
Structure-Based Virtual Screening of Tumor Necrosis Factor-α Inhibitors by Cheminformatics Approaches and Bio-Molecular Simulation


 and
and
Abstract
:1. Introduction
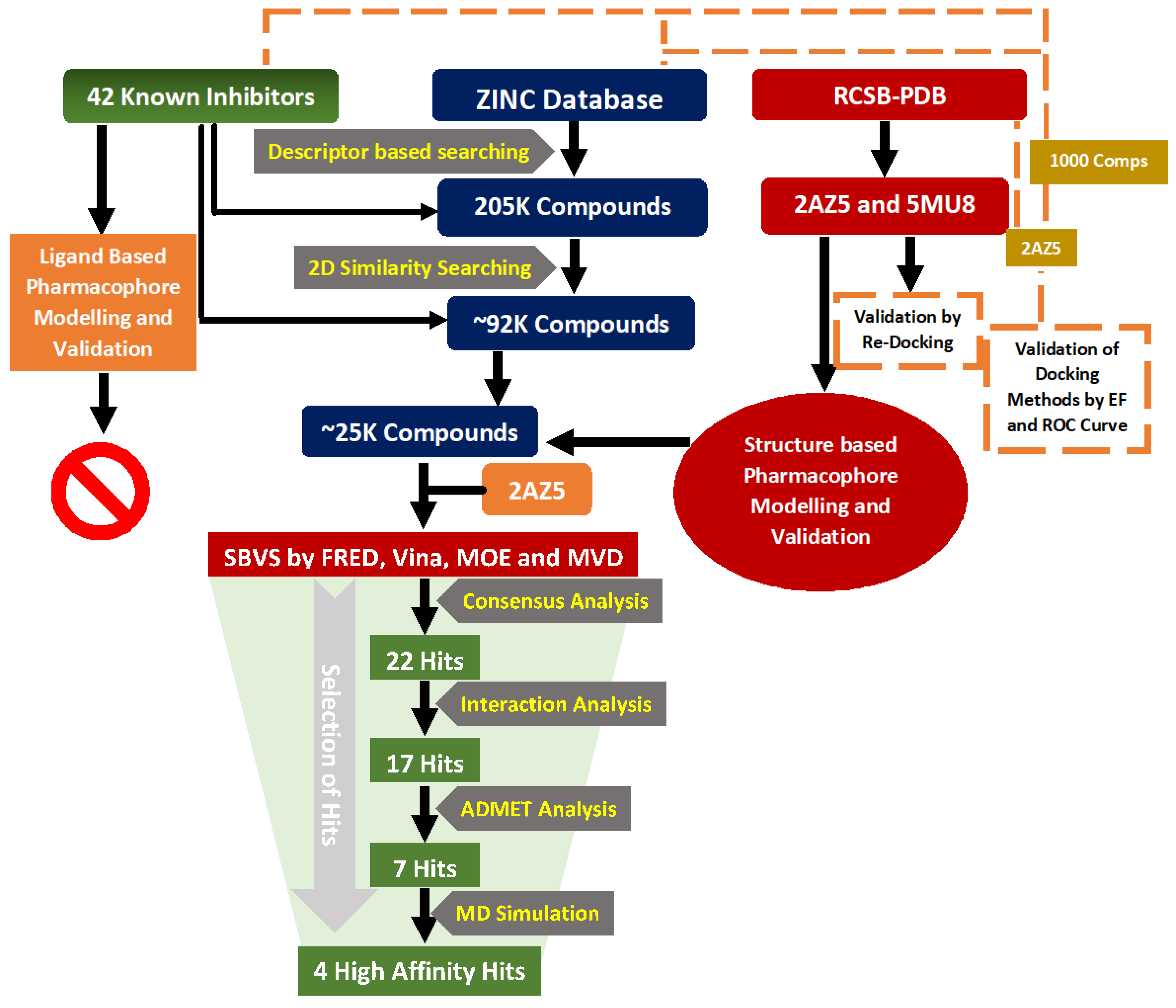
2. Material and Methods
2.1. Descriptor Based Screening of Zinc Is Not Commercial (ZINC) Database
2.2. 2D-Similarity Searching
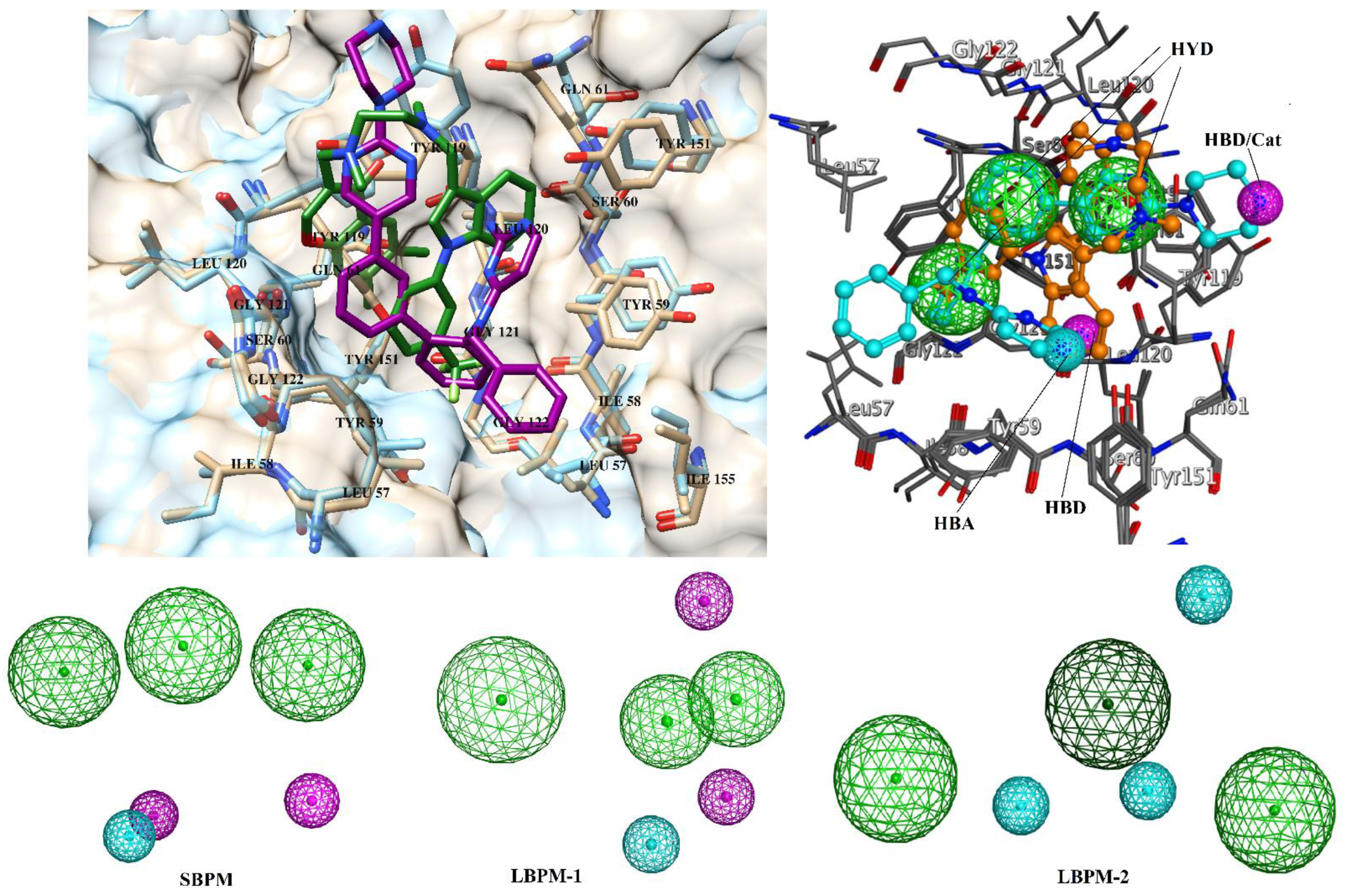
2.3. Pharmacophore Modelling
2.4. Structure-Based Virtual Screening (SBVS)
2.4.1. FRED
2.4.2. AutoDock Vina
2.4.3. Molecular Operating Environment (MOE)
2.4.4. Molegro Virtual Docker
2.5. Selection of Best Binders by Consensus Approach
2.6. ADMET Prediction
2.7. MD Simulations Protocols
2.8. Binding Free Energy Calculation
3. Results and Discussion
3.1. Ligand Based Virtual Screening
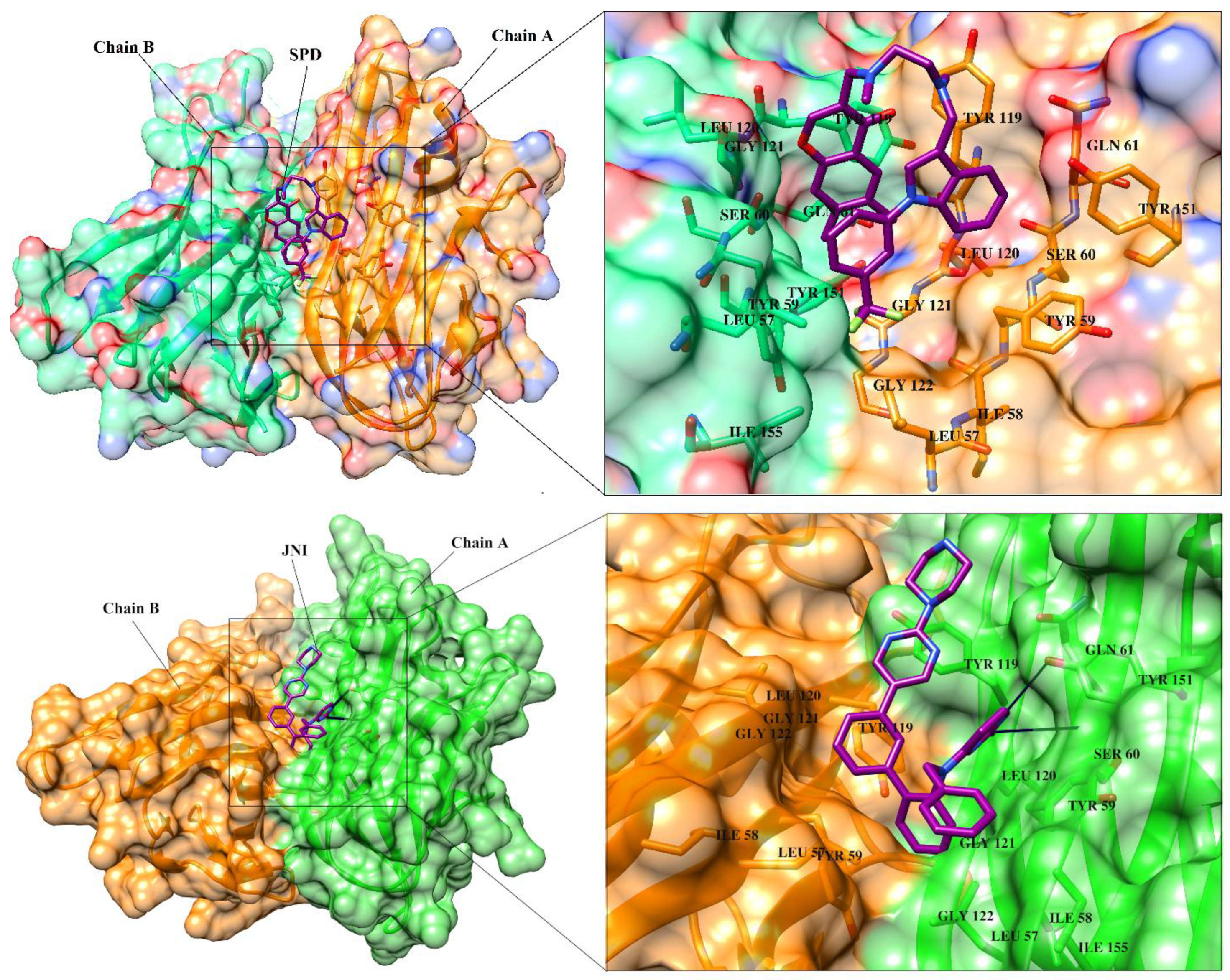
3.2. Structure-Based Virtual Screening
3.3. Selection of Best Inhibitors
3.4. ADMET Prediction
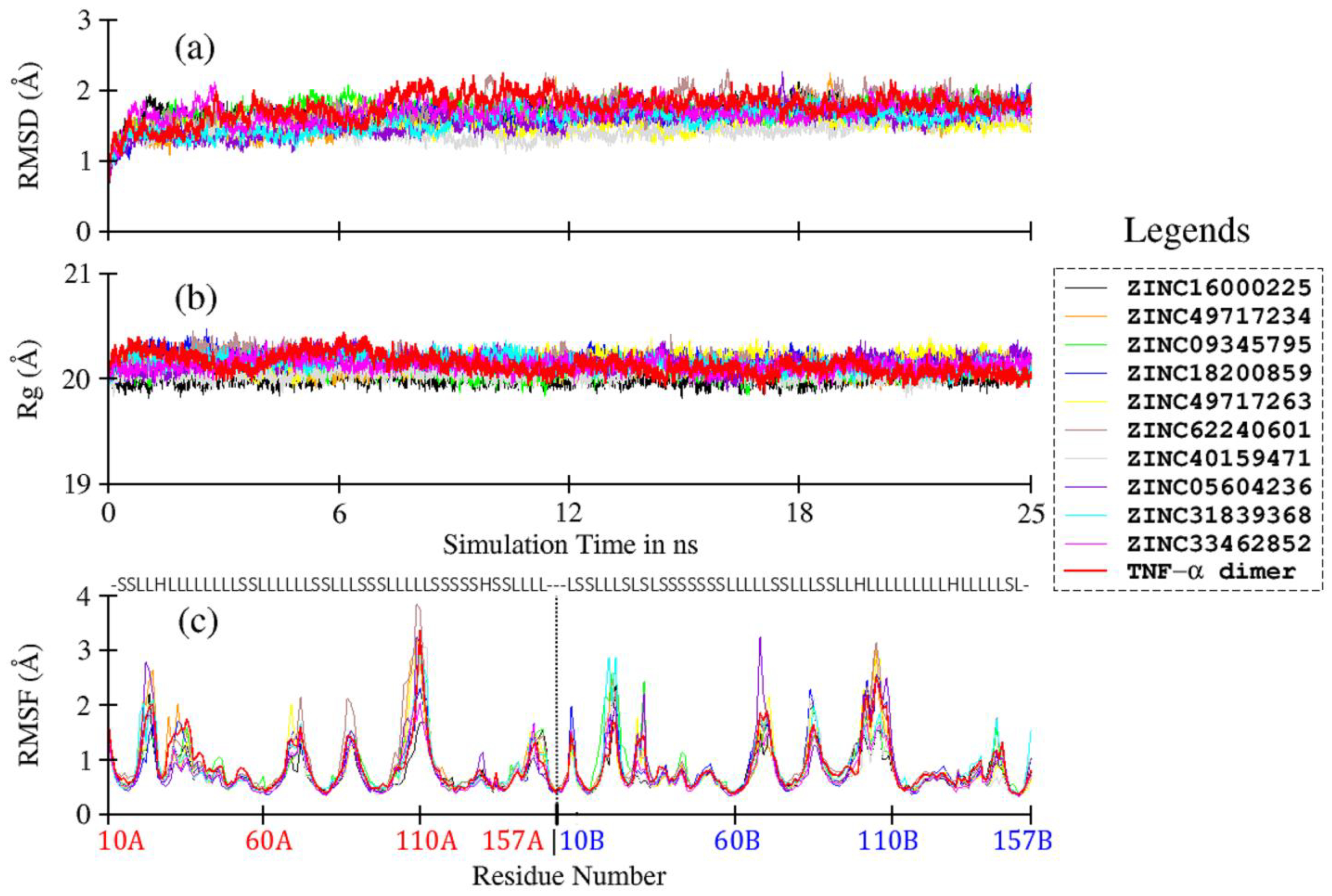
3.5. Ligand-Induced Structure and Dynamics
3.6. MM-PBSA Binding Energy Calculations
3.7. Interactions Analysis of Compounds 4, 5, 7 and 9
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADMET | Absorption, Distribution, Metabolism, Excretion and Toxicity |
| FRED | Fast Rigid Exhaustive Docking |
| MD Simulation | Molecular Dynamics Simulation |
| MM-PBSA | Molecular Mechanics: Poisson Boltzmann Surface Area |
| MOE | Molecular Operating Environment |
| MVD | Molegro Virtual Docker |
| NAMD | Nanoscale Molecular Dynamics |
| TNF-α | Tumor Necrosis Factor-α |
| TNFR | Tumor Necrosis Factor-α Receptor |
| VS | Virtual Screening |
| ZINC | Zinc Is Not Commercial |
References
- Rock, C.S.; Lowry, S.F. Tumor necrosis factor-α. J. Surg. Res. 1991, 51, 434–445. [Google Scholar] [CrossRef]
- Heffler, E.; Berry, M.; Pavord, I.D. Tumor necrosis factor-α. BioDrugs 2007, 21, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Swardfager, W.; Lanctôt, K.; Rothenburg, L.; Wong, A.; Cappell, J.; Herrmann, N. A meta-analysis of cytokines in Alzheimer’s disease. Biol. Psychiatry 2010, 68, 930–941. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Hung, M.-C.; Klostergaard, J. Human pro-Tumor Necrosis Factor Is a Homotrimer. Biochemistry 1996, 35, 8216–8225. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. 2008, 2, 149–160. [Google Scholar] [CrossRef]
- Gaur, U.; Aggarwal, B.B. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem. Pharmacol. 2003, 66, 1403–1408. [Google Scholar] [CrossRef]
- Zelova, H.; Hosek, J. TNF-α signalling and inflammation: Interactions between old acquaintances. Inflamm. Res. 2013, 62, 641–651. [Google Scholar] [CrossRef]
- Parameswaran, N.; Patial, S. Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Euk. Gene Exp. 2010, 20, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kontermann, R.E.; Maier, O. Targeting sTNF/TNFR1 Signaling as a New Therapeutic Strategy. Antibodies 2015, 4, 48–70. [Google Scholar] [CrossRef]
- Olmos, G.; Lladó, J. Tumor Necrosis Factor Alpha: A Link between Neuroinflammation and Excitotoxicity. Med. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—Past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, W.-Y.; Huang, Y.-S.; Lin, H.-H.; Luo, S.-F.; McCann, F.; McNamee, K.; Clanchy, F.; Williams, R. TNFR signalling and its clinical implications. Cytokine 2018, 101, 19–25. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Vucic, D. Intracellular regulation of TNF activity in health and disease. Cytokine 2018, 101, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.-Q. Molecular probing of TNF: From identification of therapeutic target to guidance of therapy in inflammatory diseases. Cytokine 2018, 101, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.-C.; Chen, Y.-L.; Pan, Y.-H.; Tian, F.; Xu, Y.; Zhang, X.-X.; Zhao, H.-I. The change of serum tumor necrosis factor alpha in patients with type 1 diabetes mellitus: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0176157. [Google Scholar] [CrossRef] [Green Version]
- Popa, C.; Netea, M.G.; Riel, P.L.C.M.; Van Der Meer, J.W.M.; Stalenhoef, A.F.H. The role of TNF-a in chronic inflammatory conditions, intermediary metabolism, and cardiovascular risk. J. Lipid Res. 2007, 48, 751–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Mutairi, N.; Shabaan, D. Effects of Tumor Necrosis Factor α Inhibitors Extend Beyond Psoriasis: Insulin Sensitivity in Psoriasis Patients with Type 2 Diabetes Mellitus. Cutis 2016, 97, 235–241. [Google Scholar] [PubMed]
- Sack, M.N.; Smith, R.M.; Opie, L.H. Tumor necrosis factor in myocardial hypertrophy and ischaemia—An anti-apoptotic perspective. Cardiovasc. Res. 2000, 45, 688–695. [Google Scholar] [CrossRef] [Green Version]
- Dunlay, S.M.; Weston, S.A.; Redfield, M.M.; Killian, J.M.; Roger, V.L. Tumor Necrosis Factor Alpha (TNFα) and Mortality in Heart Failure:a community study. Circulation 2008, 118, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceconi, C.; Curello, S.; Bachetti, T.; Corti, A.; Ferrari, R. Tumor necrosis factor in congestive heart failure: A mechanism of disease for the new millennium? Prog. Cardiovasc. Dis. 1998, 41, 25–30. [Google Scholar] [CrossRef]
- Scheff, N.N.; Yi, Y.; Bhattacharya, A.; MacRae, J.; Hickman, D.N.; Sharma, A.K.; Dolan, J.C.; Schmidt, B.L. Tumor necrosis factor alpha secreted from oral squamous cell carcinoma contributes to cancer pain and associated inflammation. Pain 2017, 158, 2396–2409. [Google Scholar] [CrossRef]
- Tang, D.; Tao, D.; Fang, Y.; Deng, C.; Xu, Q.; Zhou, J. TNF-Alpha Promotes Invasion and Metastasis via NF-Kappa B Pathway in Oral Squamous Cell Carcinoma. Med. Sci. Mon. Basic Res. 2017, 23, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.J. TNF-α inhibitors. Dermatol. Ther. 2007, 20, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.; Ortigosa, L.C.; Benard, G. Anti-TNF-α Agents in the Treatment of Immune-mediated Inflammatory Diseases: Mechanisms of Action and Pitfalls. Immunotherapy 2010, 2, 817–833. [Google Scholar] [CrossRef]
- Gottlieb, A.B. Tumor Necrosis Factor Blockade: Mechanism of Action. In Journal of Investigative Dermatology Symposium Proceedings; Elsevier: Amsterdam, The Netherlands, 2007; Volume 12, pp. 1–4. [Google Scholar]
- Lis, K.; Kuzawińska, O.; Bałkowiec-Iskra, E. Tumor necrosis factor inhibitors—State of knowledge. Arch. Med. Sci. 2014, 10, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Iftikhar, S.; Shahid, A.A.; Halim, S.A.; Wolters, P.J.; Vleeshouwers, V.G.; Khan, A.; Al-Harrasi, A.; Ahmad, S. Discovering Novel Alternaria solani Succinate Dehydrogenase Inhibitors by in Silico Modeling and Virtual Screening Strategies to Combat Early Blight. Front. Chem. 2017, 5, 100–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halim, S.A.; Abdalla, O.M.; Mesaik, M.A.; Wadood, A.; Kontoyianni, M. Identification of novel Interleukin-2 inhibitors through computational approaches. Mol. Divers. 2013, 17, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Halim, S.A.; Khan, A.; Csuk, R.; Al-Rawahi, A.; Al-Harrasi, A. Diterpenoids and Triterpenoids from Frankincense Are Excellent Anti-psoriatic Agents: An in silico Approach. Front. Chem. 2020, 8, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provazník, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Cur. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful Applications of Computer Aided Drug Discovery: Moving Drugs from Concept to the Clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef]
- Halim, S.A.; Ul-Haq, Z. Structure based 3D-QSAR studies of Interleukin-2 inhibitors: Comparing the quality and predictivity of 3D-QSAR models obtained from different alignment methods and charge calculations. Chem. Biol. Int. 2015, 238, 9–24. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. 1010 Sherbooke St. West, Suite 910; Chemical Computing Group ULC: Montreal, QC, Canada, 2014. [Google Scholar]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of AutoDock. J. Mol. Recog. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Thomser, R.; Mikael, H.; Christensen, J. MolDock: A New Technique for High-Accuracy Molecular Docking. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-Molecule Inhibition of TNF-α. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef]
- Jonathan, M.; Hack, B.M.D.; Herman, K.L.; Jackson, P.F.; Krawczuk, P.J.; Lebsack, A.D.; Liu, A.X.; Mirzadegan, T.; Nelen, M.I.; Patrick, A.N.; et al. Structural Basis of Small-Molecule Aggregate Induced Inhibition of a Protein–Protein Interaction. J. Med. Chem. 2017, 60, 3511–3517. [Google Scholar]
- Hawkins, P.C.D.; Nicholls, A. Conformer Generation with OMEGA: Learning from the Data Set and the Analysis of Failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for evaluating chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, M.P. Introduction to Molecular Dynamics Simulation. Comput. Soft Matter Synth. Polym. Proteins 2004, 23, 1–28. [Google Scholar]
- Skeel, R.D.; Hardy, D.J.; Phillips, J.C. Correcting mesh-based force calculations to conserve both energy and momentum in molecular dynamics simulations. J. Comput. Phys. 2007, 225, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gohlke, H.; Case, D.A. Converging free energy estimates: MM-PB(GB)SA studies on the protein–protein complex Ras–Raf. J. Comput. Chem. 2004, 25, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Bajusz, D.; Racz, A.; Heberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.-P.; Bertrand, H.-O. Virtual Screening Workflow Development Guided by the “Receiver Operating Characteristic” Curve Approach. Application to High-Throughput Docking on Metabotropic Glutamate Receptor Subtype 4. J. Med. Chem. 2005, 48, 2534–2547. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montanari, F.; Ecker, G.F. Prediction of drug-ABC-transporter interaction–Recent advances and future challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szakács, G.; Váradi, A.; Özvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Dis. Today 2008, 13, 379–393. [Google Scholar]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | ZINC ID | Chemical Structure | FRED Score | FRED Rank | Vina Score | Vina Rank | MOE Score | MOE Rank | MVD Score | MVD Rank |
|---|---|---|---|---|---|---|---|---|---|---|
| 1. | ZINC04914424 |  | −89.32 | 1 | −13.74 | 2 | −23.65 | 5 | −90.05 | 8 |
| 2. | ZINC17090251 |  | −85.29 | 2 | −13.93 | 1 | −20.23 | 11 | −81.23 | 24 |
| 3. | ZINC04914300 |  | −84.08 | 3 | −12.77 | 3 | −19.01 | 24 | −74.56 | 41 |
| 4. | ZINC16000225 |  | −82.55 | 5 | −12.34 | 7 | −23.44 | 8 | −89.88 | 11 |
| 5. | ZINC49717234 |  | −80.65 | 10 | −12.56 | 5 | −26.88 | 1 | −90.89 | 4 |
| 6. | ZINC00785143 |  | −80.26 | 11 | −12.58 | 4 | -20.05 | 15 | −77.73 | 35 |
| 7. | ZINC09345795 |  | −79.69 | 13 | −11.52 | 48 | −18.98 | 29 | −89.09 | 12 |
| 8. | ZINC18200859 |  | −79.68 | 14 | −11.81 | 26 | −20.11 | 12 | −80.6 | 29 |
| 9. | ZINC49717263 |  | −79.25 | 16 | −12.48 | 12 | −22.65 | 9 | −85.64 | 17 |
| 10. | ZINC62240601 |  | −77.64 | 23 | −12.10 | 10 | −16.01 | 55 | −72.65 | 46 |
| 11. | ZINC40159471 |  | −76.00 | 29 | −11.66 | 35 | −24.59 | 4 | −91.05 | 2 |
| 12. | ZINC31819567 |  | −75.81 | 30 | −12.33 | 8 | −15.05 | 89 | −90.85 | 5 |
| 13. | ZINC05604236 |  | −75.50 | 34 | −11.68 | 33 | −15.9 | 63 | −81.24 | 25 |
| 14. | ZINC31839368 |  | −75.19 | 36 | −11.88 | 20 | −15.55 | 71 | −80.56 | 30 |
| 15. | ZINC33462852 |  | −75.05 | 37 | −12.48 | 6 | −16.23 | 45 | −90.01 | 9 |
| 16. | ZINC04214135 |  | −74.89 | 38 | −11.58 | 41 | −19.89 | 18 | −74.22 | 42 |
| 17. | ZINC04914400 |  | −74.84 | 39 | −11.85 | 22 | −19.23 | 21 | −85.22 | 19 |
| Compounds | ZINC ID | RMSD (Å) | Rg (Å) |
|---|---|---|---|
| 4 | ZINC16000225 | 1.70 ± 0.13 | 20.01 ± 0.06 |
| 5 | ZINC49717234 | 1.59 ± 0.17 | 20.10 ± 0.06 |
| 7 | ZINC09345795 | 1.73 ± 0.15 | 20.08 ± 0.06 |
| 8 | ZINC18200859 | 1.63 ± 0.15 | 20.18 ± 0.07 |
| 9 | ZINC49717263 | 1.53 ± 0.12 | 20.18 ± 0.07 |
| 10 | ZINC62240601 | 1.75 ± 0.20 | 20.17 ± 0.08 |
| 11 | ZINC40159471 | 1.45 ± 0.14 | 20.05 ± 0.06 |
| 13 | ZINC05604236 | 1.55 ± 0.18 | 20.16 ± 0.07 |
| 14 | ZINC31839368 | 1.58 ± 0.17 | 20.15 ± 0.07 |
| 15 | ZINC33462852 | 1.70 ± 0.14 | 20.13 ± 0.06 |
| TNF-α dimer | 1.77 ± 0.19 | 20.14 ± 0.09 |
| Compounds | ZINC ID | Binding Energy ± Standard Deviation (kcal/mol) | VDW | Electrostatics | EPB | ENPOLAR |
|---|---|---|---|---|---|---|
| 4 | ZINC16000225 | −31.43 ± 3.49 | −43.07 | −15.72 | 30.96 | −3.60 |
| 5 | ZINC49717234 | −30.37 ± 3.97 | −46.47 | −9.91 | 29.89 | −3.88 |
| 7 | ZINC09345795 | −35.23 ± 3.15 | −48.51 | −6.43 | 23.64 | −3.93 |
| 8 | ZINC18200859 | −27.06 ± 3.93 | −41.82 | −6.91 | 25.22 | −3.55 |
| 9 | ZINC49717263 | −32.16 ± 4.06 | −47.36 | −12.14 | 31.30 | −3.97 |
| 10 | ZINC62240601 | −14.75 ± 2.99 | −24.44 | −66.08 | 78.55 | −2.78 |
| 11 | ZINC40159471 | −20.83 ± 3.08 | −30.42 | −2.54 | 15.13 | −3.00 |
| 13 | ZINC05604236 | −26.87 ± 2.67 | −38.87 | −14.68 | 29.78 | −3.10 |
| 14 | ZINC31839368 | −24.40 ± 3.74 | −39.63 | −6.03 | 24.99 | −3.73 |
| 15 | ZINC33462852 | −22.66 ± 2.93 | −36.62 | −8.87 | 26.28 | −3.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halim, S.A.; Sikandari, A.G.; Khan, A.; Wadood, A.; Fatmi, M.Q.; Csuk, R.; Al-Harrasi, A. Structure-Based Virtual Screening of Tumor Necrosis Factor-α Inhibitors by Cheminformatics Approaches and Bio-Molecular Simulation. Biomolecules 2021, 11, 329. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020329
Halim SA, Sikandari AG, Khan A, Wadood A, Fatmi MQ, Csuk R, Al-Harrasi A. Structure-Based Virtual Screening of Tumor Necrosis Factor-α Inhibitors by Cheminformatics Approaches and Bio-Molecular Simulation. Biomolecules. 2021; 11(2):329. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020329
Chicago/Turabian StyleHalim, Sobia Ahsan, Almas Gul Sikandari, Ajmal Khan, Abdul Wadood, Muhammad Qaiser Fatmi, René Csuk, and Ahmed Al-Harrasi. 2021. "Structure-Based Virtual Screening of Tumor Necrosis Factor-α Inhibitors by Cheminformatics Approaches and Bio-Molecular Simulation" Biomolecules 11, no. 2: 329. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020329