.; Zhu, D. Extracellular Signal–Regulated Kinase 1/2 Mitogen-Activated Protein Kinase Pathway Is Involved in Myostatin-Regulated Differentiation Repression. Cancer Res. 2006, 66, 1320–1326. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Chen, D.; Zhang, K.; Yu, B.; Chen, X.; Meng, J. Regulation of myostatin signaling by c-Jun N-terminal kinase in C2C12 cells. Cell. Signal. 2007, 19, 2286–2295. [Google Scholar] [CrossRef]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef]
- Zhang, L.; Rajan, V.; Lin, E.; Hu, Z.; Han, H.Q.; Zhou, X.; Song, Y.; Min, H.; Wang, X.; Du, J. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J. 2011, 25, 1653–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, C.L.; Kleckner, I.R.; Jatoi, A.; Schwarz, E.M.; Dunne, R.F. The Role of Systemic Inflammation in Cancer-Associated Muscle Wasting and Rationale for Exercise as a Therapeutic Intervention. JCSM Clin. Rep. 2018, 3, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Zimmers, T.A.; Fishel, M.L.; Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 28–41. [Google Scholar] [CrossRef] [Green Version]
- Poli, V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 1998, 273, 29279–29282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Gao, Q.; Han, S.; Pan, F.; Fan, W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-Twist signaling. Tumor Biol. 2015, 36, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Gupta, R.; Lee, A.C.; Ma, M.; Fang, F.; Tolbert, C.N.; Walts, A.D.; Beltran, L.E.; San, H.; Chen, G. Stat3-dependent acute Rantes production in vascular smooth muscle cells modulates inflammation following arterial injury in mice. J. Clin. Investig. 2010, 120, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J. Circulating Activin A predicts survival in cancer patients. J. Cachexia Sarcopenia Muscle 2017, 8, 768–777. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Jin, B.; Li, Y. C/EBPβ mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef] [Green Version]
- Jackman, R.W.; Floro, J.; Yoshimine, R.; Zitin, B.; Eiampikul, M.; El-Jack, K.; Seto, D.N.; Kandarian, S.C. Continuous Release of Tumor-Derived Factors Improves the Modeling of Cachexia in Muscle Cell Culture. Front. Physiol. 2017, 8, 738. [Google Scholar] [CrossRef]
- Seto, D.N.; Kandarian, S.C.; Jackman, R.W. A key role for leukemia inhibitory factor in C26 cancer cachexia. J. Biol. Chem. 2015, 290, 19976–19986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renaud, J.; Martinoli, M. Development of an insert co-culture system of two cellular types in the absence of cell-cell contact. JoVE J. Vis. Exp. 2016, e54356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadient, R.A.; Patterson, P.H. Leukemia inhibitory factor, Interleukin 6, and other cytokines using the GP130 transducing receptor: Roles in inflammation and injury. Stem Cells 1999, 17, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Edom, F.; Mouly, V.; Barbet, J.P.; Fiszman, M.Y.; Butler-Browne, G.S. Clones of human satellite cells can express in vitro both fast and slow myosin heavy chains. Dev. Biol. 1994, 164, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Mouly, V.; Cooper, R.N.; Mamchaoui, K.; Bigot, A.; Shay, J.W.; Di Santo, J.P.; Butler-Browne, G.S.; Wright, W.E. Cellular senescence in human myoblasts is overcome by human telomerase reverse transcriptase and cyclin-dependent kinase 4: Consequences in aging muscle and therapeutic strategies for muscular dystrophies. Aging Cell 2007, 6, 515–523. [Google Scholar] [CrossRef]
- Thorley, M.; Duguez, S.; Mazza, E.M.C.; Valsoni, S.; Bigot, A.; Mamchaoui, K.; Harmon, B.; Voit, T.; Mouly, V.; Duddy, W. Skeletal muscle characteristics are preserved in hTERT/cdk4 human myogenic cell lines. Skelet. Muscle 2016, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Charles, J.P.; Cappellari, O.; Spence, A.J.; Hutchinson, J.R.; Wells, D.J. Musculoskeletal geometry, muscle architecture and functional specialisations of the mouse hindlimb. PLoS ONE 2016, 11, e0147669. [Google Scholar] [CrossRef] [Green Version]
- Pekkala, S.; Wiklund, P.; Hulmi, J.J.; Pöllänen, E.; Marjomäki, V.; Munukka, E.; Pierre, P.; Mouly, V.; Mero, A.; Alén, M. Cannabinoid receptor 1 and acute resistance exercise–In vivo and in vitro studies in human skeletal muscle. Peptides 2015, 67, 55–63. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Oliveira, B.M.; Silvennoinen, M.; Hoogaars, W.M.; Ma, H.; Pierre, P.; Pasternack, A.; Kainulainen, H.; Ritvos, O. Muscle protein synthesis, mTORC1/MAPK/Hippo signaling, and capillary density are altered by blocking of myostatin and activins. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E41–E50. [Google Scholar] [CrossRef]
- Gray, A.M.; Mason, A.J. Requirement for activin A and transforming growth factor--beta 1 pro-regions in homodimer assembly. Science 1990, 247, 1328–1330. [Google Scholar] [CrossRef]
- Cotton, T.R.; Fischer, G.; Wang, X.; McCoy, J.C.; Czepnik, M.; Thompson, T.B.; Hyvönen, M. Structure of the human myostatin precursor and determinants of growth factor latency. EMBO J. 2018, 37, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Kaivo-Oja, N.; Mottershead, D.G.; Mazerbourg, S.; Myllymaa, S.; Duprat, S.; Gilchrist, R.B.; Groome, N.P.; Hsueh, A.J.; Ritvos, O. Adenoviral gene transfer allows Smad-responsive gene promoter analyses and delineation of type I receptor usage of transforming growth factor-β family ligands in cultured human granulosa luteal cells. J. Clin. Endocrinol. Metab. 2005, 90, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfman, N.M.; McPherron, A.C.; Pappano, W.N.; Davies, M.V.; Song, K.; Tomkinson, K.N.; Wright, J.F.; Zhao, L.; Sebald, S.M.; Greenspan, D.S. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc. Natl. Acad. Sci. USA 2003, 100, 15842–15846. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Zhang, G.; Sin, K.W.T.; Liu, Z.; Lin, R.; Li, M.; Li, Y. Activin A induces skeletal muscle catabolism via p38β mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Wu, Q.; Sun, S.; Li, Z.; Yang, Q.; Li, B.; Zhu, S.; Wang, L.; Wu, J.; Yuan, J.; Wang, C. Breast cancer-released exosomes trigger cancer-associated cachexia to promote tumor progression. Adipocyte 2019, 8, 31–45. [Google Scholar] [PubMed]
- Lautaoja, J.H.; Lalowski, M.; Nissinen, T.A.; Hentilä, J.J.; Shi, Y.; Ritvos, O.; Cheng, S.; Hulmi, J.J. Muscle and serum metabolomes are dysregulated in colon-26 tumor-bearing mice despite amelioration of cachexia with activin receptor type 2B ligand blockade. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E852–E865. [Google Scholar] [CrossRef]
- Kandarian, S.C.; Nosacka, R.L.; Delitto, A.E.; Judge, A.R.; Judge, S.M.; Ganey, J.D.; Moreira, J.D.; Jackman, R.W. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 1109–1120. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Moran, J.L.; Li, Y.; Hill, A.A.; Mounts, W.M.; Miller, C.P. Gene expression changes during mouse skeletal myoblast differentiation revealed by transcriptional profiling. Physiol. Genom. 2002, 10, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, S.; Stoppani, E.; Gobbo, M.; Caroli, A.; Fanzani, A. L6E9 Myoblasts Are Deficient of Myostatin and Additional TGF-β Members Are Candidates to Developmentally Control Their Fiber Formation. BioMed Res. Int. 2010, 2010, 326909. [Google Scholar]
- Ikeda, K.; Ito, A.; Imada, R.; Sato, M.; Kawabe, Y.; Kamihira, M. In vitro drug testing based on contractile activity of C2C12 cells in an epigenetic drug model. Sci. Rep. 2017, 7, 44570. [Google Scholar] [CrossRef] [Green Version]
- Ríos, R.; Carneiro, I.; Arce, V.M.; Devesa, J. Myostatin regulates cell survival during C2C12 myogenesis. Biochem. Biophys. Res. Commun. 2001, 280, 561–566. [Google Scholar] [CrossRef]
- Artaza, J.N.; Bhasin, S.; Mallidis, C.; Taylor, W.; Ma, K.; Gonzalez-Cadavid, N.F. Endogenous expression and localization of myostatin and its relation to myosin heavy chain distribution in C2C12 skeletal muscle cells. J. Cell. Physiol. 2002, 190, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.; Miyazono, K.; Ten Dijke, P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465. [Google Scholar] [CrossRef] [PubMed]
- Goodman, C.A.; McNally, R.M.; Hoffmann, F.M.; Hornberger, T.A. Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol. Endocrinol. 2013, 27, 1946–1957. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef] [Green Version]
- Sartori, R.; Milan, G.; Patron, M.; Mammucari, C.; Blaauw, B.; Abraham, R.; Sandri, M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am. J. Physiol. Cell Physiol. 2009, 296, C1248–C1257. [Google Scholar] [CrossRef] [Green Version]
- Yuzawa, H.; Koinuma, D.; Maeda, S.; Yamamoto, K.; Miyazawa, K.; Imamura, T. Arkadia represses the expression of myoblast differentiation markers through degradation of Ski and the Ski-bound Smad complex in C2C12 myoblasts. Bone 2009, 44, 53–60. [Google Scholar] [CrossRef]
- Ábrigo, J.; Campos, F.; Simon, F.; Riedel, C.; Cabrera, D.; Vilos, C.; Cabello-Verrugio, C. TGF-β requires the activation of canonical and non-canonical signalling pathways to induce skeletal muscle atrophy. Biol. Chem. 2018, 399, 253–264. [Google Scholar] [CrossRef]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Tsoli, M.; Robertson, G. Cancer cachexia: Malignant inflammation, tumorkines, and metabolic mayhem. Trends Endocrinol. Metab. 2013, 24, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ma, K.; Wang, H.; Xiao, F.; Gao, Y.; Zhang, W.; Wang, K.; Gao, X.; Ip, N.; Wu, Z. JAK1–STAT1–STAT3, a key pathway promoting proliferation and preventing premature differentiation of myoblasts. J. Cell Biol. 2007, 179, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Wang, C.; Xiao, F.; Wang, H.; Wu, Z. JAK2/STAT2/STAT3 are required for myogenic differentiation. J. Biol. Chem. 2008, 283, 34029–34036. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Li, Y. p38β MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPβ. Skelet. Muscle 2012, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Mancini, A.; El Bounkari, O.; Norrenbrock, A.F.; Scherr, M.; Schaefer, D.; Eder, M.; Banham, A.H.; Pulford, K.; Lyne, L.; Whetton, A.D. FMIP controls the adipocyte lineage commitment of C2C12 cells by downmodulation of C/EBPalpha. Oncogene 2007, 26, 1020–1027. [Google Scholar] [CrossRef]














© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lautaoja, J.H.; Pekkala, S.; Pasternack, A.; Laitinen, M.; Ritvos, O.; Hulmi, J.J. Differentiation of Murine C2C12 Myoblasts Strongly Reduces the Effects of Myostatin on Intracellular Signaling. Biomolecules 2020, 10, 695. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10050695
Lautaoja JH, Pekkala S, Pasternack A, Laitinen M, Ritvos O, Hulmi JJ. Differentiation of Murine C2C12 Myoblasts Strongly Reduces the Effects of Myostatin on Intracellular Signaling. Biomolecules. 2020; 10(5):695. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10050695
Chicago/Turabian StyleLautaoja, Juulia H., Satu Pekkala, Arja Pasternack, Mika Laitinen, Olli Ritvos, and Juha J. Hulmi. 2020. "Differentiation of Murine C2C12 Myoblasts Strongly Reduces the Effects of Myostatin on Intracellular Signaling" Biomolecules 10, no. 5: 695. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10050695