Autophagy Inhibits Grass Carp Reovirus (GCRV) Replication and Protects Ctenopharyngodon idella Kidney (CIK) Cells from Excessive Inflammatory Responses after GCRV Infection

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Fish, Cells, and Virus Infection
2.2. Plasmid Construction, Transfection, and Reagents
2.3. RNA Isolation, Library Construction, and Sequencing
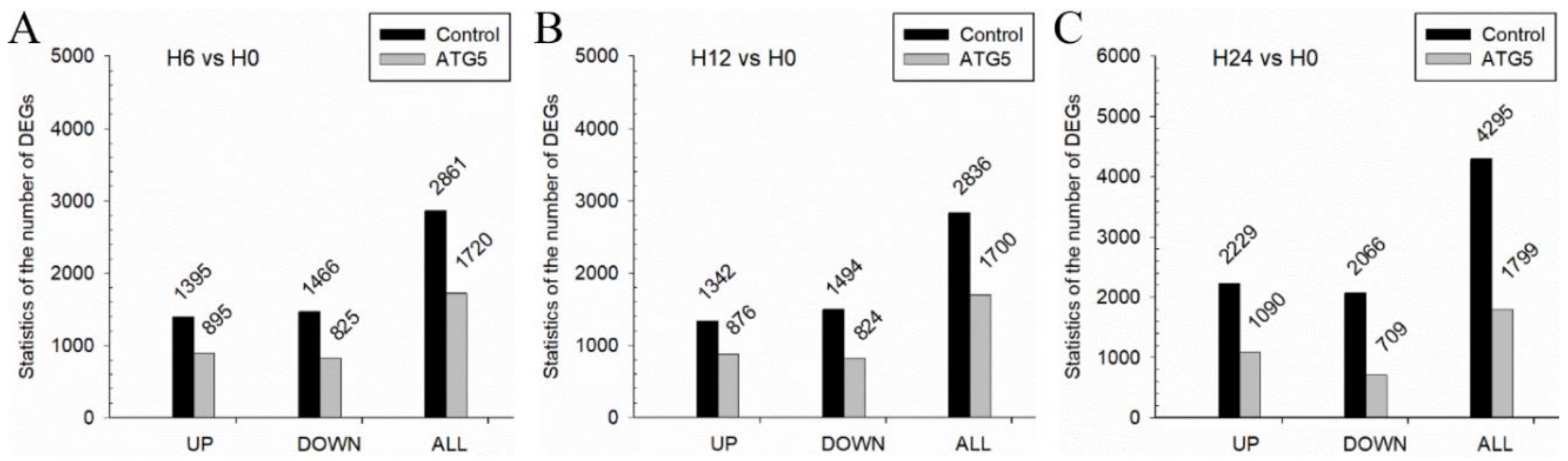
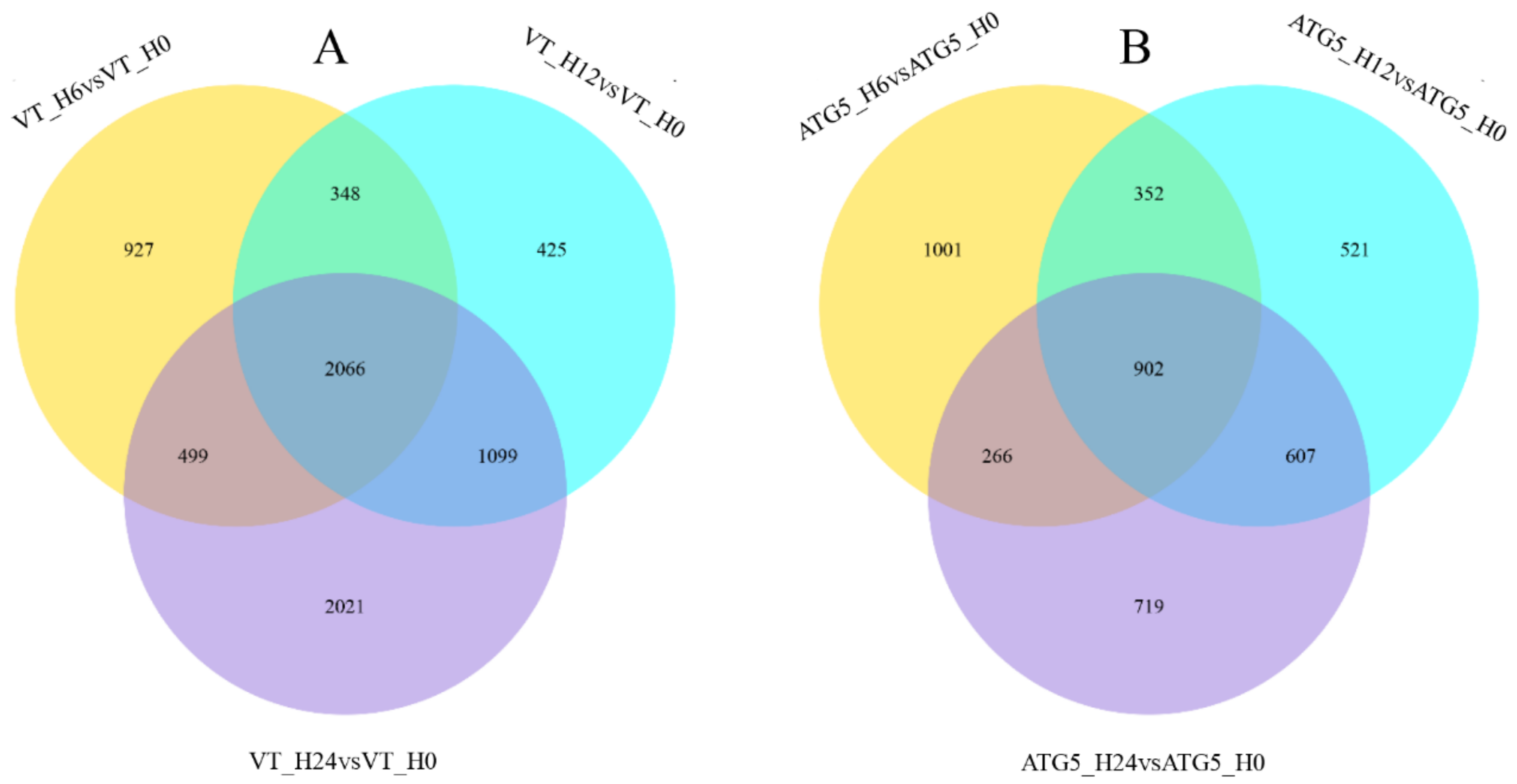
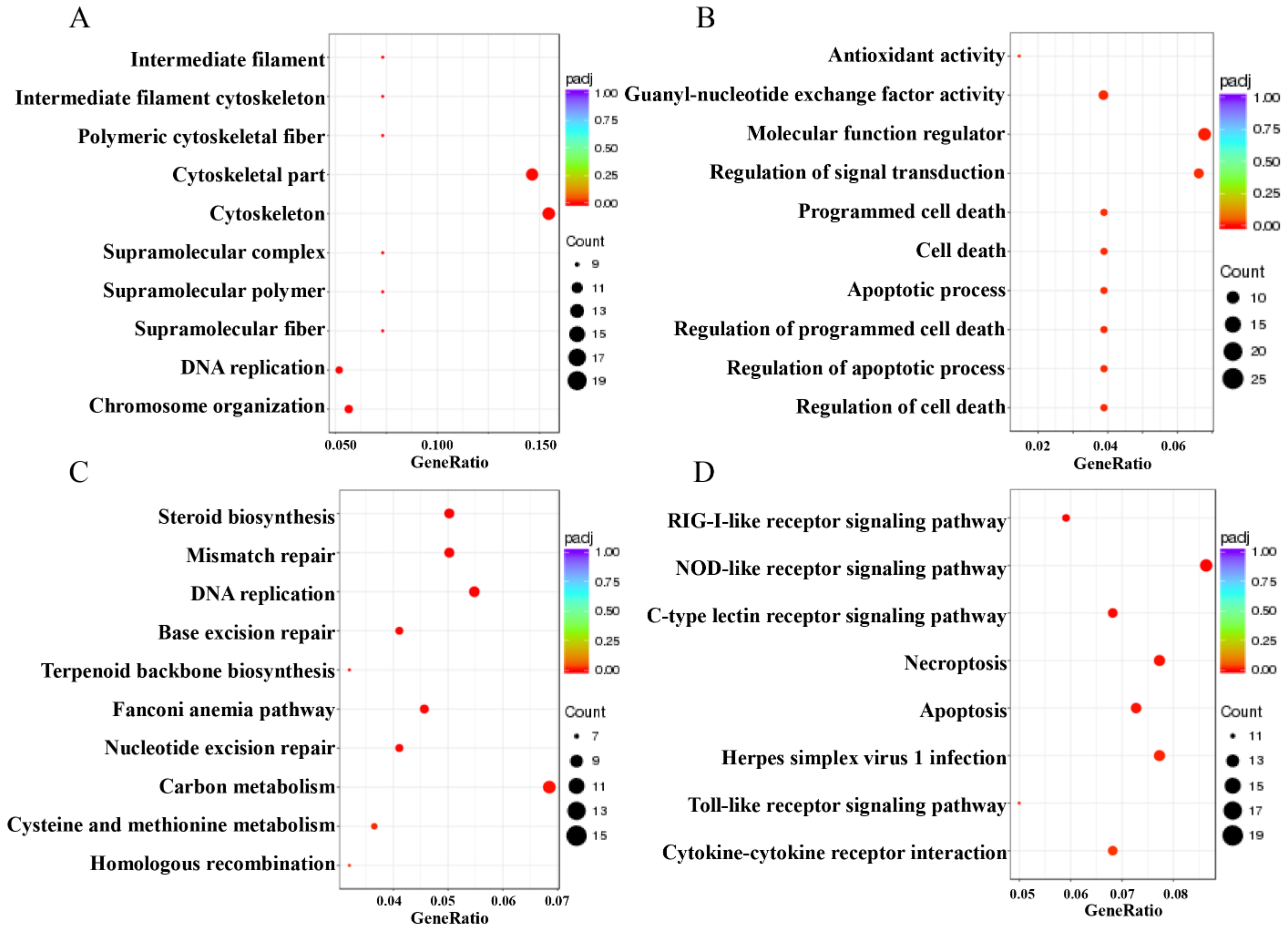
2.4. Differential Expression Analysis of Transcriptome Sequencing
2.5. Gene Expression Analysis
2.6. Immunohistochemistry and WB
2.7. Fluorescence Microscopy
2.8. Statistical Analysis
2.9. Ethics Statements
3. Results
3.1. GCRV Infection Stimulated Autophagy in the Spleen of Grass Carp
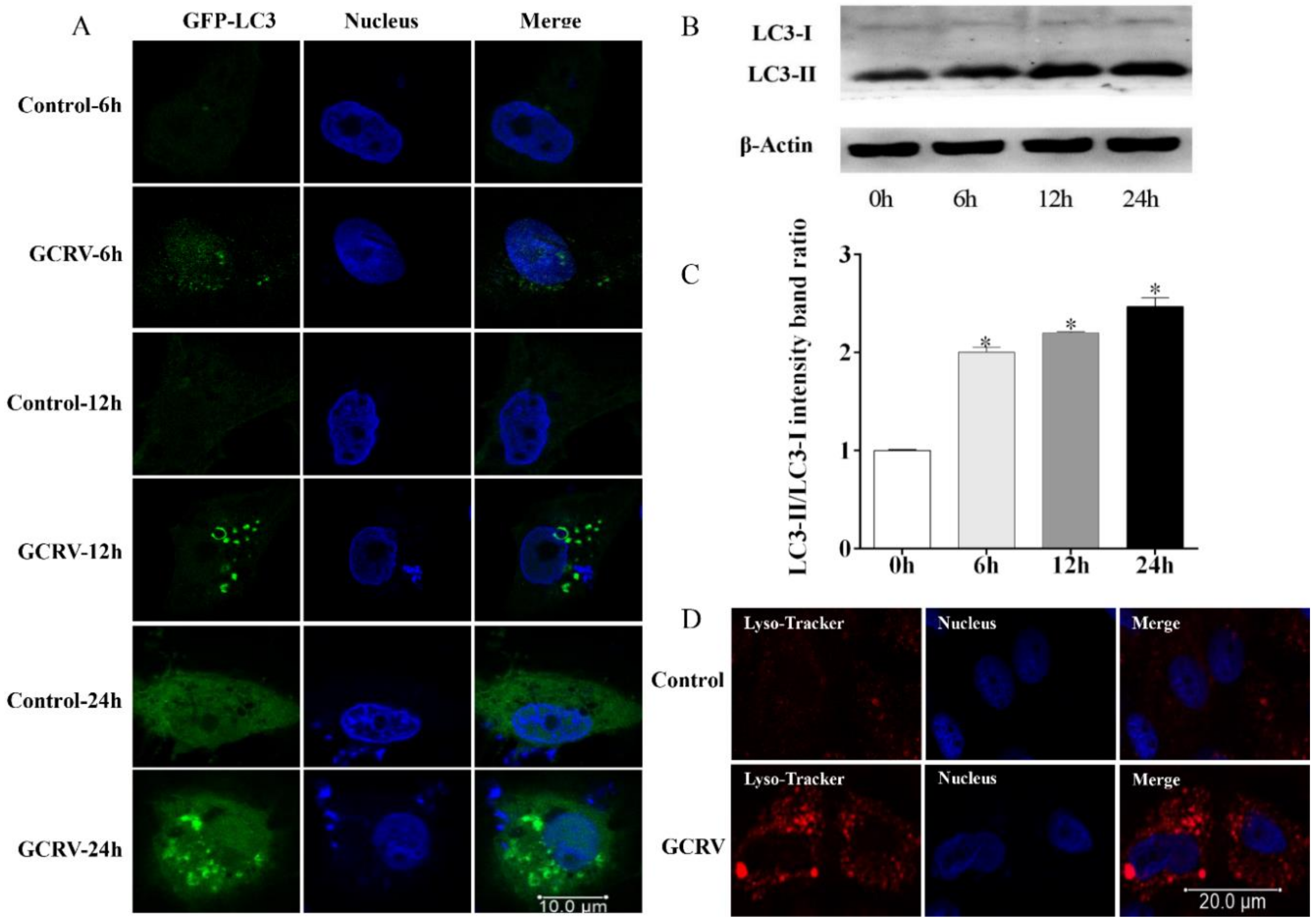
3.2. GCRV Infection Triggered Autophagy in CIK Cells
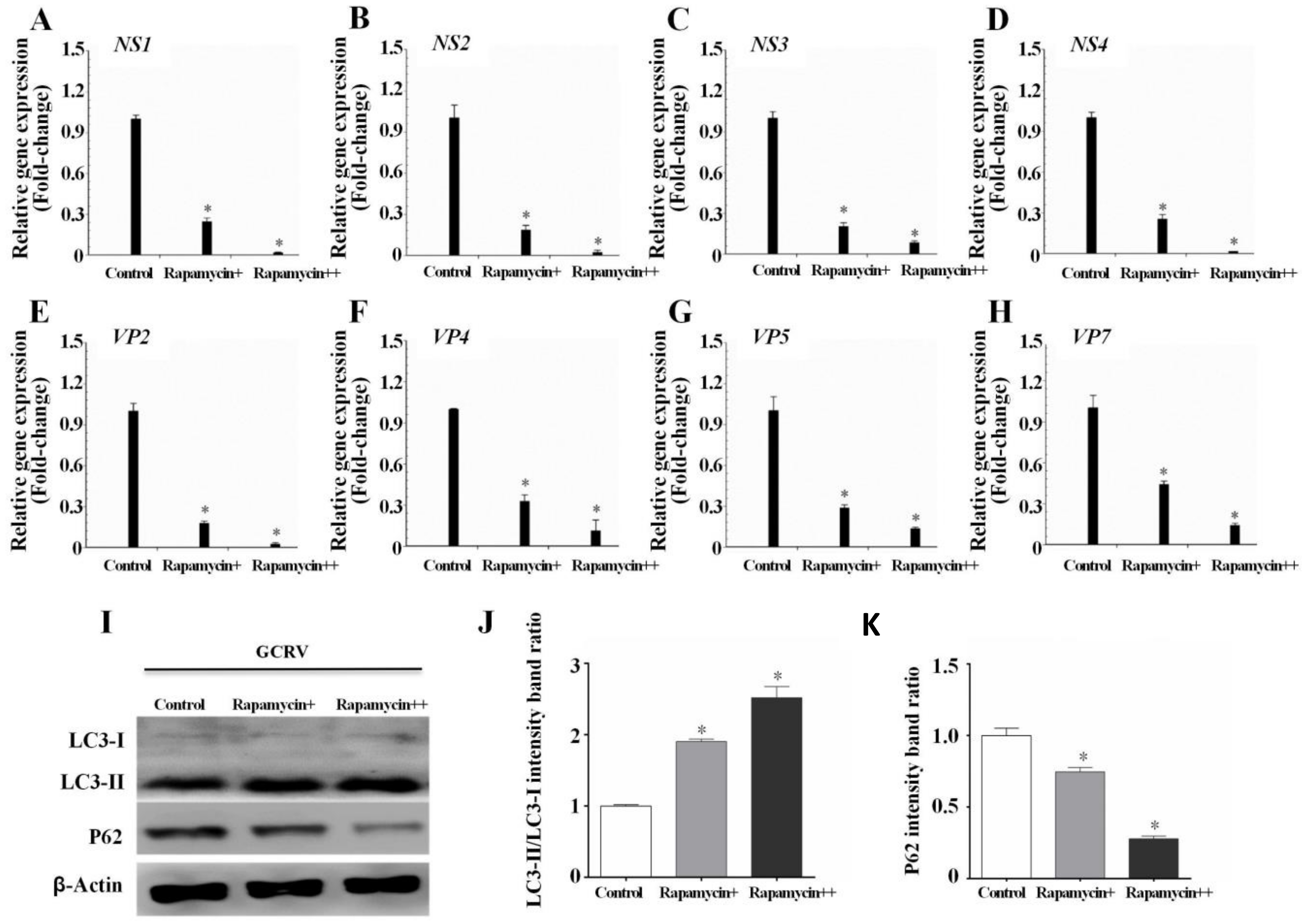
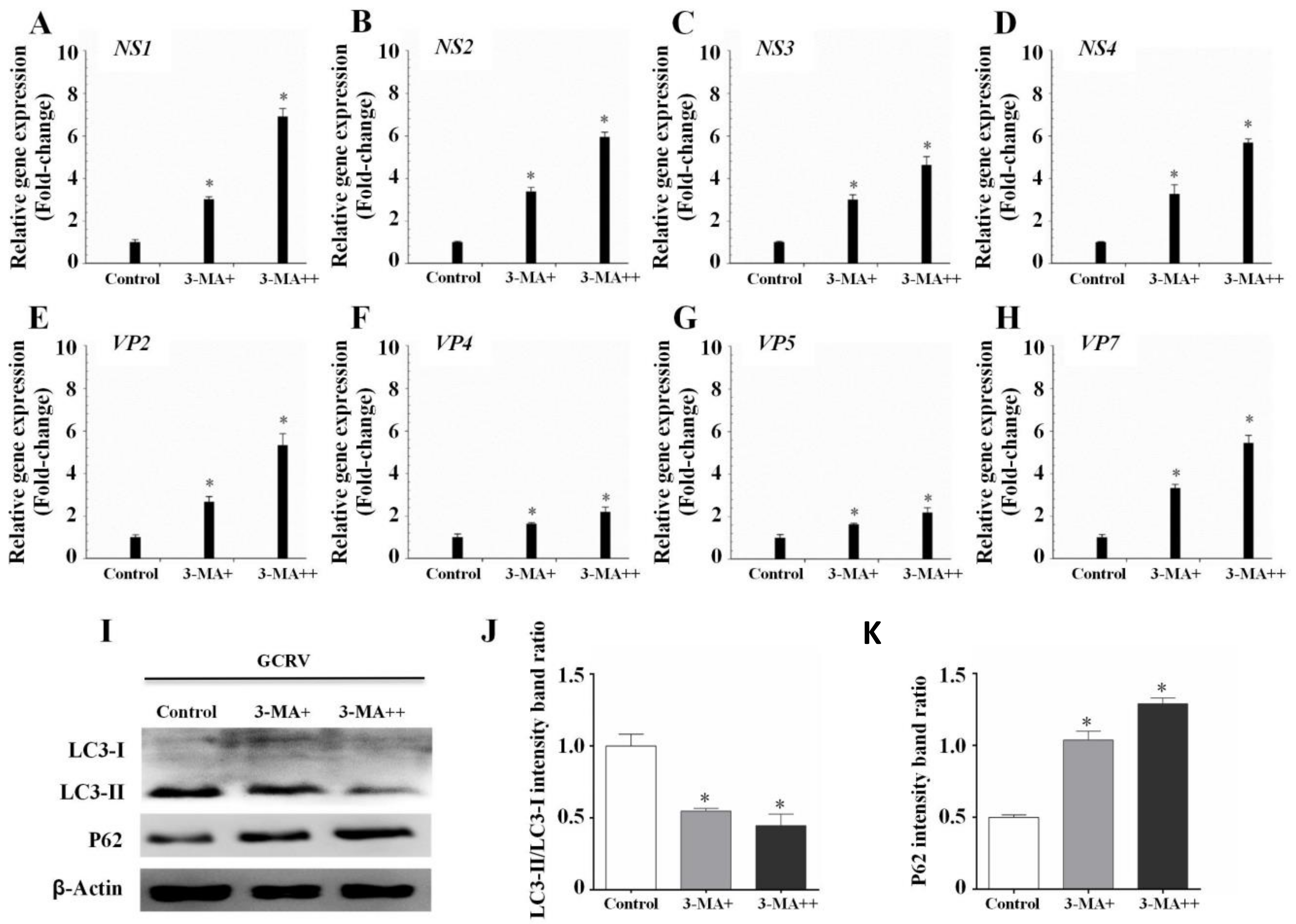
3.3. Autophagy Inhibited GCRV Replication in CIK Cells
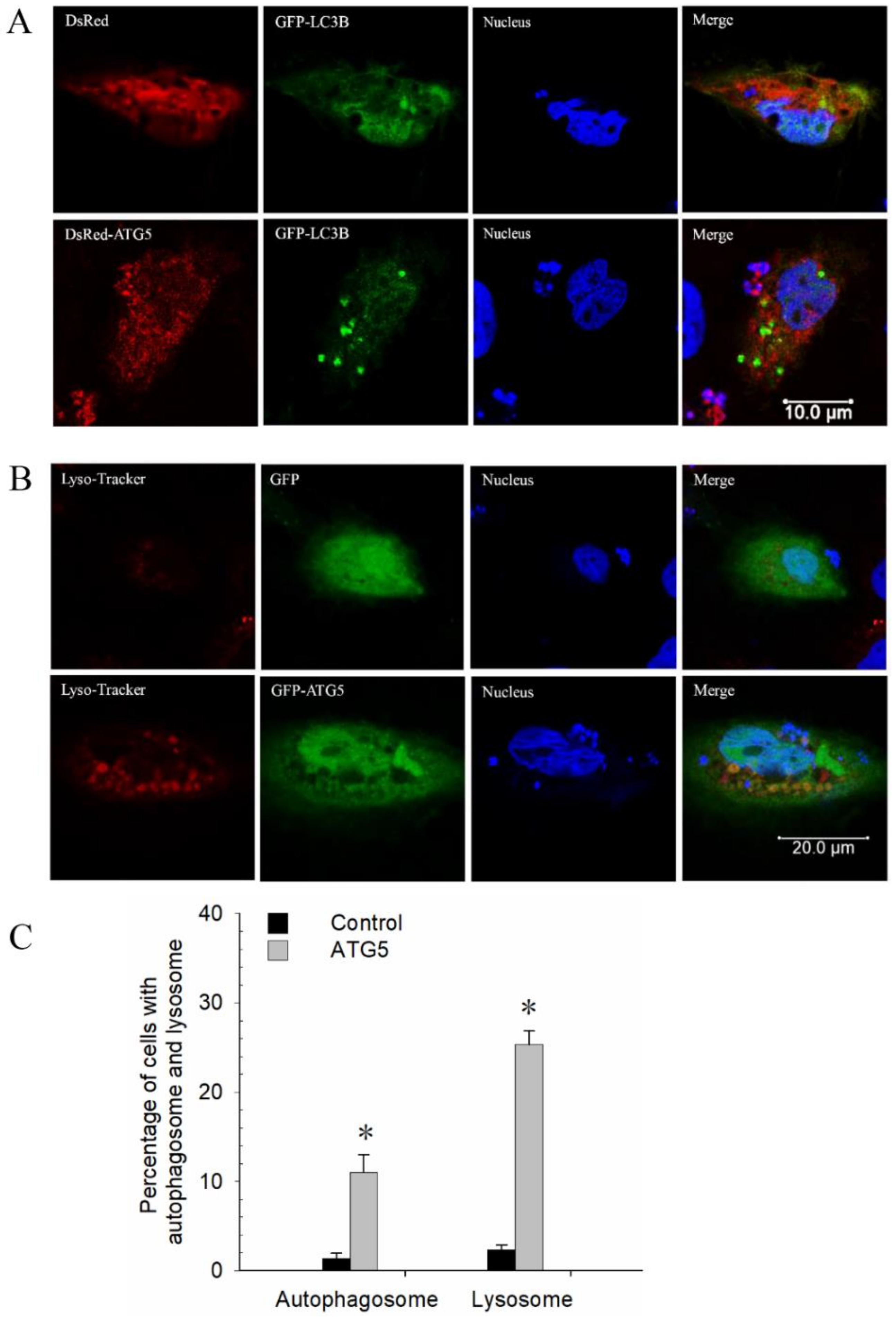
3.4. CiATG5 Promoted Autophagy in CIK Cells
3.5. CiATG5 Overexpression Inhibited GCRV Replication
3.6. GCRV Infection Caused Cell Damage and Acute Inflammatory Response
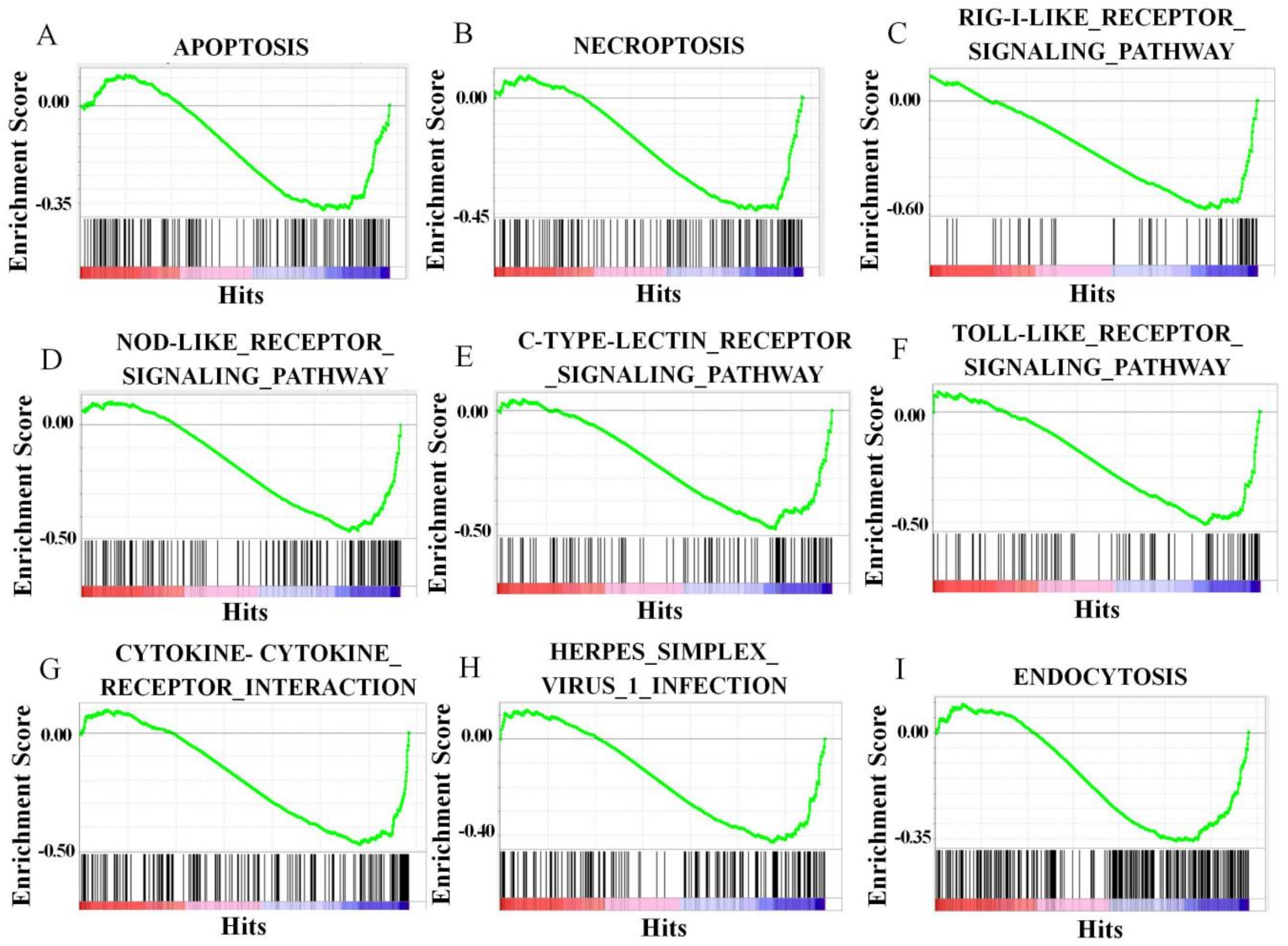
3.7. CiATG5 Weakened Host Inflammation Response to GCRV Invasion
3.8. CiATG5 Enhanced Integrity of CIK Cells against GCRV Invasion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jang, Y.J.; Kim, J.H.; Byun, S. Modulation of Autophagy for Controlling Immunity. Cells 2019, 8, 138. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef]
- Jewell, J.L.; Russel, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [Green Version]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [Green Version]
- Fujita, N.; Itoh, T.; Omori, H.; Fukuda, M.; Noda, T.; Yoshimori, T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol. Biol. Cell 2008, 19, 2092–2100. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, T.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Li, J.K.; Liang, J.J.; Liao, C.L.; Lin, Y.L. Autophagy is involved in the early step of Japanese encephalitis virus infection. Microbes Infect. 2012, 14, 159–168. [Google Scholar] [CrossRef]
- Dreux, M.; Gastaminza, P.; Wieland, S.F.; Chisari, F.V. The autophagy machinery is required to initiate hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 14046–14051. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.W.; Ducroux, A.; Jeang, K.T.; Neuveut, C. Impact of cellular autophagy on viruses: Insights from hepatitis B virus and human retroviruses. J. Biomed. Sci. 2012, 19, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Sir, D.; Tian, Y.; Chen, W.L.; Ann, D.K.; Yen, T.S.; Ou, J.H. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. USA 2010, 107, 4383–4388. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Sir, D.; Kuo, C.F.; Ann, D.F.; Ou, J.H. Autophagy required for hepatitis B virus replication in transgenic mice. J. Virol. 2011, 85, 13453–13456. [Google Scholar] [CrossRef] [Green Version]
- Dales, S.; Eggers, H.J.; Tamm, I.; Palade, G.E. Electron microscopic study of the formation of poliovirus. Virology 1965, 26, 379–389. [Google Scholar] [CrossRef]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef]
- Kim, N.; Kim, M.J.; Sung, P.S.; Bae, Y.C.; Shin, E.C.; Yoo, J.Y. Interferon-inducible protein SCOTIN interferes with HCV replication through the autolysosomal degradation of NS5A. Nat. Commun. 2016, 7, 10631–10642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valera, M.; de Armas-Rillo, L.; Barroso-González, J.; Ziglio, S.; Batisse, J.; Dubois, N.; Marrero-Hernández, S.; Borel, S.; García-Expósito, L.; Biard-Piechaczyk, M.; et al. The HDAC6/APOBEC3G complex regulates HIV-1 infectiveness by inducing Vif autophagic degradation. Retrovirology 2015, 12, 53–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, C.M.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B. Receptor usage dictates HIV-1 restriction by human TRIM5alpha in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Xia, X.J.; Wang, X.; Qin, W.H.; Jiang, J.Q.; Cheng, L.K. Emerging regulatory mechanisms and functions of autophagy in fish. Aquaculture 2019, 511, 734212–734219. [Google Scholar] [CrossRef]
- Schiotz, B.L.; Roos, N.; Rishovd, A.L.; Gjoen, T. Formation of autophagosomes and redistribution of LC3 upon in vitro infection with infectious salmon anemia virus. Virus Res. 2010, 151, 104–107. [Google Scholar] [CrossRef]
- Li, C.; Fu, X.; Lin, Q.; Liu, L.; Liang, H.; Huang, Z.; Li, N. Autophagy promoted infectious kidney and spleen necrosis virus replication and decreased infectious virus yields in CPB cell line. Fish Shellfish Immunol. 2017, 60, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, J.; Zhang, X.; Wei, S.; Huang, X.; Huang, Y.; Qin, Q. Fish Autophagy Protein 5 Exerts Negative Regulation on Antiviral Immune Response Against Iridovirus and Nodavirus. Front. Immunol. 2019, 10, 517–529. [Google Scholar] [CrossRef]
- Pereiro, P.; Romero, A.; Díaz-Rosales, P.; Estepa, A.; Figueras, A.; Novoa, B. Nucleated teleost erythrocytes play an nk-lysin-and autophagy-dependent role in antiviral immunity. Front. Immunol. 2019, 8, 1458–1472. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.; He, L.; Xiong, L.; Luo, L.; Huang, R.; Liao, L.; Li, Y.; Zhu, Z.; Wang, Y. Molecular cloning, expression analysis and localization pattern of the MST family in grass carp (Ctenopharyngodon idella). Fish Shellfish Immunol. 2018, 76, 316–323. [Google Scholar] [CrossRef]
- Zhu, D.; Huang, R.; Fu, P.; Chen, L.; Luo, L.; Chu, P.; He, L.; Li, Y.; Liao, L.; Zhu, Z.; et al. Investigating the Role of BATF3 in Grass Carp (Ctenopharyngodon idella) Immune Modulation: A Fundamental Functional Analysis. Int. J. Mol. Sci. 2019, 20, 1687. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; Fu, P.; Huang, R.; Xiong, L.; Wang, Y.; He, L.; Liao, L.; Li, Y.; Zhu, Z.; Wang, Y. Molecular characterization, tissue distribution and functional analysis of galectin 1-like 2 in grass carp (Ctenopharyngodon idella). Fish Shellfish Immunol. 2019, 94, 455–463. [Google Scholar] [CrossRef]
- Chu, P.; He, L.; Li, Y.; Huang, R.; Liao, L.; Li, Y.; Zhu, Z.; Wang, Y. Molecular cloning and functional characterisation of NLRX1 in grass carp (Ctenopharyngodon idella). Fish Shellfish Immunol. 2018, 81, 276–283. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. ClusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Cao, Y.; Tang, W.; Tang, W. Immune cell infiltration characteristics and related core genes in lupus nephritis: Results from bioinformatic analysis. BMC Immunol. 2019, 20, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [Green Version]
- Chu, P.; He, L.; Yang, C.; Zeng, W.; Huang, R.; Liao, L.; Li, Y.; Zhu, Z.; Wang, Y. Grass carp ATG5 and ATG12 promote autophagy but down-regulate the transcriptional expression levels of IFN-I signaling pathway. Fish Shellfish Immunol. 2019, 92, 600–611. [Google Scholar] [CrossRef]
- Rao, Y.; Su, J. Insights into the antiviral immunity against grass carp (Ctenopharyngodon idella) reovirus (GCRV) in grass carp. J. Immunol. Res. 2015, 2015, 670437–670454. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Zhang, A.; Pei, Y.; Chu, P.; Li, Y.; Huang, R.; Liao, L.; Zhu, Z.; Wang, Y. Differences in responses of grass carp to different types of grass carp reovirus (GCRV) and the mechanism of hemorrhage revealed by transcriptome sequencing. BMC Genom. 2017, 18, 452–466. [Google Scholar] [CrossRef]
- Smith, G.L.; Benfield, C.T.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.; Ferguson, B.J.; Sumner, R.P. Vaccinia virus immune evasion: Mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013, 94, 2367–2392. [Google Scholar] [CrossRef] [PubMed]
- Connolly, J.L.; Rodgers, S.E.; Clarke, P.; Ballard, D.W.; Kerr, L.D.; Tyler, K.L.; Dermody, T.S. Reovirus-induced apoptosis requires activation of transcription factor NF-kappa B. J. Virol. 2000, 74, 2981–2989. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.L., Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortorella, D.; Gewurz, B.E.; Furman, M.H.; Schust, D.J.; Ploegh, H.L. Viral subversion of the immune system. Annu. Rev. Immunol. 2000, 18, 861–926. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Rangel, A.A.; Rockemann, D.D.; Hetrick, F.M.; Samal, S.K. Identification of grass carp haemorrhage virus as a new genogroup of aquareovirus. J. Gen. Virol. 1999, 80, 2399–2402. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Wang, H.; Luo, L.; Li, Y.; Huang, R.; Liao, L.; Zhu, Z.; Wang, Y. Bid-deficient fish delay grass carp reovirus (GCRV) replication and attenuate GCRV-triggered apoptosis. Oncotarget 2017, 8, 76408–76422. [Google Scholar] [CrossRef]
- Cui, J.; Sim, T.H.; Gong, Z.; Shen, H.M. Generation of transgenic zebrafish with liver-specific expression of EGFP-Lc3: A new in vivo model for investigation of liver autophagy. Biochem. Biophys. Res. Commun. 2012, 422, 268–273. [Google Scholar] [CrossRef]
- Rao, Y.; Wan, Q.; Su, H.; Xiao, X.; Liao, Z.; Ji, J.; Yang, C.; Lin, L.; Su, J. ROS-induced HSP70 promotes cytoplasmic translocation of high-mobility group box 1b and stimulates antiviral autophagy in grass carp kidney cells. J. Biol. Chem. 2018, 293, 17387–17401. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Bartholomew, C.R.; Zhou, W.; Klionsky, D.J. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy 2009, 5, 520–526. [Google Scholar] [CrossRef] [Green Version]
- García-Valtanen, P.; Ortega-Villaizán Mdel, M.; Martínez-López, A.; Medina-Gali, R.; Pérez, L.; Mackenzie, S.; Figueras, A.; Coll, J.M.; Estepa, A. Autophagy-inducing peptides from mammalian VSV and fish VHSV rhabdoviral G glycoproteins (G) as models for the development of new therapeutic molecules. Autophagy 2014, 10, 1666–1680. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef]
- Nombela, I.; Requena-Platek, R.; Morales-Lange, B.; Chico, V.; Puente-Marin, S.; Ciordia, S.; Mena, M.C.; Coll, J.; Perez, L.; Mercado, L.; et al. Rainbow Trout Red Blood Cells Exposed to Viral Hemorrhagic Septicemia Virus Up-Regulate Antigen-Processing Mechanisms and MHC I&II, CD86, and CD83 Antigen-presenting Cell Markers. Cells 2019, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.C.; Rainger, G.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Johnson, B.S.; Laloraya, M. A cytokine super cyclone in COVID-19 patients with risk factors: The therapeutic potential of BCG immunization. Cytokine Growth Factor Rev. 2020. published online ahead of print. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.L.; Zhao, J.P.; Hu, Y.; Zhang, L.; Fan, G.H.; Xu, J.Y.; Gu, X.Y.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhao, B.; Qu, Y.M.; Chen, Y.R.; Xiong, J.; Feng, Y.; Men, D.; Huang, Q.C.; Liu, Y.; Yang, B.; et al. Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely correlated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.; Mafham, M.; Bell, J.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Effect of Dexamethasone in Hospitalized Patients with COVID-19: Preliminary Report. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
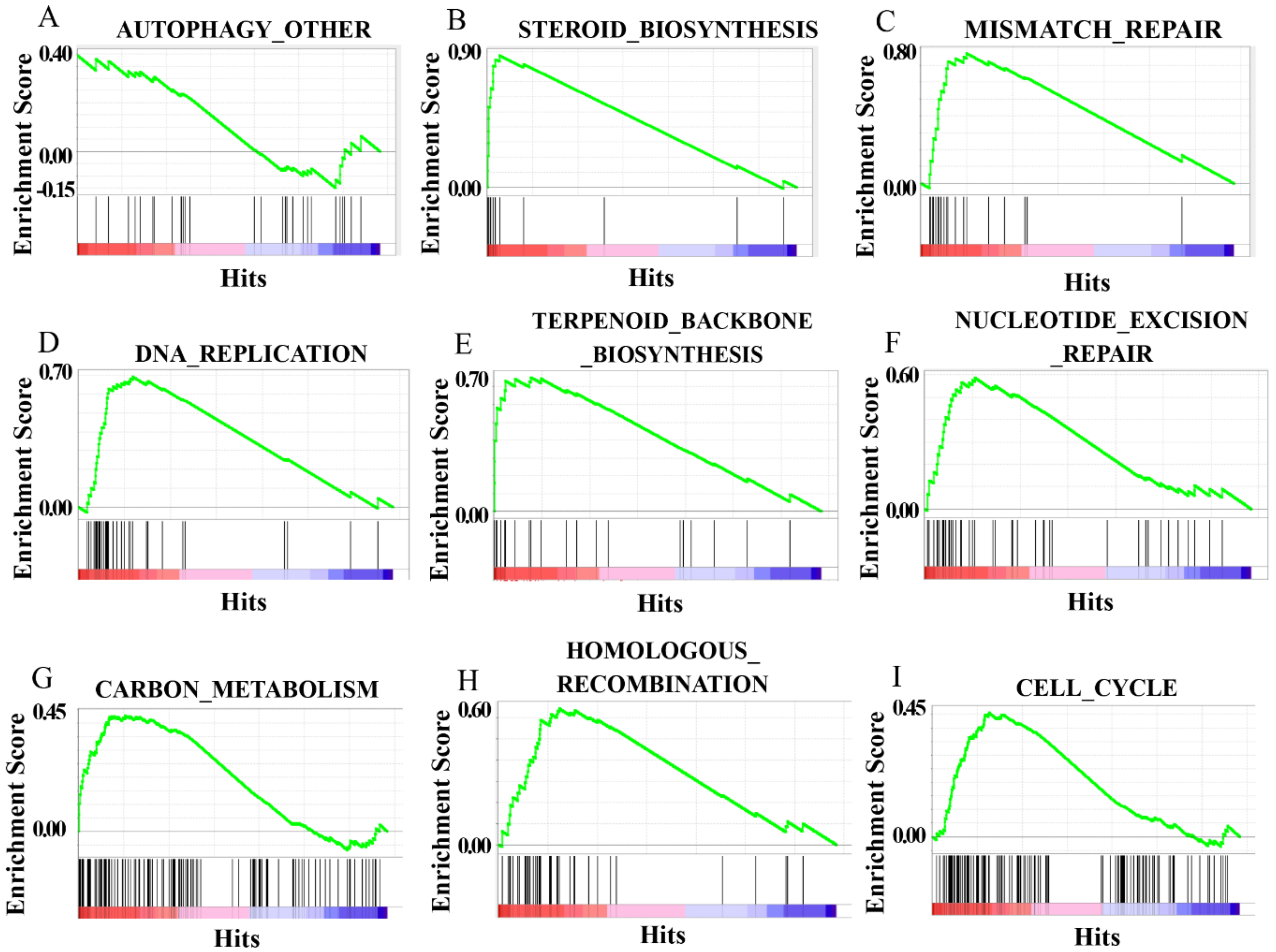{kind=link}
| Comparisons | KEGG Pathways (UP) | KEGG Pathways (DOWN) |
|---|---|---|
| H6 vs. HO | Ribosome biogenesis in eukaryotes | Lysosome |
| RNA transport | Valine, leucine and isoleucine degradation | |
| Spliceosome | Other glycan degradation | |
| Cell cycle | Autophagy—animal | |
| RNA polymerase | Carbon metabolism | |
| Protein processing in endoplasmic reticulum | Steroid biosynthesis | |
| Aminoacyl-tRNA biosynthesis | Fatty acid degradation | |
| mRNA surveillance pathway | Pyruvate metabolism | |
| Protein export | mTOR signaling pathway | |
| Cytosolic DNA-sensing pathway | Glycolysis/Gluconeogenesis | |
| H12 vs. HO | Proteasome | Lysosome |
| Protein processing in endoplasmic reticulum | Other glycan degradation | |
| DNA replication | Biosynthesis of amino acids | |
| TGF-beta signaling pathway | Carbon metabolism | |
| Spliceosome | Steroid biosynthesis | |
| Protein export | mTOR signaling pathway | |
| Cell cycle | Glycolysis/Gluconeogenesis | |
| Arachidonic acid metabolism | Aminoacyl-tRNA biosynthesis | |
| Mismatch repair | Glyoxylate and dicarboxylate metabolism | |
| Fatty acid degradation | ||
| H24 vs. HO | C-type lectin receptor signaling pathway | Oxidative phosphorylation |
| RIG-I-like receptor signaling pathway | Carbon metabolism | |
| MAPK signaling pathway | Valine, leucine and isoleucine degradation | |
| Toll-like receptor signaling pathway | Ribosome | |
| NOD-like receptor signaling pathway | Propanoate metabolism | |
| Foxo signaling pathway | Homologous recombination | |
| Herpes simplex virus 1 infection | Fatty acid degradation | |
| Apoptosis | Fatty acid metabolism | |
| Mitophagy—animal | Peroxisome | |
| Autophagy—animal | Fanconi anemia pathway |
| Comparisons | KEGG Pathways (UP) | KEGG Pathways (DOWN) |
|---|---|---|
| ATG5–H0 vs. VT–H0 | Carbon metabolism | Ribosome biogenesis in eukaryotes |
| Protein processing in endoplasmic reticulum | RNA transport | |
| Steroid biosynthesis | Aminoacyl-tRNA biosynthesis | |
| Valine, leucine and isoleucine degradation | RNA polymerase | |
| Fatty acid metabolism | Spliceosome | |
| Phagosome | RNA degradation | |
| Fatty acid degradation | Cytosolic DNA-sensing pathway | |
| ATG5–H6 vs. VT–H6 | Cell cycle | Ribosome biogenesis in eukaryotes |
| Steroid biosynthesis | Aminoacyl-tRNA biosynthesis | |
| Lysosome | Mismatch repair | |
| Apoptosis | RNA transport | |
| Terpenoid backbone biosynthesis | Nucleotide excision repair | |
| Progesterone-mediated oocyte maturation | DNA replication | |
| Fatty acid metabolism | Porphyrin and chlorophyll metabolism | |
| Oocyte meiosis | Cysteine and methionine metabolism | |
| Cellular senescence | Glutathione metabolism | |
| RNA degradation | ||
| ATG5–H12 vs. VT–H12 | Steroid biosynthesis | Glutathione metabolism |
| Terpenoid backbone biosynthesis | Arachidonic acid metabolism | |
| Cell cycle | Ferroptosis | |
| Focal adhesion | Oxidative phosphorylation | |
| Tight junction | Porphyrin and chlorophyll metabolism | |
| DNA replication | ||
| AGE-RAGE signaling pathway in diabetic complications | ||
| ATG5–H24 vs. VT–H24 | Steroid biosynthesis | RIG-I-like receptor signaling pathway |
| Mismatch repair | NOD-like receptor signaling pathway | |
| DNA replication | C-type lectin receptor signaling pathway | |
| Base excision repair | Apoptosis | |
| Terpenoid backbone biosynthesis | Necroptosis | |
| Fanconi anemia pathway | Herpes simplex virus 1 infection | |
| Nucleotide excision repair | Toll-like receptor signaling pathway | |
| Carbon metabolism | Cytokine-cytokine receptor interaction | |
| Cysteine and methionine metabolism | ||
| Homologous recombination |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, P.; He, L.; Huang, R.; Liao, L.; Li, Y.; Zhu, Z.; Hu, W.; Wang, Y. Autophagy Inhibits Grass Carp Reovirus (GCRV) Replication and Protects Ctenopharyngodon idella Kidney (CIK) Cells from Excessive Inflammatory Responses after GCRV Infection. Biomolecules 2020, 10, 1296. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10091296
Chu P, He L, Huang R, Liao L, Li Y, Zhu Z, Hu W, Wang Y. Autophagy Inhibits Grass Carp Reovirus (GCRV) Replication and Protects Ctenopharyngodon idella Kidney (CIK) Cells from Excessive Inflammatory Responses after GCRV Infection. Biomolecules. 2020; 10(9):1296. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10091296
Chicago/Turabian StyleChu, Pengfei, Libo He, Rong Huang, Lanjie Liao, Yongming Li, Zuoyan Zhu, Wei Hu, and Yaping Wang. 2020. "Autophagy Inhibits Grass Carp Reovirus (GCRV) Replication and Protects Ctenopharyngodon idella Kidney (CIK) Cells from Excessive Inflammatory Responses after GCRV Infection" Biomolecules 10, no. 9: 1296. https://0-doi-org.brum.beds.ac.uk/10.3390/biom10091296