
Anti-TNF Alpha Antibody Humira with pH-dependent Binding Characteristics: A constant-pH Molecular Dynamics, Gaussian Accelerated Molecular Dynamics, and In Vitro Study

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. System Setup
2.2. GaMD
2.3. CpHMD
2.4. Construction, Expression, and Antigen-Binding Ability of pH-Dependent Humira
3. Results
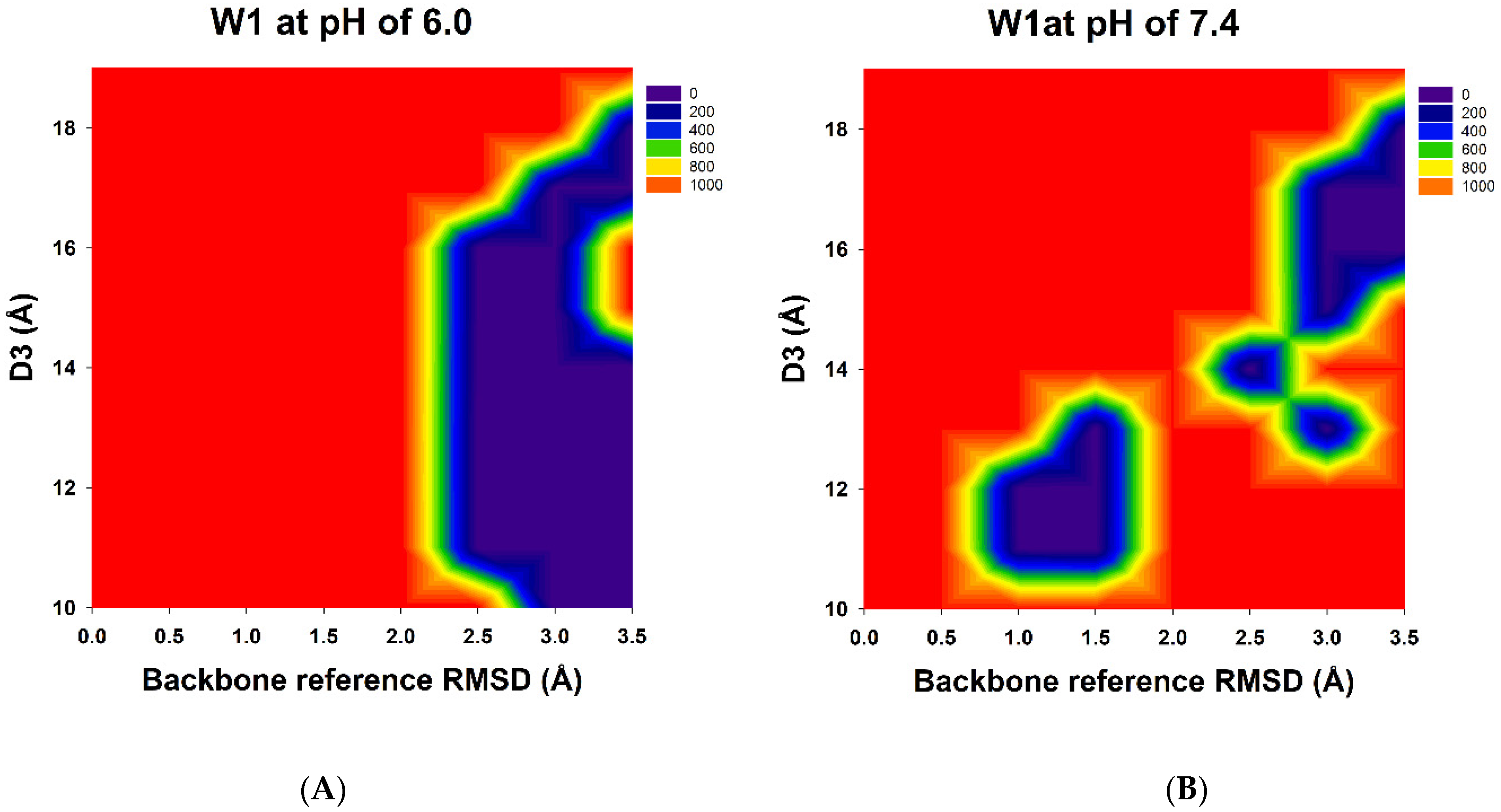
3.1. Prediction of Possible W1-Humira Conformations through GaMD/CpHMD Simulations
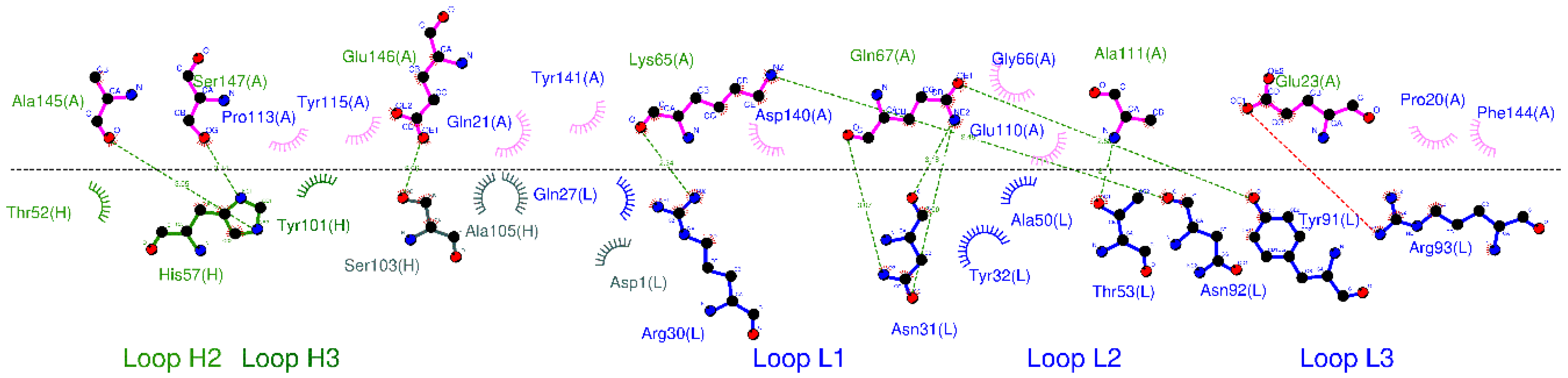
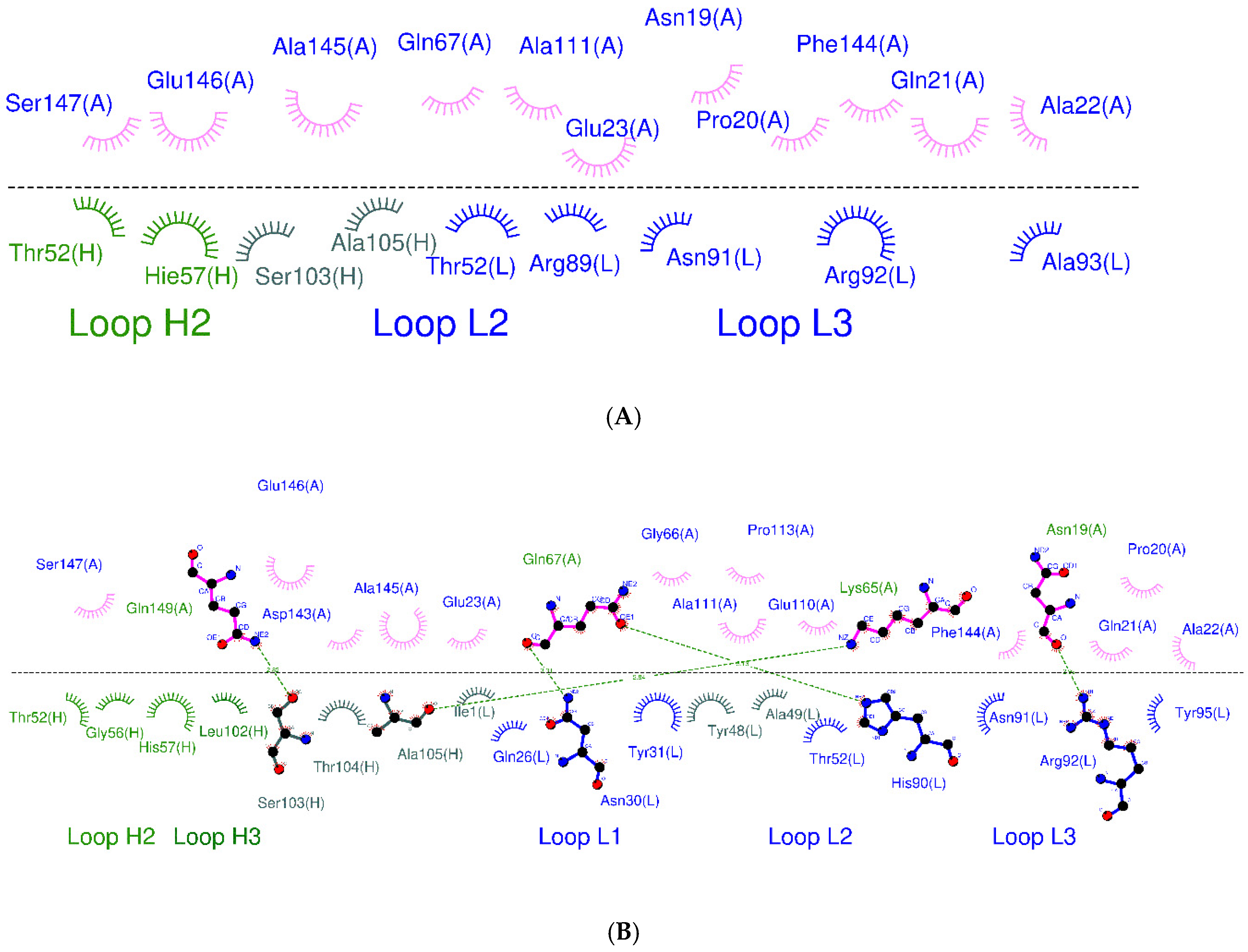
3.2. CpHMD Simulations and Binding Modes Analysis of W1-Humira and Wild-Type Humira at pH 6.0 and 7.4
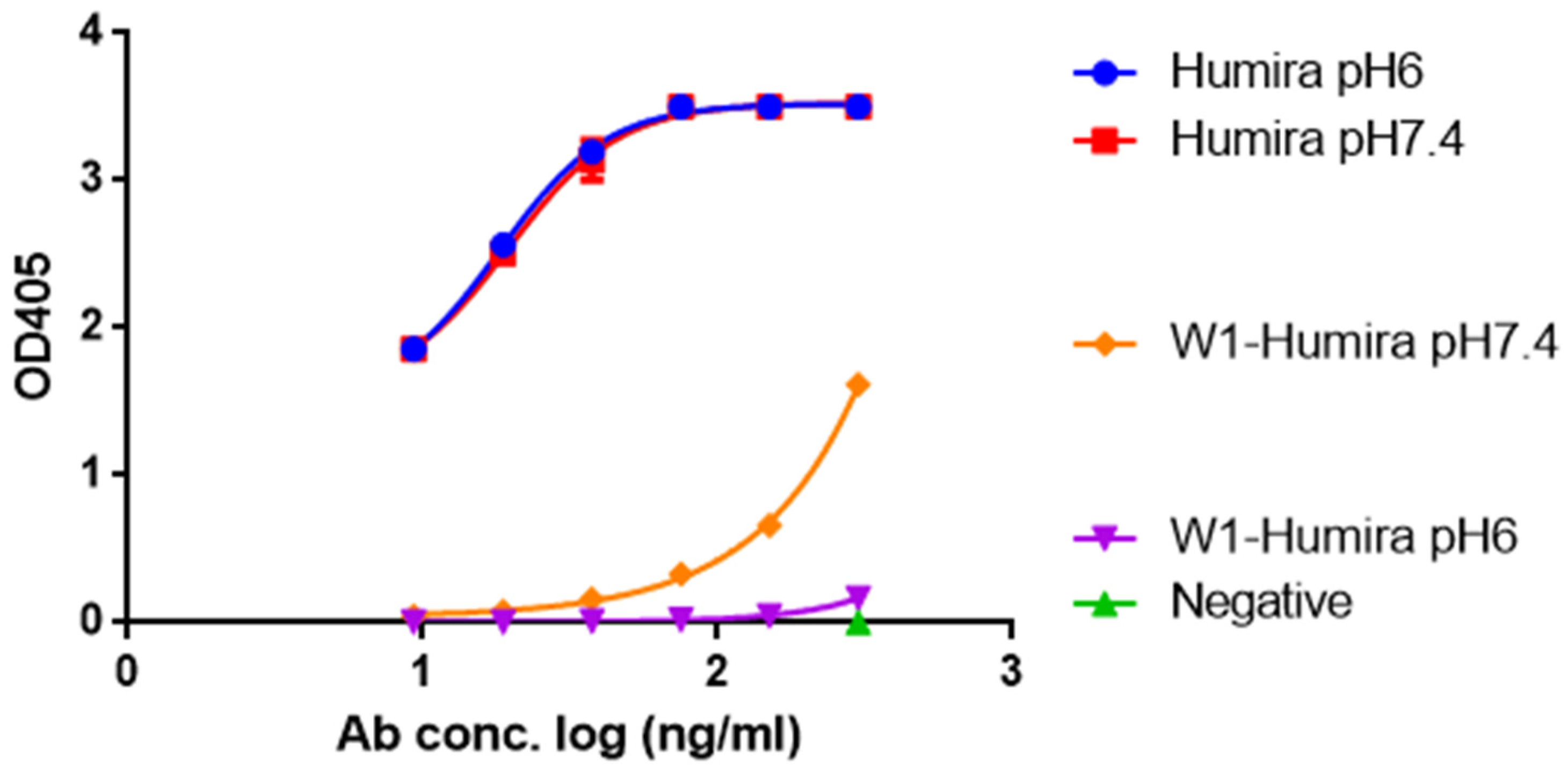
3.3. In Vitro W1-Humira and Wild-Type Humira Binding Ability Testing at pH 6.0 and 7.4
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grilo, A.L.; Mantalaris, A. The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol. 2019, 37, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tian, Z.; Thirumalai, D.; Zhang, X. Neonatal Fc receptor (FcRn): A novel target for therapeutic antibodies and antibody engineering. J. Drug Target. 2014, 22, 269–278. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Chan, Y.-H. Understanding the molecular basis of agonist/antagonist mechanism of human mu opioid receptor through gaussian accelerated molecular dynamics method. Sci. Rep. 2017, 7, 7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, Y.; Liu, J.; Wang, J.; Epps, R.E.; Kettl, D.; Marcus, K.; Seo, S.; Zhu, H.; Wang, Y. Model-Informed Drug Development Approach Supporting Approval of Adalimumab (HUMIRA) in Adolescent Patients with Hidradenitis Suppurativa: A Regulatory Perspective. AAPS J. 2019, 21, 91. [Google Scholar] [CrossRef] [PubMed]
- Aitken, D.; Laslett, L.L.; Pan, F.; Haugen, I.K.; Otahal, P.; Bellamy, N.; Bird, P.; Jones, G. A randomised double-blind placebo-controlled crossover trial of HUMira (adalimumab) for erosive hand OsteoaRthritis—The HUMOR trial. Osteoarthr. Cartil. 2018, 26, 880–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, M.C.; Glover, J.; Greenwald, M.; Porawska, W.; El Khouri, E.C.; Dokoupilova, E.; Vargas, J.I.; Stanislavchuk, M.; Kellner, H.; Baranova, E.; et al. FKB327, an adalimumab biosimilar, versus the reference product: Results of a randomized, Phase III, double-blind study, and its open-label extension. Arthritis Res. Ther. 2019, 21, 281. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.K.; Yang, Y.T.; Bennett, C.L. Why Biologics and Biosimilars Remain So Expensive: Despite Two Wins for Biosimilars, the Supreme Court’s Recent Rulings do not Solve Fundamental Barriers to Competition. Drugs 2018, 78, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- Weisman, M.H.; Moreland, L.W.; Furst, D.E.; Weinblatt, M.E.; Keystone, E.C.; Paulus, H.E.; Teoh, L.S.; Velagapudi, R.B.; Noertersheuser, P.A.; Granneman, G.R.; et al. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: A pilot study. Clin. Ther. 2003, 25, 1700–1721. [Google Scholar] [CrossRef]
- Schröter, C.; Günther, R.; Rhiel, L.; Becker, S.; Toleikis, L.; Doerner, A.; Becker, J.; Schönemann, A.; Nasu, D.; Neuteboom, B.; et al. A generic approach to engineer antibody pH-switches using combinatorial histidine scanning libraries and yeast display. mAbs 2015, 7, 138–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammers, C.M.; Stanley, J.R. Antibody Phage Display: Technique and Applications. J. Investig. Dermatol. 2014, 134, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Könning, D.; Zielonka, S.; Sellmann, C.; Schröter, C.; Grzeschik, J.; Becker, S.; Kolmar, H. Isolation of a pH-Sensitive IgNAR Variable Domain from a Yeast-Displayed, Histidine-Doped Master Library. Mar. Biotechnol. 2016, 18, 161–167. [Google Scholar] [CrossRef]
- Marillet, S.; Lefranc, M.-P.; Boudinot, P.; Cazals, F. Novel Structural Parameters of Ig–Ag Complexes Yield a Quantitative Description of Interaction Specificity and Binding Affinity. Front. Immunol. 2017, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Sankar, K.; Hoi, K.H.; Hötzel, I. Dynamics of heavy chain junctional length biases in antibody repertoires. Commun. Biol. 2020, 3, 207. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, P.; Venet, S.; Kosco-Vilbois, M.; Fischer, N. Purpose-Oriented Antibody Libraries Incorporating Tailored CDR3 Sequences. Antibodies 2015, 4, 103–122. [Google Scholar] [CrossRef]
- Lan, W.; Valente, J.J.; Ilott, A.; Chennamsetty, N.; Liu, Z.; Rizzo, J.M.; Yamniuk, A.P.; Qiu, D.; Shackman, H.M.; Bolgar, M.S. Investigation of anomalous charge variant profile reveals discrete pH-dependent conformations and conformation-dependent charge states within the CDR3 loop of a therapeutic mAb. mAbs 2020, 12, 1763138. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menger, F.M.; Yamada, K. Enzyme catalysis in water pools. J. Am. Chem. Soc. 1979, 101, 6731–6734. [Google Scholar] [CrossRef]
- Claeys, E.; De Smet, S.; Demeyer, D.; Geers, R.; Buys, N. Effect of rate of pH decline on muscle enzyme activities in two pig lines. Meat Sci. 2001, 57, 257–263. [Google Scholar] [CrossRef]
- Laidler, K.J. The influence of pH on the rates of enzyme reactions. Part 1.—General theory. Trans. Faraday Soc. 1955, 51, 528–539. [Google Scholar] [CrossRef]
- Oliveira, A.S.F.; Campos, S.R.R.; Baptista, A.M.; Soares, C.M. Coupling between protonation and conformation in cytochrome c oxidase: Insights from constant-pH MD simulations. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 759–771. [Google Scholar] [CrossRef]
- Shi, C.; Wallace, J.A.; Shen, J.K. Thermodynamic Coupling of Protonation and Conformational Equilibria in Proteins: Theory and Simulation. Biophys. J. 2012, 102, 1590–1597. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Miller, B.T.; Damjanović, A.; Brooks, B.R. Constant pH Molecular Dynamics in Explicit Solvent with Enveloping Distribution Sampling and Hamiltonian Exchange. J. Chem. Theory Comput. 2014, 10, 2738–2750. [Google Scholar] [CrossRef] [Green Version]
- Radak, B.K.; Chipot, C.; Suh, D.; Jo, S.; Jiang, W.; Phillips, J.C.; Schulten, K.; Roux, B. Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems. J. Chem. Theory Comput. 2017, 13, 5933–5944. [Google Scholar] [CrossRef]
- Khandogin, J.; Brooks, C.L. Constant pH Molecular Dynamics with Proton Tautomerism. Biophys. J. 2005, 89, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Mongan, J.; Case, D.A.; McCammon, J.A. Constant pH molecular dynamics in generalized born implicit solvent. J. Comput. Chem. 2004, 25, 2038–2048. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.C.; Shen, J. GPU-Accelerated Implementation of Continuous Constant pH Molecular Dynamics in Amber: pKa Predictions with Single-pH Simulations. J. Chem. Inf. Model. 2019, 59, 4821–4832. [Google Scholar] [CrossRef]
- Johnston, J.M.; Filizola, M. Showcasing modern molecular dynamics simulations of membrane proteins through G protein-coupled receptors. Curr. Opin. Struct. Biol. 2011, 21, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jonsson, A.L.; Beuming, T.; Shelley, J.C.; Voth, G.A. Ligand-Dependent Activation and Deactivation of the Human Adenosine A2A Receptor. J. Am. Chem. Soc. 2013, 135, 8749–8759. [Google Scholar] [CrossRef] [Green Version]
- Provasi, D.; Filizola, M. Putative Active States of a Prototypic G-Protein-Coupled Receptor from Biased Molecular Dynamics. Biophys. J. 2010, 98, 2347–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niesen, M.J.M.; Bhattacharya, S.; Vaidehi, N. The Role of Conformational Ensembles in Ligand Recognition in G-Protein Coupled Receptors. J. Am. Chem. Soc. 2011, 133, 13197–13204. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Nichols, S.E.; McCammon, J.A. Free energy landscape of G-protein coupled receptors, explored by accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2014, 16, 6398–6406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markwick, P.R.L.; McCammon, J.A. Studying functional dynamics in bio-molecules using accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2011, 13, 20053–20065. [Google Scholar] [CrossRef]
- Hamelberg, D.; de Oliveira, C.A.F.; McCammon, J.A. Sampling of slow diffusive conformational transitions with accelerated molecular dynamics. J. Chem. Phys. 2007, 127, 155102. [Google Scholar] [CrossRef]
- Pierce, L.C.T.; Salomon-Ferrer, R.; de Oliveira, C.A.F.; McCammon, J.A.; Walker, R.C. Routine Access to Millisecond Time Scale Events with Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2012, 8, 2997–3002. [Google Scholar] [CrossRef]
- Gasper, P.M.; Fuglestad, B.; Komives, E.A.; Markwick, P.R.L.; McCammon, J.A. Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities. Proc. Natl. Acad. Sci. USA 2012, 109, 21216–21222. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Markwick, P.R.L.; de Oliveira, C.A.F.; McCammon, J.A. Enhanced Lipid Diffusion and Mixing in Accelerated Molecular Dynamics. J. Chem. Theory Comput. 2011, 7, 3199–3207. [Google Scholar] [CrossRef]
- Markwick, P.R.L.; Pierce, L.C.T.; Goodin, D.B.; McCammon, J.A. Adaptive Accelerated Molecular Dynamics (Ad-AMD) Revealing the Molecular Plasticity of P450cam. J. Phys. Chem. Lett. 2011, 2, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Hamelberg, D. A statistical analysis of the precision of reweighting-based simulations. J. Chem. Phys. 2008, 129, 034103. [Google Scholar] [CrossRef] [PubMed]
- Kappel, K.; Miao, Y.; McCammon, J.A. Accelerated molecular dynamics simulations of ligand binding to a muscarinic G-protein-coupled receptor. Q. Rev. Biophys. 2015, 48, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Feher, V.A.; McCammon, J.A. Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput. 2015, 11, 3584–3595. [Google Scholar] [CrossRef]
- Miao, Y.; Feixas, F.; Eun, C.; McCammon, J.A. Accelerated molecular dynamics simulations of protein folding. J. Comput. Chem. 2015, 36, 1536–1549. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N [center-dot] log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Sinko, W.; Pierce, L.; Bucher, D.; Walker, R.C.; McCammon, J.A. Improved Reweighting of Accelerated Molecular Dynamics Simulations for Free Energy Calculation. J. Chem. Theory Comput. 2014, 10, 2677–2689. [Google Scholar] [CrossRef]
- Huang, Y.; Harris, R.C.; Shen, J. Generalized Born Based Continuous Constant pH Molecular Dynamics in Amber: Implementation, Benchmarking and Analysis. J. Chem. Inf. Model. 2018, 58, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, P.; Venet, S.; Fontaine, G.; Ravn, U.; Gueneau, F.; Kosco-Vilbois, M.; Proudfoot, A.E.; Fischer, N. De novo isolation of antibodies with pH-dependent binding properties. mAbs 2015, 7, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.; Sidhu, S.S. Chapter 1—Simplified Synthetic Antibody Libraries. In Methods in Enzymology; Wittrup, K.D., Verdine, G.L., Eds.; Academic Press: Cambridge, MA, USA, 2012; Volume 502, pp. 3–23. [Google Scholar]
- Gupta, N.; Ansari, A.; Dhoke, G.V.; Chilamari, M.; Sivaccumar, J.; Kumari, S.; Chatterjee, S.; Goyal, R.; Dutta, P.K.; Samarla, M.; et al. Computationally designed antibody–drug conjugates self-assembled via affinity ligands. Nat. Biomed. Eng. 2019, 3, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Sulea, T.; Rohani, N.; Baardsnes, J.; Corbeil, C.R.; Deprez, C.; Cepero-Donates, Y.; Robert, A.; Schrag, J.D.; Parat, M.; Duchesne, M.; et al. Structure-based engineering of pH-dependent antibody binding for selective targeting of solid-tumor microenvironment. mAbs 2020, 12, 1682866. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.Y.; Lim, T.S.; Choong, Y.S. Human IgG1 Fc pH-dependent optimization from a constant pH molecular dynamics simulation analysis. Rsc Adv. 2020, 10, 13066–13075. [Google Scholar] [CrossRef]
- Yokota, A.; Tsumoto, K.; Shiroishi, M.; Kondo, H.; Kumagai, I. The Role of Hydrogen Bonding via Interfacial Water Molecules in Antigen-Antibody Complexation: The HyHEL-10-HEL Interaction. J. Biol. Chem. 2003, 278, 5410–5418. [Google Scholar] [CrossRef] [Green Version]
- Van Oss, C.J.; Good, R.J.; Chaudhury, M.K. Nature of the antigen-antibody interaction: Primary and secondary bonds: Optimal conditions for association and dissociation. J. Chromatogr. B Biomed. Sci. Appl. 1986, 376, 111–119. [Google Scholar] [CrossRef]
- Kelow, S.P.; Adolf-Bryfogle, J.; Dunbrack, R.L. Hiding in plain sight: Structure and sequence analysis reveals the importance of the antibody DE loop for antibody-antigen binding. mAbs 2020, 12, 1840005. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Backbone Reference RMSD (BRRMSD) (Å) | Distances (D3) between the Centers of the HCDR3 and LCDR3 (Å) | PMF (Kcal/mol) |
|---|---|---|---|
| 1 | 2.5 | 11.0 | 9.2 |
| 2 | 2.5 | 12.0 | 7.6 |
| 3 | 2.5 | 13.0 | 7.6 |
| 3 | 2.5 | 14.0 | 13.9 |
| 4 | 2.5 | 15.0 | 20.9 |
| 5 | 2.5 | 16.0 | 19.1 |
| 6 | 3.0 | 10.0 | 3.1 |
| 7 | 3.0 | 11.0 | 5.3 |
| 8 | 3.0 | 12.0 | 7.6 |
| 9 | 3.0 | 13.0 | 6.8 |
| 10 | 3.0 | 14.0 | 10.1 |
| 11 | 3.0 | 15.0 | 37.5 |
| 12 | 3.0 | 16.0 | 22.0 |
| 13 | 3.0 | 17.0 | 22.8 |
| 14 | 3.5 | 10.0 | 0.0 |
| 15 | 3.5 | 11.0 | 3.9 |
| 16 | 3.5 | 12.0 | 6.1 |
| 17 | 3.5 | 13.0 | 5.6 |
| 18 | 3.5 | 14.0 | 7.1 |
| 19 | 3.5 | 17.0 | 19.1 |
| 20 | 3.5 | 18.0 | 26.4 |
| Number | Backbone Reference RMSD (BRRMSD) (Å) | Distances (D3) between the Centers of the HCDR3 and LCDR3 (Å) | PMF (Kcal/mol) |
|---|---|---|---|
| 1 | 1.0 | 11.0 | 32.0 |
| 2 | 1.0 | 12.0 | 19.1 |
| 3 | 1.5 | 11.0 | 7.6 |
| 4 | 1.5 | 12.0 | 14.2 |
| 5 | 1.5 | 13.0 | 8.9 |
| 6 | 2.5 | 14.0 | 0.0 |
| 7 | 3.0 | 13.0 | 31.7 |
| 8 | 3.0 | 15.0 | 49.8 |
| 9 | 3.0 | 16.0 | 28.4 |
| 10 | 3.0 | 17.0 | 28.5 |
| 11 | 3.5 | 16.0 | 15.8 |
| 12 | 3.5 | 17.0 | 18.5 |
| 13 | 3.5 | 18.0 | 21.8 |
| Antibody/Regions | W1-Humira at pH 7.4 | W1-Humira at pH 6.0 | Humira (PDB ID: 3WD5) |
|---|---|---|---|
| HCDR1 | Null | Null | Null |
| HCDR2 | Thr52, Gly56, His57 | Thr52 and His57 | Thr52 and His57 (HB) |
| HCDR3 | Leu102, Ser103 (HB), The104, Ala105 (HB) | Ser103 and Ala105 | Tyr101, Ser103 and Ala105 |
| LCDR1 | Gln26, Asn30 (HB) and Tyr31 | Null | Gln27, Arg30, Asn31 (HB) and Tyr32 |
| LCDR2 | Thr52 | Thr52 | Ala50 and Thr53 (HB) |
| LCDR3 | His90 (HB), Asn91, Arg92 (HB) and Tyr95 | Arg89, Asn91, Arg92, and Ala93 | Tyr91 (HB), Asn92 (HB), and Arg93 (HB) |
| Other regions (Heavy chain) | Null | Null | Null |
| Other regions (Light chain) | Ile1, Tyr48, and Ala49 | Null | Asp1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.-T.; Su, Y.-C.; Wang, Y.-J.; Cheng, T.-L.; Wang, Y.-T. Anti-TNF Alpha Antibody Humira with pH-dependent Binding Characteristics: A constant-pH Molecular Dynamics, Gaussian Accelerated Molecular Dynamics, and In Vitro Study. Biomolecules 2021, 11, 334. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020334
Hong S-T, Su Y-C, Wang Y-J, Cheng T-L, Wang Y-T. Anti-TNF Alpha Antibody Humira with pH-dependent Binding Characteristics: A constant-pH Molecular Dynamics, Gaussian Accelerated Molecular Dynamics, and In Vitro Study. Biomolecules. 2021; 11(2):334. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020334
Chicago/Turabian StyleHong, Shih-Ting, Yu-Cheng Su, Yu-Jen Wang, Tian-Lu Cheng, and Yeng-Tseng Wang. 2021. "Anti-TNF Alpha Antibody Humira with pH-dependent Binding Characteristics: A constant-pH Molecular Dynamics, Gaussian Accelerated Molecular Dynamics, and In Vitro Study" Biomolecules 11, no. 2: 334. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020334