Targeting the Id1-Kif11 Axis in Triple-Negative Breast Cancer Using Combination Therapy

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Methods
2.1. Cell Culture
2.2. Immunofluorescence
2.3. Cell Cycle Analysis
2.4. Quantitative Real-Time PCR (qRT-PCR)
2.5. Flow Cytometry
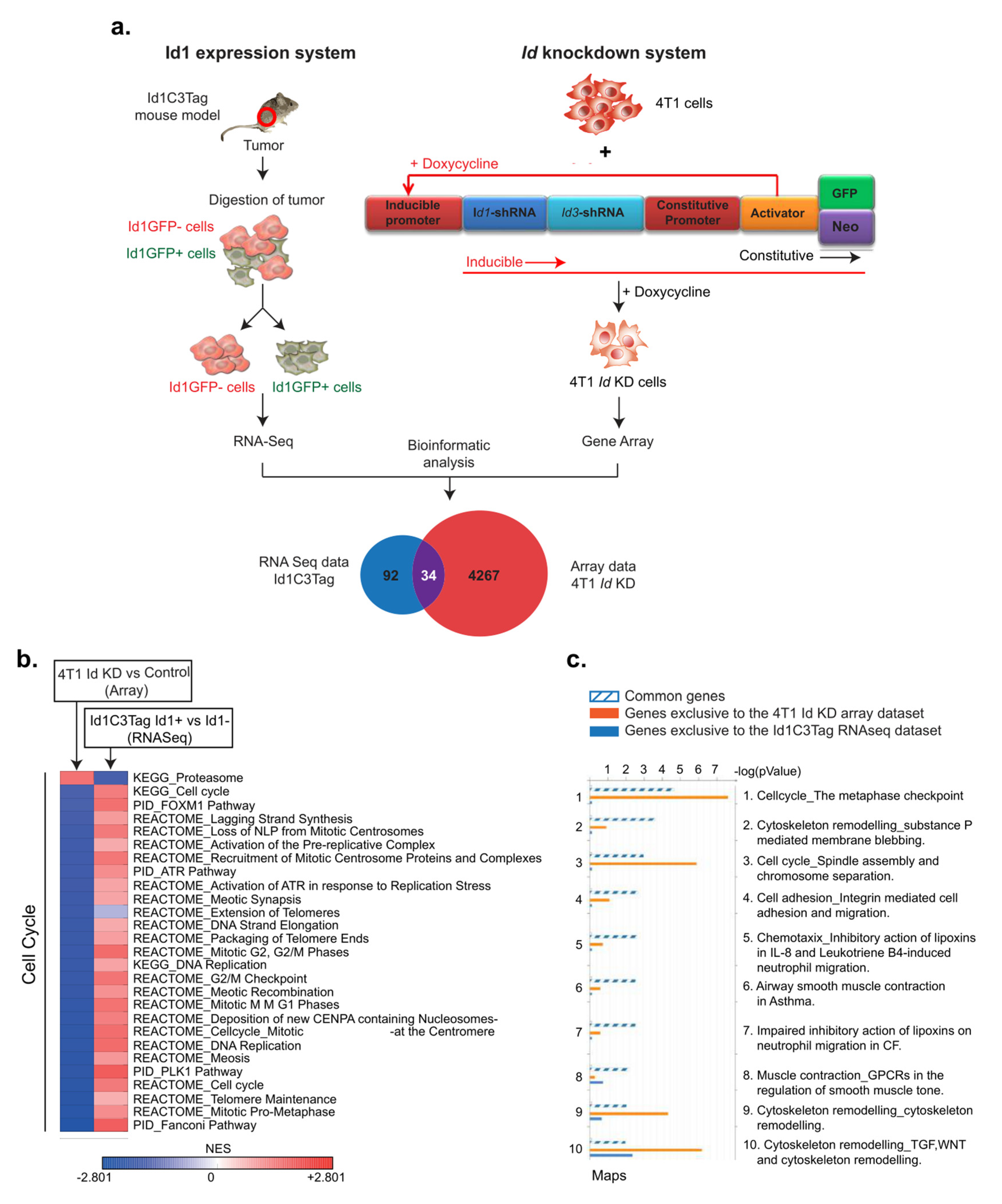
2.6. Microarray and Bioinformatics Analysis
2.7. siRNA Screen to Assess Proliferation
2.8. Western Blotting
2.9. MTT Assay
2.10. IC50 Values for Chemotherapeutic Drugs
2.11. Tumorsphere Assay
2.12. The Kaplan–Meier Plotter
2.13. Statistical Analysis
3. Results
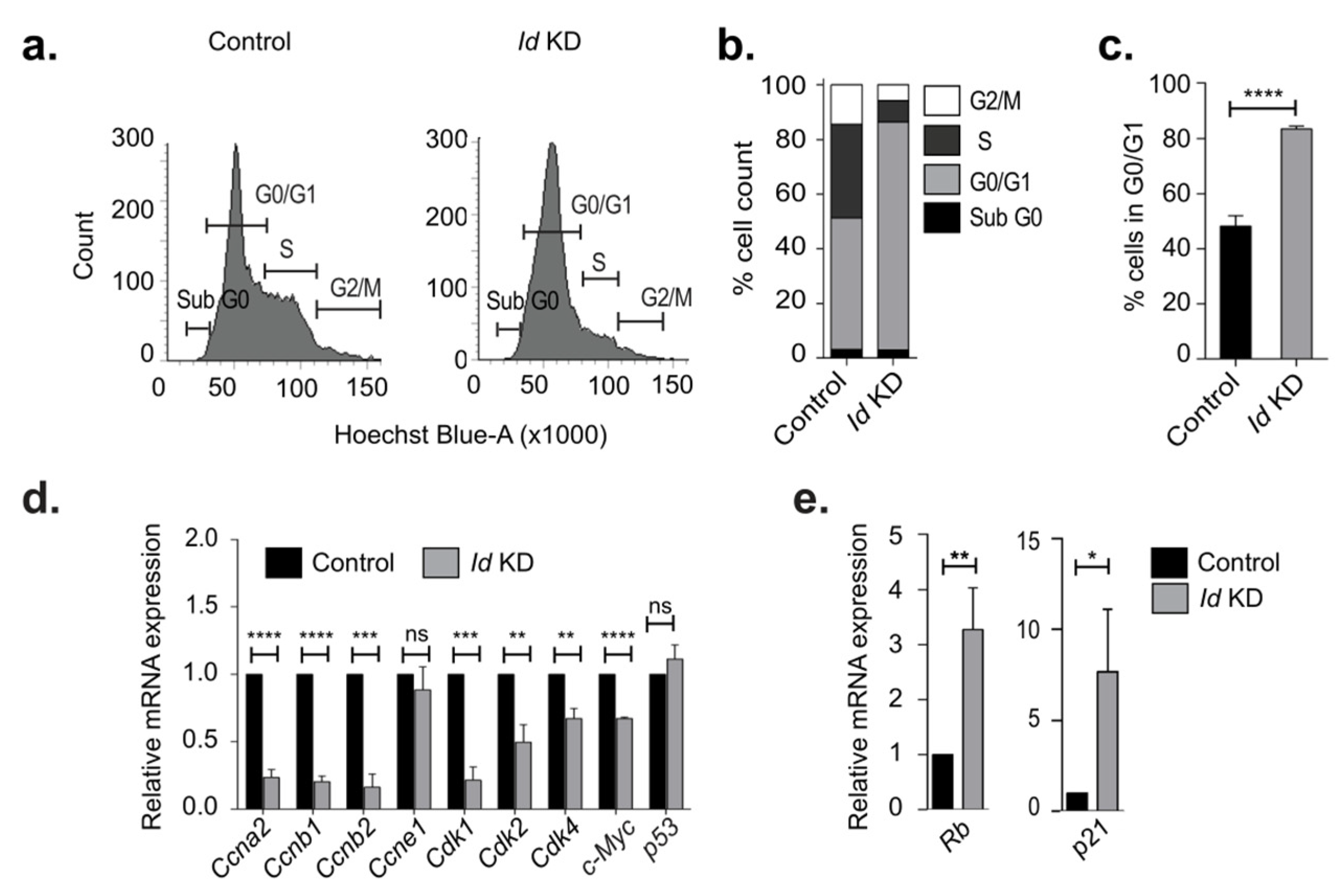
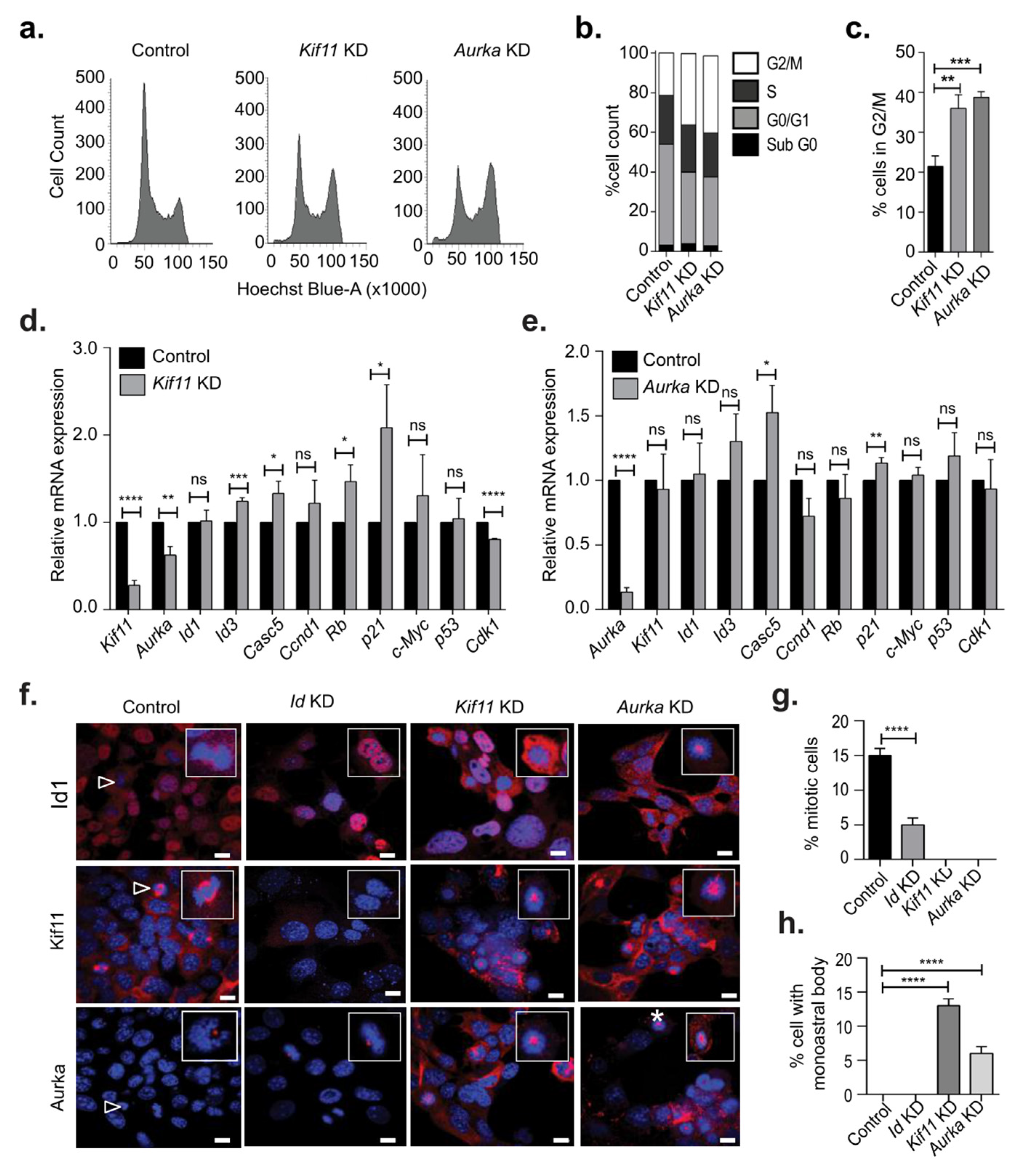
3.1. Id Depletion Leads to a G0/G1 Cell Cycle Arrest that Is Reversible
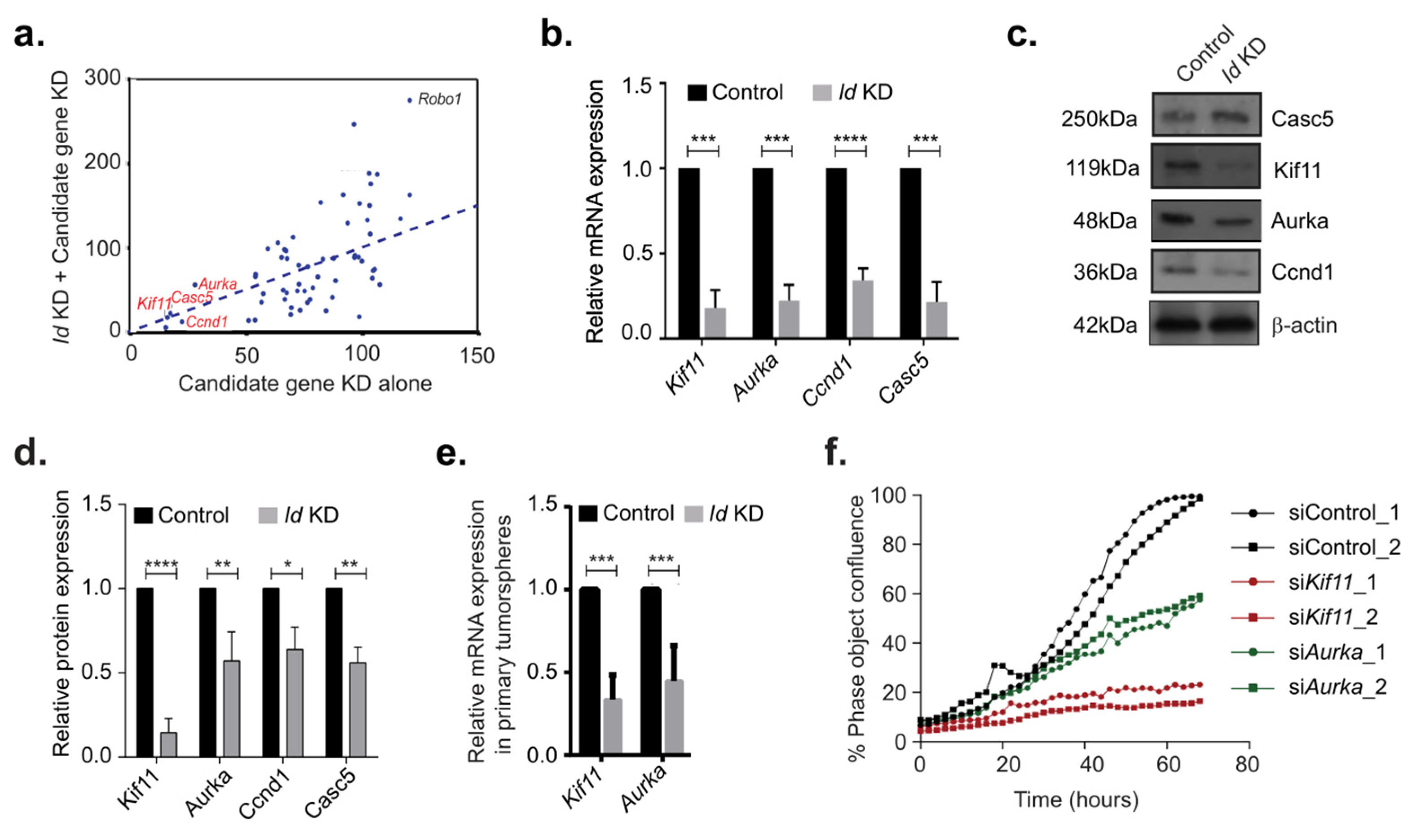
3.2. Identification of Putative Id-Regulated Genes
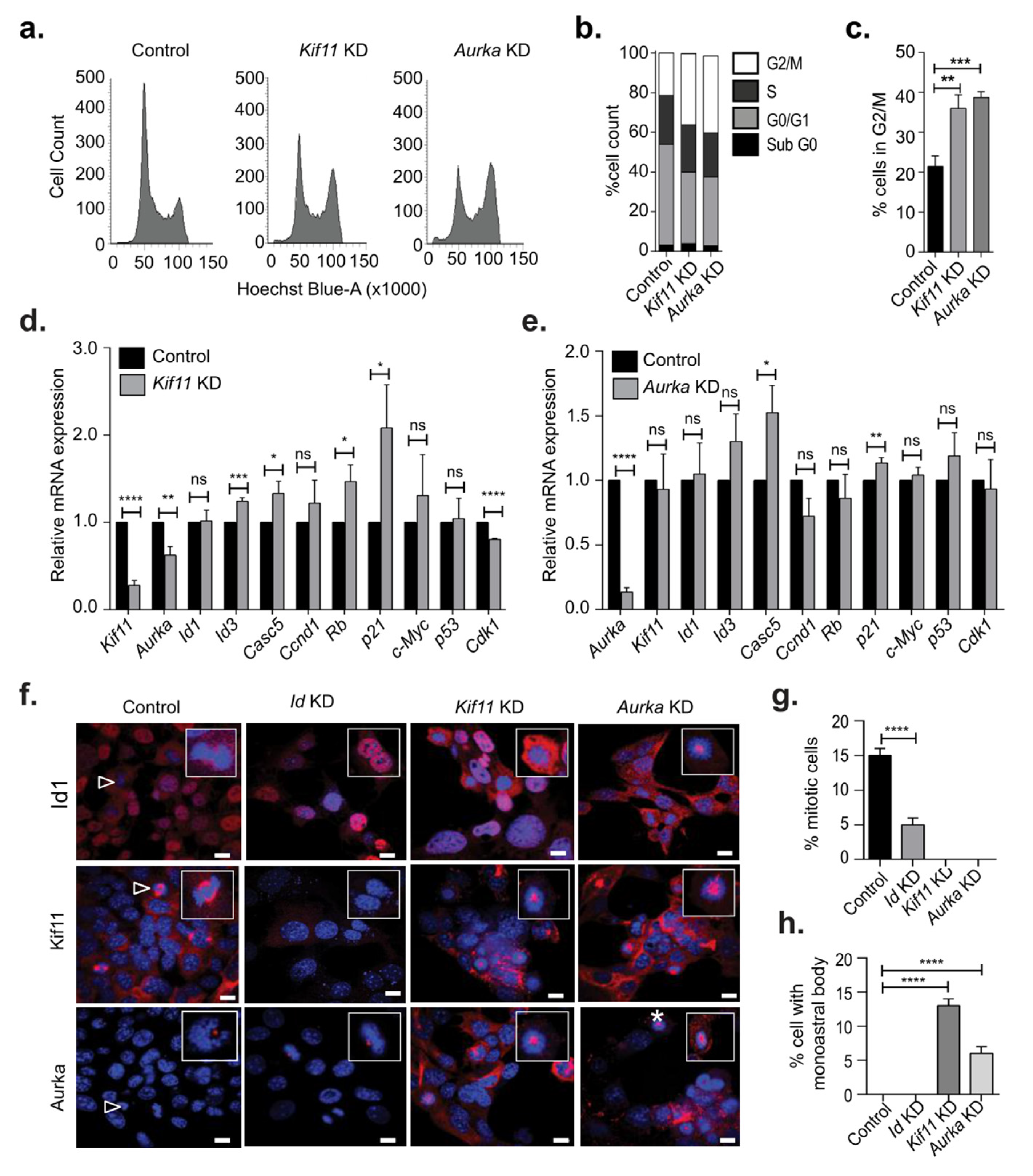
3.3. Identification of Kif11 and Aurka as Potential Id Targets
3.4. Kif11 or Aurka Depletion Did Not Phenocopy Loss of Id
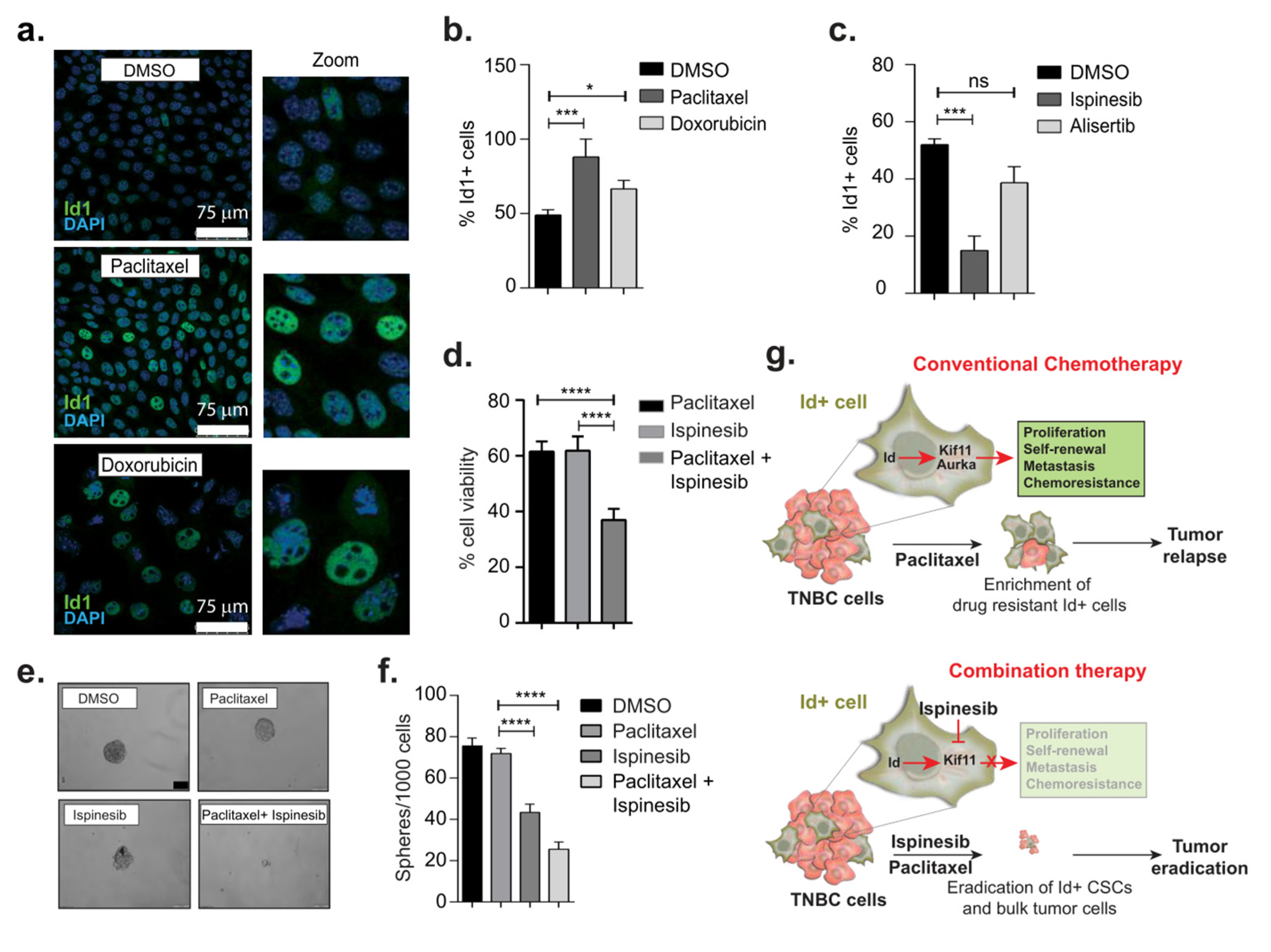
3.5. Therapeutic Targeting of CSCs Through Id–Kif11 Axis
4. Discussion
5. Conclusions
6. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CSC | cancer stem cell |
| TNBC | triple negative breast cancer |
| Id | inhibitor of differentiation |
References
- Teo, W.S.; Holliday, H.; Karthikeyan, N.; Cazet, A.S.; Roden, D.L.; Harvey, K.; Konrad, C.V.; Murali, R.; Varghese, B.A.; Archana, P.T.; et al. Id proteins promote a cancer stem cell phenotype in triple negative breast cancer via negative regulation of Robo1. Front. Cell Dev. Biol. Mol. Cellular Oncol. 2020, 8, 552. [Google Scholar] [CrossRef]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Yam, C.; Mani, S.A.; Moulder, S.L. Targeting the Molecular Subtypes of Triple Negative Breast Cancer: Understanding the Diversity to Progress the Field. Oncology 2017, 22, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P. Biology and Management of Patients with Triple-Negative Breast Cancer. Oncology 2016, 21, 1050–1062. [Google Scholar] [CrossRef] [Green Version]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.-G.; Lee, S.-H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Vila, M.; Takahashi, R.-U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Nair, R.; Roden, D.L.; Teo, W.S.; McFarland, A.; Junankar, S.; Ye, S.; Nguyen, A.; Yang, J.; Nikolić, I.; Hui, M.; et al. c-Myc and Her2 cooperate to drive a stem-like phenotype with poor prognosis in breast cancer. Oncogene 2013, 33, 3992–4002. [Google Scholar] [CrossRef] [Green Version]
- Swarbrick, A.; Åkerfeldt, M.C.; Lee, C.S.L.; Sergio, C.M.; Caldon, C.E.; Hunter, L.-J.K.; Sutherland, R.L.; Musgrove, E.A. Regulation of cyclin expression and cell cycle progression in breast epithelial cells by the helix–loop–helix protein Id1. Oncogene 2004, 24, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Győrffy, B.; Lanczky, A.; Eklund, A.C.; Denkert, C.; Budczies, J.; Li, Q.; Szállási, Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 2009, 123, 725–731. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.A.; Kreso, A.; Ryan, P.; Hermans, K.G.; Gibson, L.; Wang, Y.; Tsatsanis, A.; Gallinger, S.; Dick, J.E. ID1 and ID3 Regulate the Self-Renewal Capacity of Human Colon Cancer-Initiating Cells through p21. Cancer Cell 2012, 21, 777–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarbrick, A.; Roy, E.; Allen, T.D.; Bishop, J.M. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc. Natl. Acad. Sci. USA 2008, 105, 5402–5407. [Google Scholar] [CrossRef] [Green Version]
- Shin, K.-J.; Wall, E.A.; Zavzavadjian, J.R.; Santat, L.A.; Liu, J.; Hwang, J.-I.; Rebres, R.; Roach, T.; Seaman, W.; Simon, M.I.; et al. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 13759–13764. [Google Scholar] [CrossRef] [Green Version]
- Kaur, P.; Nagaraja, G.M.; Zheng, H.; Gizachew, D.; Galukande, M.; Krishnan, S.; Asea, A. A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC Cancer 2012, 12, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldon, C.E.; Swarbrick, A.; Lee, C.S.; Sutherland, R.L.; Musgrove, E.A. The Helix-Loop-Helix Protein Id1 Requires Cyclin D1 to Promote the Proliferation of Mammary Epithelial Cell Acini. Cancer Res. 2008, 68, 3026–3036. [Google Scholar] [CrossRef] [Green Version]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef] [Green Version]
- Hut, H.M.; Lemstra, W.; Blaauw, E.H.; Van Cappellen, G.W.; Kampinga, H.H.; Sibon, O.C. Centrosomes Split in the Presence of Impaired DNA Integrity during Mitosis. Mol. Biol. Cell 2003, 14, 1993–2004. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.; Brooks, M.; Wicha, M.S.; Brooke, M. Epithelial-Mesenchymal Plasticity of Breast Cancer Stem Cells: Implications for Metastasis and Therapeutic Resistance. Curr. Pharm. Des. 2015, 21, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [Green Version]
- Vaillant, F.; Asselin-Labat, M.-L.; Shackleton, M.; Forrest, N.C.; Lindeman, G.J.; Visvader, J.E. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res. 2008, 68, 7711–7717. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Bu, Y.; Jiang, S.; Chang, F.; Jia, F.; Xiao, X.; Song, G.; Zhang, M.; Ning, P.; Jia, Q. CCN2–MAPK–Id-1 loop feedback amplification is involved in maintaining stemness in oxaliplatin-resistant hepatocellular carcinoma. Hepatol. Int. 2019, 13, 440–453. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhuang, H.; Xia, R.; Gan, L.; Wu, Y.; Ma, J.; Sun, Y.; Zhuang, Z. KIF11 is required for proliferation and self-renewal of docetaxel resistant triple negative breast cancer cells. Oncotarget 2017, 8, 92106–92118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Stewart, A.L.; Mukherjee, S.; Huang, C.; Hartwell, K.A.; Miller, P.G.; Subramanian, R.; Carmody, L.C.; Yusuf, R.Z.; Sykes, D.B.; et al. Niche-Based Screening in Multiple Myeloma Identifies a Kinesin-5 Inhibitor with Improved Selectivity over Hematopoietic Progenitors. Cell Rep. 2015, 10, 755–770. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Id_KD_4T1_metacore | Id1C3Tag_RNASeq_metacore | ||||
|---|---|---|---|---|---|
| # | Input IDs | Signal | p-Value | Signal | p-Value |
| 1 | Adamtsl3 | −0.3927 | 0.020463 | 1.3402333 | 0.03359618 |
| 2 | Casc5 | −1.0215 | 0.001402 | 0.9961643 | 0.002269021 |
| 3 | Aspm | −1.3871 | 0.000268 | 0.9586754 | 0.000409133 |
| 4 | Aurka | −1.1447 | 0.0005806 | 0.9813335 | 0.01574964 |
| 5 | Casz1 | 0.2437 | 0.0479447 | 0.9948092 | 0.002230733 |
| 6 | Cenpf | −0.9544 | 0.001877 | 0.7718833 | 0.02742762 |
| 7 | Ctla2a | 0.4903 | 0.0023071 | 1.3066035 | 0.04998167 |
| 8 | Cxcl15 | 2.0181 | 0.0004083 | −6.916629 | 0.01322961 |
| 9 | Angptl7 | −0.631 | 0.0023779 | −1.489744 | 0.002084568 |
| 10 | Cldn6 | −0.5278 | 0.0131683 | −7.315199 | 0.004137659 |
| 11 | Gpr133 | 0.284 | 0.0448178 | −4.065336 | 0.000459154 |
| 12 | Hmga1;Hmga1-rs1 | −0.4779 | 0.0332492 | −1.080467 | 0.001750527 |
| 13 | Il6 | 1.2691 | 0.0003497 | 1.2769696 | 0.004137659 |
| 14 | Kif4 | −1.0266 | 0.0007331 | 0.7764488 | 0.01698252 |
| 15 | Kif11 | −1.3438 | 0.0002452 | 0.7487605 | 0.02222188 |
| 16 | Lphn1 | 0.4649 | 0.0027409 | 0.8679558 | 0.04907909 |
| 17 | Ltbp2;Ltbp3 | 0.2667 | 0.0244267 | 0.9104315 | 0.002134988 |
| 18 | Mylk | 0.5396 | 0.0154085 | 1.1309435 | 0.00340821 |
| 19 | Lnp;Nusap1 | −1.011 | 0.0001961 | 1.0915483 | 0.008424267 |
| 20 | Pdgfc | 1.7513 | 0.00001065 | 0.7934828 | 0.03592082 |
| 21 | Angptl2;Angptl4 | 3.5985 | 5.221 × 10−7 | −1.456264 | 1.46715 × 10−5 |
| 22 | Prc1 | −0.9945 | 0.0009206 | 0.7781182 | 0.02082709 |
| 23 | Stc2 | −1.2088 | 0.0005806 | −2.135721 | 8.46387 × 10−5 |
| 24 | Mylk | 0.5396 | 0.0154085 | 1.1309435 | 0.00340821 |
| 25 | Ube2c | −0.6684 | 0.0060464 | 1.2384745 | 0.004860396 |
| 26 | Upp1 | −0.5451 | 0.0022003 | −1.767675 | 0.000253826 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thankamony, A.P.; Murali, R.; Karthikeyan, N.; Varghese, B.A.; Teo, W.S.; McFarland, A.; Roden, D.L.; Holliday, H.; Konrad, C.V.; Cazet, A.; et al. Targeting the Id1-Kif11 Axis in Triple-Negative Breast Cancer Using Combination Therapy. Biomolecules 2020, 10, 1295. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10091295
Thankamony AP, Murali R, Karthikeyan N, Varghese BA, Teo WS, McFarland A, Roden DL, Holliday H, Konrad CV, Cazet A, et al. Targeting the Id1-Kif11 Axis in Triple-Negative Breast Cancer Using Combination Therapy. Biomolecules. 2020; 10(9):1295. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10091295
Chicago/Turabian StyleThankamony, Archana P., Reshma Murali, Nitheesh Karthikeyan, Binitha Anu Varghese, Wee S. Teo, Andrea McFarland, Daniel L. Roden, Holly Holliday, Christina Valbirk Konrad, Aurelie Cazet, and et al. 2020. "Targeting the Id1-Kif11 Axis in Triple-Negative Breast Cancer Using Combination Therapy" Biomolecules 10, no. 9: 1295. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10091295