
Actin Cytoskeleton and Regulation of TGFβ Signaling: Exploring Their Links

1
Tumor Immunology and Immunotherapy Unit, IRCCS Regina Elena National Cancer Institute, via Chianesi 53, 00144 Rome, Italy
2
Institute of Biochemistry and Cell Biology, National Research Council, via Ramarini 32, 00015 Monterotondo Scalo, Rome, Italy
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this paper.
Biomolecules 2021, 11(2), 336; https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020336
Submission received: 19 January 2021
/
Revised: 15 February 2021
/
Accepted: 20 February 2021
/
Published: 23 February 2021
(This article belongs to the Collection TGF-Beta Signaling in Tissue Fibrosis and Cancer)
Abstract
:Human tissues, to maintain their architecture and function, respond to injuries by activating intricate biochemical and physical mechanisms that regulates intercellular communication crucial in maintaining tissue homeostasis. Coordination of the communication occurs through the activity of different actin cytoskeletal regulators, physically connected to extracellular matrix through integrins, generating a platform of biochemical and biomechanical signaling that is deregulated in cancer. Among the major pathways, a controller of cellular functions is the cytokine transforming growth factor β (TGFβ), which remains a complex and central signaling network still to be interpreted and explained in cancer progression. Here, we discuss the link between actin dynamics and TGFβ signaling with the aim of exploring their aberrant interaction in cancer.
1. Introduction
Actin dynamics critically affect different aspects of human health and disease, ranging from embryonic development to wound repair, inflammation, and cancer [1]. Each of these complex processes requires that a coordinated formation of multiple actin-based structures occurs. Actin dynamics refer to dynamic rearrangements of actin-based structures whose spatial and temporal control generates the physical force that enables cell motile functions. Almost all the cells, from prokaryotic to multicellular organisms, have the ability to sense a wide range of physical and chemical signals from the surrounding environment and elaborate specific responses by adjusting their cytoskeletal organization, and hence their shape and motility [2].
A fine regulation of actin filament dynamics guarantees plasticity and dynamicity of actin cytoskeleton that allows the cell to elaborate the adequate cytoskeleton-based responses to stimuli. Although for a long time considered merely regulators of cell mechanical properties, actin dynamics have emerged as platforms for signaling pathways [3]. Notably, actin dynamics have been linked to gene transcription, pointing out mechanisms that communicate the cytoplasmic actin status to the nuclear genome [4].
The transforming growth factor β (TGFβ) family encompasses cytokines with widespread and diverse biological effects. Essentially, all cells perceive TGFβ family signals and elaborate responses that affect cell proliferation, differentiation, communication, adhesion, movement, metabolism, and death. The wide-ranging nature of biological signals elicited by TGFβ underlies the complexity of this pathway, which mediates fate decisions during development, tissue homeostasis and regeneration, tumorigenesis, fibrosis, and the immune response [5,6,7,8,9,10,11].
The effects of the cytokines of TGFβ family are mediated by a combinatorial set of ligands and receptors and can differ dramatically depending on the cellular context [12]. The TGFβ signaling pathway is critically influenced by mechanical cues including extracellular matrix (ECM) stiffness, cell–cell adhesion, cell–matrix adhesion, cell density, cell tension and cell polarity [13] all processes guaranteed by the plasticity and dynamicity of actin cytoskeleton.
While the role of TGFβ signaling in the organization of actin cytoskeleton architecture has been extensively investigated, the ways in which actin cytoskeleton dynamics modulate TGFβ signaling have just started to emerge.
Herein, we provide a brief overview of the actin dynamics and TGFβ signaling pathway and present evidence to disclose their interaction, starting from ECM, through the plasma membrane up to the nucleusall steps where actin and TGFβ signaling crosstalk and affect cancer.
2. Actin Cytoskeleton: Architecture and Signaling
2.1. Actin Cytoskeleton Architecture and Dynamics
Actin, one of the most abundant and highly conserved proteins in eukaryotic cells, is a 42 kDa adenosine-5′-triphosphate (ATP)-binding protein that cycles from monomeric globular actin (G-actin) to polymeric filamentous actin (F-actin) in a complex process governed by many different actin-binding proteins (ABPs) [14].
Actin polymerization gives rise to filaments with different types of organization: branched and cross-linked networks, parallel bundles, and anti-parallel contractile structures located in specific subcellular compartments [15]. Branched and crosslinked networks of lamellipodia at the front of the cell represent the major engine of cell movement, along with aligned bundles of filopodia. A thin layer of actin forms the cell cortex at the plasma membrane, contributing to the maintenance of cell shape. Interspersed in the rest of the cell, the actin scaffold is made up of a three-dimensional network of cross-linked filaments and contractile bundles, including stress fibers that connect the cell cytoskeleton to the extracellular matrix via focal adhesion sites. Incorporated within the actin network are filaments of the molecular motor protein myosin that produce contraction in the cell body and tension at focal adhesion [15].
This organization makes the cytoskeleton highly responsive to stimuli and regulates dynamic cell behavior [1]. The activity balance of actin assembly factors has to be achieved to ensure the main functions of the cytoskeletal system: to generate force, to create structural scaffolds, and to act as tracks for motor proteins [16]. At a molecular level, actin filament turnover proceeds through finely tuned steps: monomer sequestration or delivery, filament nucleation, elongation, capping, severing, or depolymerization [14]. The rate and direction of polymerization, as well as the shape of the newly generated filament, are determined by local intracellular concentrations of ATP-bound G-actin and by the activity of ABPs [14].
2.2. Actin Cytoskeleton Regulatory Proteins
ABPs act as key players of actin dynamics, participating in diverse processes ranging from maintaining a large pool of actin monomers available for polymerization, to initiating polymerization, nucleating assembly of new filaments, promoting elongation, capping the ends of polymers to terminate elongation, severing, and crosslinking of filaments [14]. G-actin-binding proteins maintain high concentrations of free G-actin by preventing spontaneous nucleation of new filaments while barbed end capping proteins prevent their elongation.
Profilin binds weakly to ATP-actin at the barbed ends, catalyzes nucleotide exchange by rapidly dissociating adenosine-5′-diphosphate (ADP) from newly depolymerized actin monomers, allowing further barbed end elongation [17]. To maintain the actin monomer pool, profilin cooperates with the capping protein (CP) [18]. Furthermore, profilin promotes elongation of barbed ends by delivering actin to polyproline sequences of proteins such as formins and Ena/VASP. At the same time, profilin prevents spontaneous actin nucleation and elongation at the actin pointed ends [14].
To initiate actin filament polymerization in a controlled manner, cells use an Arp2/3 complex to produce actin filament branches, formins to initiate unbranched filaments, and proteins with tandem WH-2 domains to form other filaments [14].
Severing proteins such as cofilin and members of the gelsolin family promote actin filament turnover by disassembling actin filaments, and producing free barbed ends. A large family of cross-linking proteins operate to physically connect actin filaments and stabilize higher-order structures, such as bundles of filaments in microvilli, filopodia, and cytoplasmic cables, as well as networks of actin filaments [19,20].
Among the actin cytoskeleton regulatory proteins, we focus on Ena/VASP, critical players in metastatic cancer progression. Ena/VASP proteins promote actin elongation via antagonizing actin filament capping [21]. The Ena/VASP family encompasses three mammalian family members: VASP (vasodilator-stimulated phosphoprotein), MENA (mammalian ENA homolog), and EVL (Ena-VASP-like), sharing a conserved domain structure. Each family member consists of an N-terminal EVH1 domain, a central proline-rich region, and a C-terminal EVH2 domain [22]. The EVH2 domain, which contains G-actin and F-actin-binding sites, is responsible for promoting actin polymerization [23,24,25]. In contrast, the EVH1 domain mediates intracellular targeting of Ena/VASP proteins by interacting with the “FPPPP” repeats of diverse proteins, including zyxin [26,27], and determines the recruitment of Ena/VASP proteins to specific sites within the cell [22]. The central proline-rich region harbors binding sites for SH3 and WW domain-containing proteins and the actin monomer-binding protein profilin [28]. Ena/VASP proteins are localized to the leading edge of lamellipodia and the tips of filopodia, but also to stress fibers and focal adhesions [28,29]. MENA is the only family member that contains a unique long insertion close to EVH1 domain of five-amino acid long stretch of highly charged basic and acidic amino acids (LERER) [28]. Notably, MENA binds to the C-terminal end of the cytoplasmic tail of the integrin α5 via LERER domain, affecting α5β1 signaling [30]. Differently from all other members of Ena/VASP, there are multiple splice variants of the MENA gene, enabled homolog (ENAH), which are involved in several mechanisms critical for cancer progression. ENAH contains a small coding exon 11a that is included only in epithelial cells and excluded in mesenchymal cell lines and downregulated during epithelial-mesenchymal transition (EMT) [31,32,33]. The splicing of this exon is mainly regulated by the Epithelial Splicing Regulatory Proteins 1 and 2 (ESRP1 and ESRP2) [33]. In invasive tumor cells, the hMENA11a downregulation occurs, and the human MENA (hMENA) splice variant lacking the internal exon 6 (hMENAΔv6) is upregulated [34]. ENAH splice variants containing alternative exons ++ and +++ (Mena INV), linked to cancer cell invasiveness, have also been described [35,36]. Our group has identified the two alternatively expressed isoforms, hMENA11a and hMENAΔv6, that exert opposite roles in breast, lung, and pancreatic cancer cell invasiveness [34,37,38]. Their role during cancer progression and their crosstalk with TGFβ signaling will be discussed in Section 4 and Section 5.
2.3. Actin Dynamics and Signaling: From ECM to the Nucleus
Cell–matrix contacts are dynamic, and cells sense external force and make adjustments, by modulating the cytoskeleton filaments and their linkages to transmembrane proteins [39]. At sites of cell-ECM adhesion, cells sense physical features of the microenvironment through integrins and other adhesive proteins and adjust their own tensional state through actomyosin contractility and organization of the F-actin cytoskeleton to counterbalance extracellular forces [39,40]. Thus, the mechanotransduction process relies on force generation by actin-myosin networks and force transmission through adhesive complexes. Actomyosin contractility is the result of contractile force predominantly generated by the action of myosin II motors on actin filaments. The RhoA and RhoC family members promote actomyosin contractile force generation through ROCK1- and ROCK2-mediated phosphorylation of different downstream target proteins, including LIM kinases 1 and 2 (LIMK1 and LIMK2), the myosin regulatory light chain (MLC) [41]. Force transmission at adhesive complexes relies on the activation of integrins which are clustered into macromolecular structures associated with plasma membrane at the cytoplasmic side, namely focal adhesions, that provide a direct physical link between intracellular actin cytoskeleton and ECM. Integrin activation results from a conformational reorganization of the α-β integrin dimer that critically increases its affinity to the matrix ligand. Proteins such as talins and kindlins bind to β integrin cytoplasmic domains, connecting them to the actin cytoskeleton [2]. A reciprocal regulation between integrins and F-actin exists: integrins promote bundling of actin filaments to generate tension within a cell. Conversely, actin regulatory proteins, actin polymerization, and spatial organization affect integrin function [39].
By playing a key part in the matrix-sensing machinery, actin cytoskeleton, for a long time considered the skeleton of the cell, now emerges as a platform for signal transduction [4], able to transmit tensile stresses generated at the interface between the cell and ECM to the nucleus, thus affecting nuclear properties and gene expression programs [42].
LINC (linker of nucleoskeleton and cytoskeleton) complex is one illustrative player of the transduction of mechanical signals to the nucleus through cytoskeletal physical links [43]. LINC complex, located at the nuclear envelope (NE), enables dynamic rearrangements of the cytoskeletal actin to be communicated to the nucleus. The nuclear membrane proteins Nesprins are components of the LINC complex and sustain the physical connection between the cytoskeleton and nuclear lamina, allowing cells to maintain tension between the plasma membrane and nucleus, essential for both organelle and cytoskeletal organization [44]. Nesprins are characterized by a C-terminal transmembrane domain, the KASH-domain, which anchors them in the nuclear membrane and a N-terminal domain by which they connect the nucleus to the actin cytoskeleton (Nesprin-1 and -2). Connections within the nucleus are mediated by nuclear lamina (lamins A, B1, B2, and C) and associated proteins like emerin.
To the actin cytoskeleton-mediated nuclear transduction also contribute transcription factors such as Yes-associated protein (YAP) and Myocardin-Related Transcription Factors (MRTFs), whose shuttling between the cytoplasm and the nucleus through the nuclear pore complex (NPC) is regulated by actin dynamics.
YAP and TAZ (transcriptional co-activator with PDZ-binding motif) are critical players in the sensing and transduction of mechanical cues to gene expression [45]. They elicit biological effects in response to ECM elasticity by acting as transcriptional cofactors that shuttle from the cytoplasm to the nucleus [45]. YAP and TAZ play key roles not only in physiological settings, but also in tumorigenesis, cancer stem cell induction, cancer-associated fibroblast (CAF) activation, and chemoresistance [46,47]. YAP/TAZ localize to the nucleus and are transcriptionally active in cells plated on large and stiff substrates, which display high ROCK-and non-muscle-myosin-II-driven cytoskeletal tension. Conversely, YAP/TAZ are excluded from the nucleus when cells are plated on softer or smaller substrates, which impose cell rounding and reduced adhesive area [48]. Noteworthy, YAP/TAZ activity is regulated by the conformation and tension of the F-actin cytoskeleton. Rather than F-actin total levels, YAP/TAZ is regulated by the F-actin subcellular organization, fine structure, tension, and resistance offered by molecular structures within the cytoskeleton and by the whole nucleus [48].
The MRTF-SRF circuit is activated by the Rho GTPase family, which regulates essentially every aspect of actin cytoskeleton function [49]. Rho GTPases actively participate in processes by which actin dynamics are transmitted up to the nucleus by regulating effector proteins that modulate the equilibrium of G-actin and F-actin in the cytoplasm. Actin polymerization, downstream to Rho GTPases thus affects the MRTF-SRF (serum response factor) circuit. In the presence of high levels of cytoplasmic G-actin, MRTFs, able to bind G-actin, are retained in the cytoplasm. Changes in actin dynamics may imply the incorporation of G-actin into the F-actin filaments, causing a reduction of G-actin concentration, thus leaving MRTFs free to enter the nucleus and to interact with the transcription factor SRF, which drives the expression of its target genes [50].
3. TGFβ Biology and Signaling in Cancer
Herein, we briefly summarize basics of TGFβ biology and signaling that are covered more extensively in excellent reviews [53,54,55,56,57].
3.1. Brief Overview of TGFβ Isoforms and Their Role in Normal and Pathological Context
Transforming growth factor β factors (TGFβ1, TGFβ2, and TGFβ3) are multi-tasking cytokines implicated in the regulation of a broad range of cellular functions and different biological processes i.e., embryogenesis, immune regulation, fibrosis-associated diseases, and tumor progression [12,58].
Encoded by different genes and uniquely expressed in mammals [59], TGFβ isoforms exert peculiar and not-redundant functions in vivo, as suggested by the diversity of phenotypes of knockout mice for each isoform [60,61,62,63]. TGFβ ligands, alone or in the context of other environmental cues, balance the self-renewal and differentiation process of stem cells and define cell fate during embryonic lineage specification and differentiation [8]. A body of evidence has indicated TGFβ factors as crucial in controlling the immune system [64], during development and maturation of immune cells, in maintaining immune tolerance and homeostasis and controlling innate, as well as in the adaptive immune system [65,66,67]. Moreover, context-dependent pro- or anti-inflammatory effects have been clearly demonstrated [67].
Following tissue damage, TGFβ1 becomes a master regulator of the repair process, providing a rapid restoration of tissue integrity, by reprogramming and suppressing excessive tissue inflammation [58,64,68,69,70].
The deregulation of TGFβ activity may shift the physiological repair to pathological condition such as fibrosis, an aberrant repair response which strongly affects organ functionality. TGFβ implication in the pathogenesis of fibrosis-associated diseases has been extensively sustained by experimental and in vivo models as well as clinical evidence [71,72,73,74,75]. On the other hand, therapeutic implementation of anti-TGFβ approaches in fibrosis-associated diseases is still hampered by the pleiotropic multifunctional role of TGFβ, leaving opened many questions to design effective therapies [58].
In cancer, TGFβ exerts both tumor-suppressive and tumor-promoting functions, referred to as “TGFβ Paradox”, which represents the most critical and cryptic issue of the physio-pathological TGFβ role [76,77]. To suppress tumor, TGFβ induces apoptosis in pre-malignant cells and inhibits cancer cell proliferation, whereas in the late stage of tumorigenesis, it sustains tumor progression. The switch from tumor-suppressive to tumor-promoting function of TGFβ is intimately linked to the triggering of EMT program, closely related to actin cytoskeleton modifications, as further detailed.
In the tumor microenvironment, stromal cells are an abundant source of TGFβ [78], with cancer-associated fibroblasts (CAFs) the most significant producers [79,80]. Activated TGFβ signaling leads to CAF activation, CAF-mediated cancer progression [81] and may differently influence CAF subsets [82].
Noteworthy, CAF-derived TGFβ may contribute to immunosurveillance and escape and may participate in therapy resistance, including immunotherapy with immune checkpoint blockade [83,84,85], also by excluding CD8+ T lymphocytes from the tumor site [86,87]. A recent paper suggests that TGFβ neutralization targets only selected CAF subtypes and, in turn, promotes CAF immunomodulatory properties and the formation of an immune-permissive tumor microenvironment (TME) by regulating ECM density [82].
3.2. Basics of TGFβ Signaling
TGFβ ligands signal to the nucleus, mainly through serine/threonine kinase receptors tetrameric complexes composed of two type II receptors (TβRII) and two type I receptors (TβRI). The receptor activation initiates signaling via both canonical SMAD2/3 and non-canonical pathways.
In the canonical TGFβ-SMAD pathway, upon TGFβ binding, TβRII trans-phosphorylates TβRI and activates its kinase activity resulting in phosphorylation and mobilization of SMAD proteins which include SMAD1, 2, 3, 5 and 8 [88]. SMAD2/3 phosphorylation is required for their association with SMAD4 in mammalian cells [89]. This heterotrimeric complex translocates into the nucleus where it activates or represses the transcription of several genes [88,90]. The interaction of SMAD protein complex with other transcription factors and/or with additional co-activators and co-repressors as well as with chromatin-modifying enzymes ensures the context-dependent cellular response to TGFβ signaling [57].
In the non-canonical pathways, TGFβ can activate the mitogen-activated protein kinases (MAPKs), the extracellular signal-regulated kinase (ERK), phosphatidylinositide 3-kinase (PI3K)/AKT, TNF receptor-associated factor 4/6 (TRAF 4/6) and Rho-like GTPases (Rho) [56,91,92,93,94,95]. Notably, TGFβ has been reported as able to crosstalk with several other signaling pathways (i.e., Wnt, Notch, Hippo signaling, and with tyrosine kinase receptors) to elicit a context-dependent signaling [12,96].
4. TGFβ-Induced Actin Reorganization in Physio-and Pathological Contexts
The actin cytoskeleton reorganization is one of the earliest cellular response to TGFβ signaling, which may generate rapid and long-term modifications of actin dynamics, thus modulating morphology, adhesion, growth, motility, and invasiveness of different cell types. In both non-transformed and transformed epithelial cells, TGFβ can rapidly induce membrane ruffling and actin polymerization at the cell edges [97], whereas a prolonged stimulus results in stable actin filament bundles (stress fibers) [98]. In the short-term response, TGFβ induces rapid activation of GTPases Rho family, including Rac, Cell Division Cycle 42 (CDC42), and RhoA [98], followed by ROCK⁄LIMK⁄cofilin pathway activation and actin cytoskeleton polymerization [99]. On the contrary, the long-term actin cytoskeleton response involves the SMAD pathway, and leads to transcriptional regulation of RhoB and α-SMA in fibroblasts [100].
TGFβ induces myofibroblast differentiation by increasing Rho-dependent cytoskeletal stress fiber formation and Rho/actin/MRTF-A/SRF signaling pathway [101].
TGFβ signaling controls cellular plasticity and is essential in EMT promotion, a physiological process crucial in tissue and organ formation during development, but also acts as a facilitator of tumor progression [102]. Remodeling of the cytoskeleton is a hallmark of EMT, and notably, the actin regulatory genes are the most highly upregulated during TGFβ-induced EMT [103,104,105,106,107]. Several actin cytoskeleton-associated proteins, including hMENA and hMENAΔv6, have been reported to be upregulated during EMT [38,103,104,105,108,109]. Functionally, as a consequence of the TGFβ-induced cytoskeletal remodeling, actin stress fibers are formed in cancer cells, affecting cell shape and function and favoring cancer cell invasion and tissue rigidity [107].
5. Actin-Related Regulatory Functions that Control TGFβ Signaling
Whether TGFβ signaling affects actin cytoskeleton is well established, the reciprocal contribution of actin dynamics on TGFβ signaling regulation is still not enough explored. In this section, we will discuss the specific interactions of actin-related functions with TGFβ pathway, from ECM to the nucleus (Figure 1) passing through the receptor trafficking.
5.1. TGFβ Ligand Activation: Mechanical Cues
Differently from most of the growth factors that are ready to function upon secretion, TGFβ is typically secreted and stored in the ECM as a latent non-active form. This includes a signal peptide (N-terminal latency-associated peptide LAP), and a C-terminal mature TGFβ, which corresponds to the mature active cytokine monomer [110,111]. The presence of LAP prevents binding of TGFβ to its receptors, impeding signaling function. After secretion, both LAP and mature TGFβ1 homodimers are non-covalently associated in the small latent TGFβ complex (SLC). In most cases, LAP is further covalently associated in the ECM with a latent TGFβ-binding protein (LTBP), to create the large latent complex (LLC), an ECM reservoir of TGFβ. The activation of TGFβ requires the release of the LLC from the ECM and further proteolysis/deformation of LAP to release active TGFβ [13,112].
The best-understood mechanism of active TGFβ release from LLC involves the interaction with a specific subset of integrins, and several RGD-binding integrins are able to activate latent TGFβ through binding to this site [113,114]. Contractile force generated by actin cytoskeleton is required for αvβ6 integrin-mediated activation of TGFβ1 [115]. Tensile force is transmitted by cytoskeleton to integrins, which shift towards an active configuration that favors TGFβ1 release [116]. The inhibitor of actin polymerization cytochalasin D abrogates αvβ6-mediated TGFβ1 activation [115], whereas G-protein coupled receptor agonists induce αvβ6-mediated TGFβ activation. Downstream to G-protein coupled receptor agonists, RhoA and Rho kinase are activated, leading to cytoskeletal reorganization that generates cellular tension, which is transmitted to the cytoplasmic domains of the αvβ6 integrin, and, in turn, activates the large latent TGFβ complex [117,118,119].
TGFβ activation engages a dynamic reciprocity with ECM, and a stiffer ECM increases the free available TGFβ and its activation (Figure 1a), in turn driving a positive feedback by inducing matrix synthesis and stiffness, with pathological consequences [120]. Mechanistically, increased stiffness induces integrin clustering, and, in turn, the activation of Rho family GTPases, which stimulate actin remodeling. This integrin-actin linkage enables cell to sense ECM stiffness and regulates TGFβ signaling (Figure 1a) [40].
5.2. ECM-Driven SMAD Intracellular Localization and Transcriptional Activity
Noteworthy, the dynamic remodeling of the actin cytoskeleton is also involved in the localization and activation of SMADs (Figure 1b–e).
In the basal state, SMAD proteins constantly shuttle between the cytoplasm and the nucleus. Once phosphorylated by the receptors, nuclear SMADs activate or repress the transcription of several genes. The constant shuttle of SMAD proteins in to and out of the nucleus also relies on their interaction with the cytoskeleton. Below, some mechanisms of cytoskeleton-related SMAD shuttle are described.
The lamin-binding protein LEMD3 (MAN-1) inhibits TGFβ signaling by binding SMAD2/3 and promoting their dephosphorylation and nuclear export [121,122]. An interesting study by Chambers et al. has shown that the interaction of LEMD3 with SMAD2/3 is negatively regulated by ECM stiffness and antagonized by actin polymerization, highlighting that a stiffened ECM regulates cell response to TGFβ [123].
Nesprin-2 Giant isoform of the Nesprin family proteins is a component of the LINC complex at NE and participates to the mechanical force generation dependent on F-actin network rearrangements, by binding F-actin through the terminal actin-binding domain (ABD) [124]. Studies of wound healing in nesprin-2 Giant deficient mice have reported delayed wound closure and defects in keratinocyte and fibroblast migration and proliferation [125]. Notably, nesprin-2G deficient fibroblasts show defects in cytoskeleton, with F-actin filaments less regular around the nucleus, and a delayed nuclear SMAD accumulation upon TGFβ stimulation [125].
Actin-mediated mechanotransduction intercepts TGFβ signaling also via YAP/TAZ, effectors of the Hippo pathway, critically linked to actin cytoskeleton dynamics as discussed above. TAZ binds and retains SMAD complexes into the nucleus, coupling SMADs to transcriptional machinery. Cytoplasmic retention of phosphorylated TAZ prevents SMAD2/3-SMAD4 complex from accumulating in the nucleus, and TAZ knockdown leads to TGFβ signaling inhibition [126] (Figure 1d). An interesting study shows a multi-step mechanism integrating epithelial polarity with TGFβ signaling. In polarizing epithelial cells, Hippo pathway activation is an early event that promotes cytoplasmic sequestration of TAZ/YAP and suppression of TGFβ-SMAD activity, while prolonged stimulus leads to the basal-lateral restriction of TGFβ receptors, thus reducing SMAD activation. TGFβ receptor sequestration and YAP/TAZ cytoplasmic retention are distinct events that regulate TGFβ signaling in polarized epithelia [127].
Consistent with the involvement of actin cytoskeleton in cellular response to mechanical cues, several studies support a link between matrix rigidity, Rho-Actin-MRTF signatures and TGFβ pathway (Figure 1e). Although the details of this crosstalk are elusive, MRTFs, and in particular MRTF-A, are key regulators of cytoskeletal genes involved in matrix rigidity and TGFβ1-induced EMT. Indeed, TGFβ induces the translocation of MRTFs in to the nucleus and MRTF/SMAD3 complex activates slug transcription [128]. Noteworthy, MRTF-A is implicated in TGFβ1-mediated myofibroblastic differentiation in various cell systems and CAF functions [129,130].
In addition, matrix stiffness regulates cytoskeletal architecture [131], and induces the nuclear localization of MRTF-A [132,133], required for the activation of colon and pulmonary fibroblasts [134,135]. The sum of these findings indicates that ECM stiffness and Actin-MRTF-A signaling are crucial in TGFβ1-induced EMT.
5.3. Intracellular Trafficking of TGFβ Receptors
Different mechanisms regulating TGFβ receptor availability on the cell surface have been reported [136]. Membrane localization and trafficking of receptors are dynamically regulated by two distinct endocytic pathways: clathrin-dependent internalization into the early endosome, important for propagating signal through the SMAD-dependent pathway, and internalization in caveolin-1-positive lipid rafts that sequester TGFβ receptors for their degradation [137].
Actin dynamics, mainly by regulating intracellular vesicle transport, take part in the complex regulation of TGFβ receptor trafficking via numerous actin regulatory proteins (Figure 2).
Fascin actin-bundling protein 1 (FSCN1), a direct TGFβ/Nodal target gene, is specifically required for the trafficking of TβRI from clathrin-coated vesicles to early endosomes, promoting TGFβ signaling. In particular, FSCN1 specifically interacts with TβRI, and its depletion disrupts the association between receptors and actin filaments and sequesters the internalized receptors into clathrin-coated vesicles (Figure 2a) [138]. Notably, FSCN1 is upregulated by TGFβ/Nodal signaling also in a range of tumor cells and overexpressed in different carcinomas where correlates with the clinical aggressiveness [139], suggesting that FSCN1 sustains TGFβ signaling also during tumor progression.
The monomeric small GTPase Rab11 contributes to TGFβ receptor recycling occurring in early endosome compartments, a process that requires the actin-binding protein VASP. In an experimental liver metastasis mouse model, VASP, by regulating Rab11-dependent TβRII recycling to the plasma membrane, sustains TGFβ signaling activation. Reciprocally, TGFβ stimulation results in VASP upregulation in hepatic stellate cells (HSCs). Overall, TGFβ-mediated activation of HSCs within the hepatic tumor microenvironment is a process essential for metastatic tumor growth in the liver, and VASP takes part to this process by sensitizing hepatic stellate cells to TGFβ effects [140] (Figure 2b).
Unconventional myosins, known to participate in the endocytic trafficking and tether membranes or transport them along the actin cytoskeleton, [141] have been linked to the regulation of TGFβ receptor trafficking. Their specific inhibition reduces cell surface expression of TβRII and promotes receptor degradation (Figure 2c) [142].
TβRII recycling is also affected by Arf GAP with GTP-binding protein-like domain, Ankyrin repeat, and PH domain 2 isoform 2 (AGAP2), a member of Arf GAP (ADP-ribosylation factor GTPase activating proteins) family, critical regulators of membrane trafficking and remodeling of actin cytoskeleton. In the pathophysiology of liver cancer, the depletion of AGAP2 inhibits the recycling of TβRII back to the plasmatic membrane with an important implication in TGFβ-driven pro-fibrotic effects [143]. Proliferation and migration of HSC induced by TGFβ are reduced by AGAP2 depletion and loss of AGAP2 also interferes with TGFβ-dependent collagen type I production (Figure 2d).
5.4. SMAD-Dependent Transcriptional Activation
The dynamic of actin cytoskeleton also influences the expression of the negative regulators of TGFβ signaling, Ski (Sloan-Kettering Institute proto-oncogene) and SnoN (Ski-related novel gene) proteins [144]. Ski and SnoN are able to interact with SMAD2/3 and SMAD4 in the cytoplasm and to recruit elements of the repressor machinery on TGFβ target gene promoters [145,146,147]. Notably, the TGFβ/Smad pathway and co-regulators Ski and SnoN clearly control each other by activating positive and negative feedback mechanisms. Ski and SnoN are upregulated by TGFβ signaling and, in turn, they act in a negative feedback manner. Moreover, TGFβ regulates Ski and SnoN levels by inducing their degradation via the ubiquitin-proteasome system [148,149]. The alteration in these regulatory mechanisms controls the magnitude and duration of the TGFβ signal and may lead to disease development [150]. Remarkably, changes in actin cytoskeleton dynamics control Ski protein stability and subcellular localization induced by TGFβ in normal hepatocytes mechanisms suggested to be lost in hepatoma cells (Figure 3) [151]. Interestingly, while the inhibition of actin-cytoskeleton rearrangements by cytochalasin D treatment induces a rapid and strong degradation of Ski protein, it stabilizes SnoN protein. This differential actin-cytoskeleton-mediated regulation of the two proteins controls different sets of TGFβ target genes, as demonstrated in normal hepatocytes. On the contrary, in hepatoma cells the actin-mediated modulation of Ski and SnoN protein stability is lost or deeply modified (Figure 3) [152].
5.5. Actin-Dynamic Regulators that Control TGFβ Signaling
Several actin regulatory proteins affect TGFβ signaling at different levels. The roles of Rho GTPase family have been extensively described. Herein, we focus on the role of the actin cytoskeleton regulatory protein hMENA, which regulates TGFβ signaling. Of note, ENAH gene splicing is regulated by TGFβ. Furthermore, a number of other actin regulatory proteins are reported and summarized in Table 1.
The Rho family of small GTPases, by controlling the polymerization and depolymerization of actin filaments, regulates specific actin cytoskeletal structures, including stress fibers, lamellipodia, and filopodia [49,169]. Rho proteins exert both a positive and negative regulatory role of SMAD-dependent TGFβ signaling. The complete inhibition of Rho activity by C3 exotoxin attenuated SMAD-mediated transactivation and the ectopic expression of a dominant-negative RhoA mutant in neural crest stem cells blocked SMAD2 and SMAD3 phosphorylation and their translocation to the nucleus [153]. On the other hand, the overexpression of RhoB in epithelial cells has been shown to inhibit TGFβ transcriptional program [154]. RhoB but not RhoA overexpression in HaCaT keratinocytes and pancreatic carcinoma cells decreases the expression levels of TβRII and antagonizes the TGFβ-mediated anti-proliferative responses.
Interestingly, antagonistic functions in the regulation of TGFβ signaling have also been reported for the two alternatively spliced isoforms of Rho, ras-related C3 botulinum toxin substrate 1 (RAC1), and RAC1b proteins. Most of the studies have shown a positive interaction between RAC1 and TGFβ signaling [170,171,172], since RAC1 can be activated by TGFβ and may promote the activation of SMAD2. Surprisingly, despite its high structural similarity with RAC1, RAC1b has been shown to be a negative regulator of SMAD signaling, acting as an endogenous inhibitor of RAC1. RAC1b maintains a differentiated epithelial phenotype and prevents the RAC1-driven EMT process [173,174] and cell migration [175]. RAC1b also antagonizes TGFβ dependent EMT by inhibiting p38 and MEK/ERK signaling. Recently, Ungefroren et al. have suggested that RAC1b confers anti-oncogenic properties to pancreatic carcinoma cells not only by acting as an antagonist of RAC1, but also by directly affecting the regulation of main components of TGFβ signal pathway [176]. Ungefroren et al. have also recently revealed an unexpected role of RAC1B in the regulation of TGFβ secretion implicated in cell motility suppression [158]. It is interesting to note that RAC1b increases malignant transformation in response to other EMT-inducers, such as MMP3 [177,178]. Noteworthy, the knockdown of the master regulator of EMT, ESRP1, but not ESRP2, increases the level of RAC1b in head and neck squamous cancer cells [179]. Considering that RAC1 and RAC1b control TGFβ responses in cancer cells in an antagonistic manner, the final outcome of TGFβ signal transduction, in a given tissue or cell type, appears to be strictly dependent on RAC1b/RAC1 ratio [176,180].
Downstream of the Rho GTPases, WAVE3, belonging to the WASP/WAVE family of proteins, activates the Arp2/3 complex, leading to actin polymerization and assembly of actin filaments [181], playing a critical role in cell motility and invasion [182]. WAVE3 expression is highly upregulated by TGFβ in metastatic triple-negative breast cancer cells (TNBCs), where is required for TGFβ-mediated EMT [160]. Noteworthy, the Authors suggest that the TGFβ-induced increase of WAVE3 expression correlates with the functional conversion of TGFβ from a suppressor to a promoter of TNBC development, proposing that therapeutic targeting of WAVE3 may restore the cytostatic activities of TGFβ in late-stage TNBCs [160]. hMENA and its isoforms critically support malignant transformation and progression in different tumors [37,38,183,184,185,186]. Our studies indicated that the alternative splicing of ENAH gene generates the alternatively expressed epithelial hMENA11a and mesenchymal hMENAΔv6 isoforms, which contribute to isoform-specific actin cytoskeleton organization, crucial in activating signaling pathways related to immunosuppressive TME such as TGFβ, β1 integrin, and AXL-GAS6 signaling [38,186,187]. hMENA spliced variants have been linked to TGFβ-induced EMT process and TGFβ is involved in the regulation of splicing of ENAH gene. TGFβ inhibits the expression of the epithelial splicing factor epithelial splicing regulatory proteins 1/2 (ESRP1/2) and primes the switching of hMENA isoforms by excluding the exon 11a in mammary epithelial and breast cancer cells [33,34,188,189,190]. In addition, TGFβ induces the expression of polypyrimidine tract-binding protein 1 (PTBP1), involved in exon 11a skipping, enhancing migration and invasion of lung cancer cells as well as EMT features in A549 cells [191]. Interestingly, loss of CDC-like kinase 2 (CLK2), kinase linked to the splicing factor RBFOX2, has been shown to activate EMT and TGFβ signaling pathway [192]. Notably, our group has demonstrated that TGFβ1 treatment downregulates hMENA11a expression in pancreatic cancer cell lines, which we have proposed as a prerequisite for EMT [38]. Differently, TGFβ1 upregulates hMENA and the mesenchymal pro-invasive hMENAΔv6 isoform, implicated in SMAD2 phosphorylation and TGFβ1-induced EMT [38]. Noteworthy, hMENA/hMENAΔv6 overexpression defines a CAF subtype with pro-tumoral functions and hMENA expression in CAFs is able to regulate both autocrine and paracrine TGFβ signaling, leading to the regulation of TGFβ-mediated crosstalk between CAFs and tumor cells (unpublished data). We also demonstrated that hMENA isoforms interact with ECM/β1 integrin axis by modulating the expression and activation of β1 integrin and the composition of ECM. Indeed, depletion of all hMENA isoforms in lung cancer cells results in abrogation of stress fibers, actin reorganization, and a dramatic cell shape change, affecting the balance between actin polymerization and depolymerization, that results in F-actin decrease. As a consequence, hMENA affects MRTF-A subcellular localization, SRF activity, and in turn the expression of its target gene β1 integrin.
From a clinical point of view, the pattern of hMENA isoform expression associates with clinical outcome in pancreatic and lung cancer patients [37,38,193].
We have demonstrated that in tumor tissues of early NSCLC patients, the presence of the epithelial-associated isoform, hMENA11a is associated with a low expression of fibronectin in the stroma, and that these tumor features identify patients with better prognosis [186].
The actin regulatory protein Zyxin directly interacts with-actinin and Ena/VASP proteins to dock them to actin filaments [194,195]. Zyxin primarily localizes to focal adhesion, where serves as a docking protein involved in the regulation of cell-extracellular matrix adhesion and in the mechano-transduction process [196]. Several studies pointed at a significant role of zyxin in both physiological and pathological TGFβ1-mediated EMT. In a context of pathological EMT, zyxin has been shown to be not only a functional target of TGFβ, but also an effector of the TGFβ signaling regulation. Indeed, in lung cancer cells, it controls cell motility through the modulation of cell adhesion and expression of integrin α5β1 [161]. Zyxin-knockdown in MDA-MB-231 breast cancer cells abrogates the TGFβ-mediated E-cadherin downregulation and impairs TGFβ-induced cell motility, supporting the notion that zyxin controls TGFβ-induced migration [162]. Recently, high zyxin expression has been associated with a poor prognosis in glioblastoma multiforme patients [197].
A tight association between actin cytoskeleton dynamics and TGFβ1-induced EMT program has also been evidenced for cofilin-1 activity, crucial for the regulation of cell migration and invasion [198]. Cofilin is directly involved in the actin polymerization and remodeling dynamics in response to extracellular signals, including TGFβ and has been firstly identified as a SMAD-independent intracellular effector of TGFβ signaling in prostate cancer cells [199]. Collazo et al. found that Cofilin1 activity coordinates the responses to TGFβ needed for cancer cell migration and metastasis in murine and human prostate cancer [163]. In gastric cancer cells, Wang et al., proved the role of Cofilin in actin-mediated TGFβ-induced EMT [164]. In addition, in colorectal cancer (CRC) Cofilin-1 has been shown to augment the cell–cell disassembly, migration, invasion, and focal adhesion organization during TGFβ-induced EMT. Interestingly, in urothelial cancer, Hensley et al., found a close association of high nuclear localization of Cofilin-1 with increased tumor stage and progression, and have suggested that Cofilin-1 involvement in EMT may be due to its ability to control gene expression by regulating actin organization in the nucleus [200].
Filamin has been identified as a SMAD-binding protein, and Filamin-deficient melanoma cells showed an impaired SMAD2 phosphorylation [166]. Its role as a scaffold to connect the actin cytoskeleton with a multitude of proteins involved in signal transduction has been extensively reported [201,202,203,204]. Due to its ability to interact with SMAD proteins, Filamin has been proposed to function as an anchor protein able to control SMAD protein localization near the cell surface receptors or to keep SMAD conformation, allowing TβRI-mediated phosphorylation [166]. Although Filamin has been originally identified as a tumor promoting factor, prognostic value is mainly dependent on its subcellular localization and binding with different proteins [205]. High Filamin levels have been shown to be predictors of poor patient outcome in several tumors including melanoma, breast, and glioblastoma multiforme [206]. Whereas, the Filamin A isoform has been shown to inhibit tumor progression by regulating breast cancer type 1 susceptibility protein (BRCA1) expression in human breast cancer [207].
An interesting role for actin-binding proteins in the epigenetic regulation of TGFβ signaling has been shown for Profilin 2 (Pfn2). It prevents nuclear translocation of HDAC1 and suppresses its recruitment to SMAD2 and SMAD3 promoters, thus leading to transcriptional activation of SMAD2 and SMAD3. Moreover, Pfn2 correlates with SMAD3 expression in human lung cancers, where its overexpression promotes lung cancer growth and metastasis [167]. On the other hand, the loss of profilin 2 contributes to enhanced EMT and metastasis of colorectal cancer. Thus, the prognostic value of Pfn2 is still controversial [208,209].
Among actin regulatory proteins which nucleate the assembly of unbranched actin filaments, the Formins and in particular the two Formin family members, diaphanous-related formin 1 and 3 (DIAPH1 and DIAPH3) have a crucial role in TGFβ-induced EMT in several tumor cells (i.e., lung, mammary, and renal epithelial cells) [168,210]. The inhibition of Formins, and not of Arp2/3 complex, completely blocked both TGFβ-mediated cell transcription and morphological changes as suggested by their ability to increase the activity of SRF [211,212].
6. Conclusions and Perspectives
Herein, we highlight how actin dynamics and TGFβ signaling act in tissue homeostasis and how their deregulation leads to fibrosis and cancer. Key proteins involved in the actin cytoskeleton described in this review are linked to TGFβ signaling and TGFβ-mediated EMT. Our effort was to envision how the deregulation of pathways from ECM to the nucleus involves actin dynamics, contributing to TGFβ-mediated cancer progression. While substantial insights in the field were obtained, new layers of complexity and regulation continue to be discovered.
For progress to occur in the understanding of the crucial role of actin dynamics in cancer, a significant effort needs to be made to identify master regulators of TGFβ pro-tumor activity mediated by actin cytoskeleton dynamics. The comprehension of these mechanisms during cancer progression and overall in therapy resistance will be a future cornerstone for novel TGFβ-directed therapies in the new era of immuno-oncology.
Author Contributions
R.M. and P.T. designed and drafted the manuscript; A.T. prepared the figures; P.N. designed and revised the manuscript and critically discussed the content. All authors have read and agreed to the published version of the manuscript.
Funding
The work of P.N. laboratory is supported by the Italian Association for Cancer Research AIRC: 5 × 1000, 12182 (P.N.) and IG 19822 (P.N.).
Acknowledgments
The Authors thank all components of the Laboratory for their support and in particular Francesca Di Modugno for her critical comments and suggestions. The Authors apologize to those authors whose important studies have not been cited due to space limitations. Figures were created with BioRender (http://biorender.com/, accessed on 22 February 2021).
Conflicts of Interest
The Authors declare no conflict of interest.
References
- Davidson, A.J.; Wood, W. Unravelling the Actin Cytoskeleton: A New Competitive Edge? Trends Cell Biol. 2016, 26, 569–576. [Google Scholar] [CrossRef] [Green Version]
- Geiger, B.; Spatz, J.P.; Bershadsky, A.D. Environmental sensing through focal adhesions. Nat. Rev. Mol. Cell Biol. 2009, 10, 21–33. [Google Scholar] [CrossRef]
- Moujaber, O.; Stochaj, U. The Cytoskeleton as Regulator of Cell Signaling Pathways. Trends Biochem. Sci. 2020, 45, 96–107. [Google Scholar] [CrossRef]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, B.J.; Kan, O.K.; Loveland, K.L.; Elias, J.A.; Bardin, P.G. In the Shadow of Fibrosis: Innate Immune Suppression Mediated by Transforming Growth Factor-beta. Am. J. Respir. Cell Mol. Biol. 2016, 55, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Benke, K.; Ágg, B.; Szilveszter, B.; Tarr, F.; Nagy, Z.B.; Pólos, M.; Daróczi, L.; Merkely, B.; Szabolcs, Z. The role of transforming growth factor-beta in Marfan syndrome. Cardiol. J. 2013, 20, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Shovlin, C.L. Molecular Defects in Rare Bleeding Disorders: Hereditary Haemorrhagic Telangiectasia. Thromb. Haemost. 1997, 78, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Wrana, J.L. TGF-beta Family Signaling in Embryonic and Somatic Stem-Cell Renewal and Differentiation. Cold Spring Harb. Perspect Biol. 2017, 9, a022186. [Google Scholar] [CrossRef] [PubMed]
- Beyer, T.A.; Narimatsu, M.; Weiss, A.; David, L.; Wrana, J.L. The TGFbeta superfamily in stem cell biology and early mammalian embryonic development. Biochim. Biophys. Acta 2013, 1830, 2268–2279. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Ten Dijke, P. Immunoregulation by members of the TGFbeta superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Massague, J. Contextual determinants of TGFbeta action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin Dynamics, Architecture, and Mechanics in Cell Motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Campellone, K.G.; Welch, M.D. A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 2010, 11, 237–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mockrin, S.C.; Korn, E.D. Acanthamoeba profilin interacts with G-actin to increase the rate of exchange of actin-bound adenosine 5′-triphosphate. Biochemistry 1980, 19, 5359–5362. [Google Scholar] [CrossRef]
- Edwards, M.; Zwolak, A.; Schafer, D.A.; Sept, D.; Dominguez, R.; Cooper, J.A. Capping protein regulators fine-tune actin assembly dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10, a018267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsudaira, P. Actin crosslinking proteins at the leading edge. Semin. Cell Biol. 1994, 5, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Bear, J.E.; Gertler, F.B. Ena/VASP: Towards resolving a pointed controversy at the barbed end. J. Cell Sci. 2009, 122 Pt 12, 1947–1953. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Dent, E.W.; Bear, J.E.; Loureiro, J.J.; Gertler, F.B. Ena/VASP Proteins: Regulators of the Actin Cytoskeleton and Cell Migration. Annu. Rev. Cell Dev. Biol. 2003, 19, 541–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzik, M.; Kotova, T.I.; Higgs, H.N.; Hazelwood, L.; Hanein, D.; Gertler, F.B.; Schafer, D.A. Ena/VASP Proteins Enhance Actin Polymerization in the Presence of Barbed End Capping Proteins. J. Biol. Chem. 2005, 280, 28653–28662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Urbanke, C.; Resch, G.P.; Small, J.V.; Faix, J. Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J. 2008, 27, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Vinzenz, M.; Stradal, T.E.B.; Small, J.V.; Curth, U.; Dickinson, R.B.; Faix, J. Molecular mechanism of Ena/VASP-mediated actin-filament elongation. EMBO J. 2011, 30, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, K.; Ebel, F.; Frank, R.; Reinhard, M.; Domann, E.; Carl, U.D.; Walter, U.; Gertler, F.B.; Wehland, J.; Chakraborty, T. A novel proline-rich motif present in ActA of Listeria monocytogenes and cytoskeletal proteins is the ligand for the EVH1 domain, a protein module present in the Ena/VASP family. EMBO J. 1997, 16, 5433–5444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bear, J.E.; Loureiro, J.J.; Libova, I.; Fässler, R.; Wehland, J.; Gertler, F.B. Negative Regulation of Fibroblast Motility by Ena/VASP Proteins. Cell 2000, 101, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Gertler, F.B.; Niebuhr, K.; Reinhard, M.; Wehland, J.; Soriano, P. Mena, a Relative of VASP and Drosophila Enabled, Is Implicated in the Control of Microfilament Dynamics. Cell 1996, 87, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, M.; Halbrugge, M.; Scheer, U.; Wiegand, C.; Jockusch, B.M.; Walter, U. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. EMBO J. 1992, 11, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- Gupton, S.L.; Riquelme, D.; Hughes-Alford, S.K.; Tadros, J.; Rudina, S.S.; Hynes, R.O.; Lauffenburger, D.; Gertler, F.B. Mena binds alpha5 integrin directly and modulates alpha5beta1 function. J. Cell Biol. 2012, 198, 657–676. [Google Scholar] [CrossRef] [Green Version]
- Di Modugno, F.; DeMonte, L.; Balsamo, M.; Bronzi, G.; Nicotra, M.R.; Alessio, M.; Jager, E.; Condeelis, J.S.; Santoni, A.; Natali, P.G.; et al. Molecular cloning of hMena (ENAH) and its splice variant hMena+11a: Epidermal growth factor increases their expression and stimulates hMena+11a phosphorylation in breast cancer cell lines. Cancer Res. 2007, 67, 2657–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pino, M.S.; Balsamo, M.; Di Modugno, F.; Mottolese, M.; Alessio, M.; Melucci, E.; Milella, M.; McConkey, D.J.; Philippar, U.; Gertler, F.B.; et al. Human Mena+11a Isoform Serves as a Marker of Epithelial Phenotype and Sensitivity to Epidermal Growth Factor Receptor Inhibition in Human Pancreatic Cancer Cell Lines. Clin. Cancer Res. 2008, 14, 4943–4950. [Google Scholar] [CrossRef] [Green Version]
- Warzecha, C.C.; Jiang, P.; Amirikian, K.; Dittmar, K.A.; Lu, H.; Shen, S.; Guo, W.; Xing, Y.; Carstens, R.P. An ESRP-regulated splicing programme is abrogated during the epithelial–mesenchymal transition. EMBO J. 2010, 29, 3286–3300. [Google Scholar] [CrossRef] [PubMed]
- Di Modugno, F.; Iapicca, P.; Boudreau, A.; Mottolese, M.; Terrenato, I.; Perracchio, L.; Carstens, R.P.; Santoni, A.; Bissell, M.J.; Nisticò, P. Splicing program of human MENA produces a previously undescribed isoform associated with invasive, mesenchymal-like breast tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 19280–19285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goswami, S.; Philippar, U.; Sun, D.; Patsialou, A.; Avraham, J.; Wang, W.; Di Modugno, F.; Nistico, P.; Gertler, F.B.; Condeelis, J.S. Identification of invasion specific splice variants of the cytoskeletal protein Mena present in mammary tumor cells during invasion in vivo. Clin. Exp. Metastasis 2009, 26, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Philippar, U.; Roussos, E.T.; Oser, M.; Yamaguchi, H.; Kim, H.-D.; Giampieri, S.; Wang, Y.; Goswami, S.; Wyckoff, J.B.; Lauffenburger, D.A.; et al. A Mena Invasion Isoform Potentiates EGF-Induced Carcinoma Cell Invasion and Metastasis. Dev. Cell 2008, 15, 813–828. [Google Scholar] [CrossRef] [Green Version]
- Bria, E.; Di Modugno, F.; Sperduti, I.; Iapicca, P.; Visca, P.; Alessandrini, G.; Antoniani, B.; Pilotto, S.; Ludovini, V.; Vannucci, J.; et al. Prognostic impact of alternative splicing-derived hMENA isoforms in resected, node-negative, non-small-cell lung cancer. Oncotarget 2014, 5, 11054–11063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchionna, R.; Iapicca, P.; Di Modugno, F.; Trono, P.; Sperduti, I.; Fassan, M.; Cataldo, I.; Rusev, B.C.; Lawlor, R.T.; Diodoro, M.G.; et al. The pattern of hMENA isoforms is regulated by TGF-beta1 in pancreatic cancer and may predict patient outcome. Oncoimmunology 2016, 5, e1221556. [Google Scholar] [CrossRef] [Green Version]
- Vogel, V.; Sheetz, M.P. Local force and geometry sensing regulate cell functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 265–275. [Google Scholar] [CrossRef]
- Dufort, C.C.; Paszek, M.J.; Weaver, V.M. Balancing forces: Architectural control of mechanotransduction. Nat. Rev. Mol. Cell Biol. 2011, 12, 308–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alisafaei, F.; Jokhun, D.S.; Shivashankar, G.V.; Shenoy, V.B. Regulation of nuclear architecture, mechanics, and nucleocytoplasmic shuttling of epigenetic factors by cell geometric constraints. Proc. Natl. Acad. Sci. USA 2019, 116, 13200–13209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 216, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellad, J.A.; Warren, D.T.; Shanahan, C.M. Nesprins LINC the nucleus and cytoskeleton. Curr. Opin. Cell Biol. 2011, 23, 47–54. [Google Scholar] [CrossRef]
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Hall, A. Rho GTPases and the Actin Cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Posern, G.; Treisman, R. Actin’ together: Serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol. 2006, 16, 588–596. [Google Scholar] [CrossRef]
- Serebryannyy, L.; de Lanerolle, P. Nuclear actin: The new normal. Mutat. Res. 2020, 821, 111714. [Google Scholar] [CrossRef]
- Spencer, V.A.; Costes, S.; Inman, J.L.; Xu, R.; Chen, J.; Hendzel, M.J.; Bissell, M.J. Depletion of nuclear actin is a key mediator of quiescence in epithelial cells. J. Cell Sci. 2010, 124, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Padua, D.; Massague, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFbeta pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, M.; Massague, J. SMADs: Mediators and regulators of TGF-beta signaling. Curr. Opin. Genet. Dev. 1998, 8, 103–111. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. The regulation of TGFbeta signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P.; Mueller, T.D.; Springer, T.A. Structural Biology and Evolution of the TGF-beta Family. Cold Spring Harb. Perspect Biol. 2016, 8, a022103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Dickson, M.C.; Martin, J.S.; Cousins, F.M.; Kulkarni, A.B.; Karlsson, S.; Akhurst, R.J. Defective haematopoiesis and vasculogenesis in transforming growth factor-beta 1 knock out mice. Development 1995, 121, 1845–1854. [Google Scholar]
- Proetzel, G.; Pawlowski, S.A.; Wiles, M.V.; Yin, M.; Boivin, G.P.; Howles, P.N.; Ding, J.; Ferguson, M.W.; Doetschman, T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 1995, 11, 409–414. [Google Scholar] [CrossRef]
- Kulkarni, A.B.; Thyagarajan, T.; Letterio, J.J. Function of cytokines within the TGF-beta superfamily as determined from transgenic and gene knockout studies in mice. Curr. Mol. Med. 2002, 2, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-beta activation and function in immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanjabi, S.; Oh, S.A.; Li, M.O. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb. Perspect Biol. 2017, 9, a022236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Bartram, U.; Speer, C.P. The role of transforming growth factor beta in lung development and disease. Chest 2004, 125, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.; Jenkins, R.H.; Fraser, D.J. MicroRNAs, transforming growth factor beta-1, and tissue fibrosis. J. Pathol. 2012, 229, 274–285. [Google Scholar] [CrossRef]
- Ignotz, R.A.; Kelly, B.; Davis, R.J.; Massagué, J. Biologically active precursor for transforming growth factor type alpha, released by retrovirally transformed cells. Proc. Natl. Acad. Sci. USA 1986, 83, 6307–6311. [Google Scholar] [CrossRef] [Green Version]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnylal, S.; Denton, C.P.; Zheng, B.; Keene, D.R.; He, R.; Adams, H.P.; VanPelt, C.S.; Geng, Y.J.; Deng, J.M.; Behringer, R.R.; et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007, 56, 334–344. [Google Scholar] [CrossRef]
- Sanderson, N.; Factor, V.; Nagy, P.; Kopp, J.; Kondaiah, P.; Wakefield, L.; Roberts, A.B.; Sporn, M.B.; Thorgeirsson, S.S. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc. Natl. Acad. Sci. USA 1995, 92, 2572–2576. [Google Scholar] [CrossRef] [Green Version]
- Anscher, M.S.; Peters, W.P.; Reisenbichler, H.; Petros, W.P.; Jirtle, R.L. Transforming growth factor beta as a predictor of liver and lung fibrosis after autologous bone marrow transplantation for advanced breast cancer. N. Engl. J. Med. 1993, 328, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.M.; Padilla, C.M.; McLaughlin, S.R.; Mathes, A.L.; Ziemek, J.; Goummih, S.; Nakerakanti, S.; York, M.; Farina, G.; Whitfield, M.L.; et al. Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Investig. 2015, 125, 2795–2807. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Schiemann, W.P. The TGF-beta paradox in human cancer: An update. Future Oncol. 2009, 5, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakowlew, S.B. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006, 25, 435–457. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.F.; et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012, 22, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.F.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Calon, A.; Tauriello, D.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Grauel, A.L.; Nguyen, B.; Ruddy, D.; Laszewski, T.; Schwartz, S.; Chang, J.; Chen, J.; Piquet, M.; Pelletier, M.; Yan, Z.; et al. TGFbeta-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat. Commun. 2020, 11, 6315. [Google Scholar] [CrossRef]
- Nakazawa, N.; Yokobori, T.; Kaira, K.; Turtoi, A.; Baatar, S.; Gombodorj, N.; Handa, T.; Tsukagoshi, M.; Ubukata, Y.; Kimura, A.; et al. High Stromal TGFBI in Lung Cancer and Intratumoral CD8-Positive T Cells were Associated with Poor Prognosis and Therapeutic Resistance to Immune Checkpoint Inhibitors. Ann. Surg. Oncol. 2019, 27, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Vanpouille-Box, C.; Formenti, S.C. Dual Transforming Growth Factor-beta and Programmed Death-1 Blockade: A Strategy for Immune-Excluded Tumors? Trends Immunol. 2018, 39, 435–437. [Google Scholar] [CrossRef]
- Endo, E.; Okayama, H.; Nakajima, S.; Saito, K.; Nakano, H.; Endo, E.; Kase, K.; Ito, M.; Yamauchi, N.; Yamada, L.; et al. A TGFbeta-Dependent Stromal Subset Underlies Immune Checkpoint Inhibitor Efficacy in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Colorectal Cancer. Mol. Cancer Res. 2020, 18, 1402–1413. [Google Scholar] [CrossRef]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Abdollah, S.; Macias-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997, 272, 27678–27685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.H.; Derynck, R. Specificity and versatility in tgf-beta signaling through Smads. Annu. Rev. Cell. Dev. Biol. 2005, 21, 659–693. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; Von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landström, M. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Zhang, Y.E. Mechanistic insight into contextual TGF-beta signaling. Curr. Opin. Cell. Biol. 2018, 51, 1–7. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Tang, J.; Overstreet, J.M.; Anorga, S.; Lian, F.; Arnouk, A.; Goldschmeding, R.; Higgins, P.J.; Samarakoon, R. Rac-GTPase promotes fibrotic TGF-beta1 signaling and chronic kidney disease via EGFR, p53, and Hippo/YAP/TAZ pathways. FASEB J. 2019, 33, 9797–9810. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Stournaras, C. Regulation of actin organisation by TGF-beta in H-ras-transformed fibroblasts. J. Cell Sci. 1999, 112, 1169–1179. [Google Scholar]
- Edlund, S.; Landstrom, M.; Heldin, C.H.; Aspenstrom, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell. 2002, 13, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Vardouli, L.; Moustakas, A.; Stournaras, C. LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J. Biol. Chem. 2005, 280, 11448–11457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardouli, L.; Vasilaki, E.; Papadimitriou, E.; Kardassis, D.; Stournaras, C. A novel mechanism of TGFbeta-induced actin reorganization mediated by Smad proteins and Rho GTPases. FEBS J. 2008, 275, 4074–4087. [Google Scholar] [CrossRef]
- Sandbo, N.; Dulin, N. Actin cytoskeleton in myofibroblast differentiation: Ultrastructure defining form and driving function. Transl. Res. 2011, 158, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahata, K.; Dadras, M.S.; Moustakas, A. TGF-beta Family Signaling in Epithelial Differentiation and Epithelial-Mesenchymal Transition. Cold Spring Harb. Perspect. Biol. 2018, 10, a022194. [Google Scholar] [CrossRef] [Green Version]
- Zavadil, J.; Bitzer, M.; Liang, D.; Yang, Y.C.; Massimi, A.; Kneitz, S.; Piek, E.; Böttinger, E.P. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc. Natl. Acad. Sci. USA 2001, 98, 6686–6691. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Law, B.K.; Aakre, M.E.; Edgerton, M.; Shyr, Y.; Bhowmick, N.A.; Moses, H.L. Transforming growth factor beta-regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res. 2003, 5, R187–R198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-beta and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell. 2005, 16, 1987–2002. [Google Scholar] [CrossRef] [Green Version]
- Keshamouni, V.G.; Michailidis, G.; Grasso, C.S.; Anthwal, S.; Strahler, J.R.; Walker, A.; Arenberg, D.A.; Reddy, R.C.; Akulapalli, S.; Thannickal, V.J.; et al. Differential Protein Expression Profiling by iTRAQ−2DLC−MS/MS of Lung Cancer Cells Undergoing Epithelial-Mesenchymal Transition Reveals a Migratory/Invasive Phenotype. J. Proteome Res. 2006, 5, 1143–1154. [Google Scholar] [CrossRef]
- Gladilin, E.; Ohse, S.; Boerries, M.; Busch, H.; Xu, C.; Schneider, M.; Meister, M.; Eils, R. TGFbeta-induced cytoskeletal remodeling mediates elevation of cell stiffness and invasiveness in NSCLC. Sci. Rep. 2019, 9, 7667. [Google Scholar] [CrossRef] [PubMed]
- Haynes, J.; Srivastava, J.; Madson, N.; Wittmann, T.; Barber, D.L. Dynamic actin remodeling during epithelial–mesenchymal transition depends on increased moesin expression. Mol. Biol. Cell 2011, 22, 4750–4764. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, J.S.; Harpel, J.G.; Gleizes, P.E.; Mazzieri, R.; Nunes, I.; Rifkin, D.B. Latent transforming growth factor-beta: Structural features and mechanisms of activation. Kidney Int. 1997, 51, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Barcellos-Hoff, M.H. Latency and activation in the control of TGF-beta. J. Mammary Gland. Biol. Neoplasia 1996, 1, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Sheppard, D. Cross talk among TGF-beta signaling pathways, integrins, and the extracellular matrix. Cold Spring Harb. Perspect. Biol. 2011, 3, a005017. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Lodyga, M.; Hinz, B. TGF-beta1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.G.; Su, X.; Su, G.; Scotton, C.J.; Camerer, E.; Laurent, G.J.; Davis, G.E.; Chambers, R.C.; Matthay, M.A.; Sheppard, D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J. Clin. Invest. 2006, 116, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Porte, J.; Knox, A.J.; Weinreb, P.H.; Maher, T.M.; Violette, S.M.; McAnulty, R.J.; Sheppard, D.; Jenkins, G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 2009, 174, 1264–1279. [Google Scholar] [CrossRef] [Green Version]
- Tatler, A.L.; Jenkins, G. TGF-beta activation and lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 130–136. [Google Scholar] [CrossRef]
- Wipff, P.J.; Rifkin, D.B.; Meister, J.J.; Hinz, B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J. Cell. Biol. 2007, 179, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Lin, F.; Morrison, J.M.; Wu, W.; Worman, H.J. MAN1, an integral protein of the inner nuclear membrane, binds Smad2 and Smad3 and antagonizes transforming growth factor-beta signaling. Hum. Mol. Genet. 2005, 14, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, B.; Gilquin, B.; Tellier-Lebègue, C.; Östlund, C.; Wu, W.; Pérez, J.; El Hage, P.; Lallemand, F.; Worman, H.J.; Zinn-Justin, S. Inhibition of TGF-beta signaling at the nuclear envelope: Characterization of interactions between MAN1, Smad2 and Smad3, and PPM1A. Sci. Signal. 2013, 6, ra49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, D.M.; Moretti, L.; Zhang, J.J.; Cooper, S.W.; Chambers, D.M.; Santangelo, P.J.; Barker, T.H. LEM domain-containing protein 3 antagonizes TGFbeta-SMAD2/3 signaling in a stiffness-dependent manner in both the nucleus and cytosol. J. Biol. Chem. 2018, 293, 15867–15886. [Google Scholar] [CrossRef] [Green Version]
- Zhen, Y.-Y.; Libotte, T.; Munck, M.; Noegel, A.A.; Korenbaum, E. NUANCE, a giant protein connecting the nucleus and actin cytoskeleton. J. Cell Sci. 2002, 115, 3207–3222. [Google Scholar]
- Rashmi, R.; Eckes, B.; Eichinger, L.; Noegel, A.A.; Glöckner, G.; Groth, M.; Neumann, S.; Gloy, J.; Sellin, L.; Walz, G.; et al. The nuclear envelope protein Nesprin-2 has roles in cell proliferation and differentiation during wound healing. Nucleus 2012, 3, 172–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varelas, X.; Sakuma, R.; Samavarchi-Tehrani, P.; Peerani, R.; Rao, B.M.; Dembowy, J.; Yaffe, M.B.; Zandstra, P.W.; Wrana, J.L. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 2008, 10, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, M.; Samavarchi-Tehrani, P.; Varelas, X.; Wrana, J.L. Distinct polarity cues direct Taz/Yap and TGFbeta receptor localization to differentially control TGFbeta-induced Smad signaling. Dev. Cell. 2015, 32, 652–656. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Mayanagi, T.; Sobue, K. Dual roles of myocardin-related transcription factors in epithelial–mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 2007, 179, 1027–1042. [Google Scholar] [CrossRef]
- Crider, B.J.; Risinger, G.M., Jr.; Haaksma, C.J.; Howard, E.W.; Tomasek, J.J. Myocardin-related transcription factors A and B are key regulators of TGF-beta1-induced fibroblast to myofibroblast differentiation. J. Invest. Dermatol. 2011, 131, 2378–2385. [Google Scholar] [CrossRef] [Green Version]
- Werner, S.; Lützkendorf, J.; Müller, T.; Müller, L.P.; Posern, G. MRTF-A controls myofibroblastic differentiation of human multipotent stromal cells and their tumour-supporting function in xenograft models. Sci. Rep. 2019, 9, 11725. [Google Scholar] [CrossRef] [Green Version]
- Yeung, T.; Georges, P.C.; Flanagan, L.A.; Marg, B.; Ortiz, M.; Funaki, M.; Zahir, N.; Ming, W.; Weaver, V.; Janmey, P.A. Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskelet. 2004, 60, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.W.; Chen, Q.K.; Gjorevski, N.; Nelson, C.M. Tissue geometry patterns epithelial-mesenchymal transition via intercellular mechanotransduction. J. Cell. Biochem. 2010, 110, 44–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjorevski, N.; Boghaert, E.; Nelson, C.M. Regulation of Epithelial-Mesenchymal Transition by Transmission of Mechanical Stress through Epithelial Tissues. Cancer Microenviron. 2011, 5, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Yang, N.; Fiore, V.F.; Barker, T.H.; Sun, Y.; Morris, S.W.; Ding, Q.; Thannickal, V.J.; Zhou, Y. Matrix Stiffness–Induced Myofibroblast Differentiation Is Mediated by Intrinsic Mechanotransduction. Am. J. Respir. Cell Mol. Biol. 2012, 47, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.A.; Rodansky, E.S.; Haak, A.J.; Larsen, S.D.; Neubig, R.R.; Higgins, P.D. Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-beta-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel. Dis. 2014, 20, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Yakymovych, I.; Yakymovych, M.; Heldin, C.H. Intracellular trafficking of transforming growth factor beta receptors. Acta Biochim. Biophys. Sin. 2018, 50, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Guglielmo, G.M.; Le Roy, C.; Goodfellow, A.F.; Wrana, J.L. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat. Cell. Biol. 2003, 5, 410–421. [Google Scholar] [CrossRef]
- Liu, Z.; Ning, G.; Xu, R.; Cao, Y.; Meng, A.; Wang, Q. Fscn1 is required for the trafficking of TGF-beta family type I receptors during endoderm formation. Nat. Commun. 2016, 7, 12603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Kim, D.J.; Adams, J.C. The roles of fascins in health and disease. J. Pathol. 2011, 224, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.; Li, J.; Shah, V.H.; Kang, N.; Verma, V.K.; Liu, C.; Billadeau, D.D.; Lamprecht, G.; Xiang, X.; Guo, L.; et al. Vasodilator-stimulated phosphoprotein promotes activation of hepatic stellate cells by regulating Rab11-dependent plasma membrane targeting of transforming growth factor beta receptors. Hepatology 2014, 61, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Batters, C.; Veigel, C. Mechanics and Activation of Unconventional Myosins. Traffic 2016, 17, 860–871. [Google Scholar] [CrossRef] [Green Version]
- Shih-Wei, W.; Chih-Ling, C.; Kao, Y.C.; Martin, R.; Knölker, H.J.; Shiao, M.S.; Chen, C.L. Pentabromopseudilin: A myosin V inhibitor suppresses TGF-beta activity by recruiting the type II TGF-beta receptor to lysosomal degradation. J. Enzyme Inhib. Med. Chem. 2018, 33, 920–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-Corcuera, A.; Ansorena, E.; Montiel-Duarte, C.; Iraburu, M.J. AGAP2: Modulating TGFbeta1-Signaling in the Regulation of Liver Fibrosis. Int. J. Mol. Sci. 2020, 21, 1400. [Google Scholar] [CrossRef] [Green Version]
- Deheuninck, J.; Luo, K. Ski and SnoN, potent negative regulators of TGF-beta signaling. Cell Res. 2009, 19, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabata, T.; Kokura, K.; Ten Dijke, P.; Ishii, S. Ski co-repressor complexes maintain the basal repressed state of the TGF-beta target gene, SMAD7, via HDAC3 and PRMT. Genes Cells 2009, 14, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Denissova, N.G.; Liu, F. Repression of Endogenous Smad7 by Ski. J. Biol. Chem. 2004, 279, 28143–28148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tecalco-Cruz, A.C.; Sosa-Garrocho, M.; Vázquez-Victorio, G.; Ortiz-García, L.; Domínguez-Hüttinger, E.; Macías-Silva, M. Transforming growth factor-beta/SMAD Target gene SKIL is negatively regulated by the transcriptional cofactor complex SNON-SMAD. J. Biol. Chem. 2012, 287, 26764–26776. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T.; Oshima, Y.; Hikita, A. Regulation of TGF-beta family signalling by ubiquitination and deubiquitination. J. Biochem. 2013, 154, 481–489. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liu, X.; Ng-Eaton, E.; Lodish, H.F.; Weinberg, R.A. SnoN and Ski protooncoproteins are rapidly degraded in response to transforming growth factor beta signaling. Proc. Natl. Acad. Sci. USA 1999, 96, 12442–12447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tecalco-Cruz, A.C.; Rios-Lopez, D.G.; Vázquez-Victorio, G.; Rosales-Alvarez, R.E.; Macías-Silva, M. Transcriptional cofactors Ski and SnoN are major regulators of the TGF-beta/Smad signaling pathway in health and disease. Signal. Transduct. Target. Ther. 2018, 3, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Victorio, G.; Caligaris, C.; Del Valle-Espinosa, E.; Sosa-Garrocho, M.; González-Arenas, N.R.; Reyes-Cruz, G.; Briones-Orta, M.A.; Macías-Silva, M. Novel Regulation of Ski Protein Stability and Endosomal Sorting by Actin Cytoskeleton Dynamics in Hepatocytes. J. Biol. Chem. 2015, 290, 4487–4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caligaris, C.; Vázquez-Victorio, G.; Sosa-Garrocho, M.; Ríos-López, D.G.; Marín-Hernández, A.; Macías-Silva, M. Actin-cytoskeleton polymerization differentially controls the stability of Ski and SnoN co-repressors in normal but not in transformed hepatocytes. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 1832–1841. [Google Scholar] [CrossRef]
- Chen, S.; Crawford, M.; Day, R.M.; Briones, V.R.; Leader, J.E.; Jose, P.A.; Lechleider, R.J. RhoA modulates Smad signaling during transforming growth factor-beta-induced smooth muscle differentiation. J. Biol. Chem. 2006, 281, 1765–1770. [Google Scholar] [CrossRef] [Green Version]
- Engel, M.E.; Datta, P.K.; Moses, H.L. RhoB is stabilized by transforming growth factor beta and antagonizes transcriptional activation. J. Biol. Chem. 1998, 273, 9921–9926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adnane, J.; Seijo, E.; Chen, Z.; Bizouarn, F.; Leal, M.; Sebti, S.M.; Muñoz-Antonia, T. RhoB, not RhoA, represses the transcription of the transforming growth factor beta type II receptor by a mechanism involving activator protein. J. Biol. Chem. 2002, 277, 8500–8507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Groth, S.; Sebens, S.; Lehnert, H.; Gieseler, F.; Fändrich, F. Differential roles of Smad2 and Smad3 in the regulation of TGF-beta1-mediated growth inhibition and cell migration in pancreatic ductal adenocarcinoma cells: Control by Rac. Mol. Cancer. 2011, 10, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Sebens, S.; Giehl, K.; Helm, O.; Groth, S.; Fändrich, F.; Röcken, C.; Sipos, B.; Lehnert, H.; Gieseler, F. Rac1b negatively regulates TGF-beta1-induced cell motility in pancreatic ductal epithelial cells by suppressing Smad signalling. Oncotarget 2014, 5, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Otterbein, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.U. RAC1B Regulation of TGFB1 Reveals an Unexpected Role of Autocrine TGFbeta1 in the Suppression of Cell Motility. Cancers 2020, 12, 3570. [Google Scholar] [CrossRef]
- Witte, D.; Otterbein, H.; Förster, M.; Giehl, K.; Zeiser, R.; Lehnert, H.; Ungefroren, H. Negative regulation of TGF-beta1-induced MKK6-p38 and MEK-ERK signalling and epithelial-mesenchymal transition by Rac1b. Sci. Rep. 2017, 7, 17313. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.A.; Davuluri, G.; Parvani, J.G.; Schiemann, B.J.; Wendt, M.K.; Plow, E.F.; Schiemann, W.P.; Sossey-Alaoui, K. Upregulated WAVE3 expression is essential for TGF-beta-mediated EMT and metastasis of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2013, 142, 341–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mise, N.; Savai, R.; Yu, H.; Schwarz, J.; Kaminski, N.; Eickelberg, O. Zyxin Is a Transforming Growth Factor-β (TGF-β)/Smad3 Target Gene That Regulates Lung Cancer Cell Motility via Integrin α5β1. J. Biol. Chem. 2012, 287, 31393–31405. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Cheng, H.; Gao, R.; Mu, C.; Chen, L.; Wu, S.; Chen, Q.; Zhu, Y. Zyxin-Siah2-Lats2 axis mediates cooperation between Hippo and TGF-beta signalling pathways. Nat. Commun. 2016, 7, 11123. [Google Scholar] [CrossRef] [Green Version]
- Collazo, J.; Zhu, B.; Larkin, S.; Martin, S.K.; Pu, H.; Horbinski, C.; Koochekpour, S.; Kyprianou, N. Cofilin drives cell-invasive and metastatic responses to TGF-beta in prostate cancer. Cancer Res. 2014, 74, 2362–2373. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Tao, L.; Jin, F.; Gu, H.; Dai, X.; Ni, T.; Feng, J.; Ding, Y.; Xiao, W.; Qian, Y.; et al. Cofilin 1 induces the epithelial-mesenchymal transition of gastric cancer cells by promoting cytoskeletal rearrangement. Oncotarget 2017, 8, 39131–39142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa-Squiavinato, A.C.M.; Rocha, M.R.; Barcellos-de-Souza, P.; de Souza, W.F.; Morgado-Diaz, J.A. Cofilin-1 signaling mediates epithelial-mesenchymal transition by promoting actin cytoskeleton reorganization and cell-cell adhesion regulation in colorectal cancer cells. Biochim. Biophys. Acta Mol. Cell. Res. 2019, 1866, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, A.; Masuda, Y.; Ohta, Y.; Ikeda, K.; Watanabe, K. Filamin associates with Smads and regulates transforming growth factor-beta signaling. J. Biol. Chem. 2001, 276, 17871–17877. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.-N.; Ding, W.-Q.; Guo, X.-J.; Yuan, X.-W.; Wang, D.-M.; Song, J.-G. Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 2015, 6, 8230. [Google Scholar] [CrossRef] [PubMed]
- Rana, M.K.; Aloisio, F.M.; Choi, C.; Barber, D.L. Formin-dependent TGF-beta signaling for epithelial to mesenchymal transition. Mol. Biol. Cell. 2018, 29, 1465–1475. [Google Scholar] [CrossRef]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef]
- Santibanez, J.F.; Kocic, J.; Fabra, A.; Cano, A.; Quintanilla, M. Rac1 modulates TGF-beta1-mediated epithelial cell plasticity and MMP9 production in transformed keratinocytes. FEBS Lett. 2010, 584, 2305–2310. [Google Scholar] [CrossRef]
- Wang, S.E.; Yu, Y.; Criswell, T.L.; DeBusk, L.M.; Lin, P.C.; Zent, R.; Johnson, D.H.; Ren, X.; Arteaga, C.L. Oncogenic mutations regulate tumor microenvironment through induction of growth factors and angiogenic mediators. Oncogene 2010, 29, 3335–3348. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.-J.; Sun, Y.-H.; Zhang, S.-J.; Jiang, J.-X.; Dong, X.-W.; Jia, Y.-L.; Shen, J.; Guan, Y.; Zhang, L.-H.; Li, F.-F.; et al. Cigarette smoke-induced alveolar epithelial–mesenchymal transition is mediated by Rac1 activation. Biochim. Biophys. Acta 2014, 1840, 1838–1849. [Google Scholar] [CrossRef]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A Guardian of the Epithelial Phenotype and Protector Against Epithelial-Mesenchymal Transition. Cells 2019, 8, 1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungefroren, H.; Otterbein, H.; Fiedler, C.; Mihara, K.; Hollenberg, M.D.; Gieseler, F.; Lehnert, H.; Witte, D. RAC1B Suppresses TGF-β1-Dependent Cell Migration in Pancreatic Carcinoma Cells through Inhibition of the TGF-β Type I Receptor ALK. Cancers 2019, 11, 691. [Google Scholar] [CrossRef] [Green Version]
- Otterbein, H.; Lehnert, H.; Ungefroren, H. Negative Control of Cell Migration by Rac1b in Highly Metastatic Pancreatic Cancer Cells Is Mediated by Sequential Induction of Nonactivated Smad3 and Biglycan. Cancers 2019, 11, 1959. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Wellner, U.F.; Keck, T.; Lehnert, H.; Marquardt, J.-U. The Small GTPase RAC1B: A Potent Negative Regulator of-and Useful Tool to Study-TGFβ Signaling. Cancers 2020, 12, 3475. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Nieto, M.A.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nat. Cell Biol. 2005, 436, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Stallings-Mann, M.L.; Waldmann, J.; Zhang, Y.; Miller, E.; Gauthier, M.L.; Visscher, D.W.; Downey, G.P.; Radisky, E.S.; Fields, A.P.; Radisky, D.C. Matrix Metalloproteinase Induction of Rac1b, a Key Effector of Lung Cancer Progression. Sci. Transl. Med. 2012, 4, 142ra95. [Google Scholar] [CrossRef] [Green Version]
- Ishii, H.; Saitoh, M.; Sakamoto, K.; Kondo, T.; Katoh, R.; Tanaka, S.; Motizuki, M.; Masuyama, K.; Miyazawa, K. Epithelial Splicing Regulatory Proteins 1 (ESRP1) and 2 (ESRP2) Suppress Cancer Cell Motility via Different Mechanisms. J. Biol. Chem. 2014, 289, 27386–27399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; Hass, R.; von der Ohe, J.; Lehnert, H.; Ungefroren, H. The role of TGF-beta and its crosstalk with RAC1/RAC1b signaling in breast and pancreas carcinoma. Cell Commun. Signal. 2017, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Takenawa, T.; Suetsugu, S. The WASP-WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell. Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef]
- Martin, T.A.; Frugtniet, B.; Jiang, W.G. Role of the WASP and WAVE family proteins in breast cancer invasion and metastasis. Breast Cancer: Targets Ther. 2015, 7, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Gertler, F.; Condeelis, J. Metastasis: Tumor cells becoming MENAcing. Trends Cell Biol. 2011, 21, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Di Modugno, F.; Mottolese, M.; Di Benedetto, A.; Conidi, A.; Novelli, F.; Perracchio, L.; Venturo, I.; Botti, C.; Jager, E.; Santoni, A.; et al. The cytoskeleton regulatory protein hMena (ENAH) is overexpressed in human benign breast lesions with high risk of transformation and human epidermal growth factor receptor-2-positive/hormonal receptor-negative tumors. Clin. Cancer Res. 2006, 12, 1470–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.-D.; Jin, Q.; Wang, L.-L.; Han, S.-F.; Chen, Y.-B.; Sun, G.-D.; Sun, S.-F.; Sun, S.-W.; Wang, T.; Liu, F.-J.; et al. The significance of ENAH in carcinogenesis and prognosis in gastric cancer. Oncotarget 2017, 8, 72466–72479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Modugno, F.; Spada, S.; Palermo, B.; Visca, P.; Iapicca, P.; Di Carlo, A.; Antoniani, B.; Sperduti, I.; Di Benedetto, A.; Terrenato, I.; et al. hMENA isoforms impact NSCLC patient outcome through fibronectin/beta1 integrin axis. Oncogene 2018, 37, 5605–5617. [Google Scholar] [CrossRef]
- Melchionna, R.; Spada, S.; Di Modugno, F.; D’Andrea, D.; Di Carlo, A.; Panetta, M.; Mileo, A.M.; Sperduti, I.; Antoniani, B.; Gallo, E.; et al. The actin modulator hMENA regulates GAS 6- AXL axis and pro-tumor cancer/stromal cell cooperation. EMBO Rep. 2020, 21, e50078. [Google Scholar] [CrossRef]
- Horiguchi, K.; Sakamoto, K.; Koinuma, D.; Semba, K.; Inoue, A.; Inoue, S.; Fujii, H.; Yamaguchi, A.; Miyazawa, K.; Miyazono, K.; et al. TGF-beta drives epithelial-mesenchymal transition through deltaEF1-mediated downregulation of ESRP. Oncogene 2012, 31, 3190–3201. [Google Scholar] [CrossRef]
- Saitoh, M.; Miyazawa, K. Transcriptional and post-transcriptional regulation in TGF-beta-mediated epithelial-mesenchymal transition. J. Biochem. 2012, 151, 563–571. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, N.; Ashok, C.; Natua, S.; Pant, D.; Cherian, A.; Pandkar, M.R.; Yadav, P.; Narayanan, S.S.V.; Mishra, J.; Samaiya, A.; et al. Hypoxia-induced TGF-beta-RBFOX2-ESRP1 axis regulates human MENA alternative splicing and promotes EMT in breast cancer. NAR Cancer. 2020, 2, zcaa021. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, L.; Huang, L.; Lei, S.; Cai, X.; Breitzig, M.; Zhang, B.; Yang, A.; Ji, W.; Huang, M.; et al. PTBP1 enhances exon11a skipping in Mena pre-mRNA to promote migration and invasion in lung carcinoma cells. Biochim. Biophys. Acta Gene. Regul. Mech. 2019, 1862, 858–869. [Google Scholar] [CrossRef]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Di Modugno, F.; Caprara, V.; Chellini, L.; Tocci, P.; Spadaro, F.; Ferrandina, G.; Sacconi, A.; Blandino, G.; Nisticò, P.; Bagnato, A.; et al. hMENA is a key regulator in endothelin-1/beta-arrestin1-induced invadopodial function and metastatic process. Proc. Natl. Acad. Sci. USA 2018, 115, 3132–3137. [Google Scholar] [CrossRef] [Green Version]
- Schmeichel, K.L.; Beckerle, M.C. The LIM domain is a modular protein-binding interface. Cell 1994, 79, 211–219. [Google Scholar] [CrossRef]
- Crawford, A.W.; Michelsen, J.W.; Beckerle, M.C. An interaction between zyxin and alpha-actinin. J. Cell Biol. 1992, 116, 1381–1393. [Google Scholar] [CrossRef]
- Yoshigi, M.; Hoffman, L.M.; Jensen, C.C.; Yost, H.J.; Beckerle, M.C. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J. Cell Biol. 2005, 171, 209–215. [Google Scholar] [CrossRef] [Green Version]
- Wen, X.-M.; Luo, T.; Jiang, Y.; Wang, L.-H.; Luo, Y.; Chen, Q.; Yang, K.; Yuan, Y.; Luo, C.; Zhang, X.; et al. Zyxin (ZYX) promotes invasion and acts as a biomarker for aggressive phenotypes of human glioblastoma multiforme. Lab. Investig. 2020, 100, 812–823. [Google Scholar] [CrossRef]
- Bravo-Cordero, J.J.; Magalhaes, M.A.O.; Eddy, R.J.; Hodgson, L.; Condeelis, J. Functions of cofilin in cell locomotion and invasion. Nat. Rev. Mol. Cell Biol. 2013, 14, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Fukada, K.; Zhu, H.; Kyprianou, N. Prohibitin and cofilin are intracellular effectors of transforming growth factor beta signaling in human prostate cancer cells. Cancer Res. 2006, 66, 8640–8647. [Google Scholar] [CrossRef] [Green Version]
- Hensley, P.J.; Zetter, D.; Horbinski, C.M.; Strup, S.E.; Kyprianou, N. Association of epithelial-mesenchymal transition and nuclear cofilin with advanced urothelial cancer. Hum. Pathol. 2016, 57, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Razinia, Z.; Mäkelä, T.; Ylänne, J.; Calderwood, D.A. Filamins in Mechanosensing and Signaling. Annu. Rev. Biophys. 2012, 41, 227–246. [Google Scholar] [CrossRef] [Green Version]
- Ehrlicher, A.J.; Nakamura, F.; Hartwig, J.H.; Weitz, D.A.; Stossel, T.P. Mechanical strain in actin networks regulates FilGAP and integrin binding to filamin A. Nature 2011, 478, 260–263. [Google Scholar] [CrossRef]
- Zhou, A.-X.; Hartwig, J.H.; Akyürek, L.M. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010, 20, 113–123. [Google Scholar] [CrossRef]
- Luo, T.; Mohan, K.; Iglesias, P.A.; Robinson, D.N. Molecular mechanisms of cellular mechanosensing. Nat. Mater. 2013, 12, 1064–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoy, R.M.; Ghosh, P.M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer 2013, 20, R341–R356. [Google Scholar] [CrossRef] [Green Version]
- Kamil, M.; Shinsato, Y.; Higa, N.; Hirano, T.; Idogawa, M.; Takajo, T.; Minami, K.; Shimokawa, M.; Yamamoto, M.; Kawahara, K.; et al. High filamin-C expression predicts enhanced invasiveness and poor outcome in glioblastoma multiforme. Br. J. Cancer 2019, 120, 819–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.; Li, M.; Bai, G.; Li, X.; Sun, Z.; Yang, J.; Wang, L.; Sun, J. Filamin A inhibits tumor progression through regulating BRCA1 expression in human breast cancer. Oncol. Lett. 2018, 16, 6261–6266. [Google Scholar] [CrossRef]
- Cui, X.-B.; Zhang, S.-M.; Xu, Y.-X.; Dang, H.-W.; Liu, C.-X.; Wang, L.-H.; Yang, L.; Hu, J.-M.; Liang, W.-H.; Jiang, J.-F.; et al. PFN2, a novel marker of unfavorable prognosis, is a potential therapeutic target involved in esophageal squamous cell carcinoma. J. Transl. Med. 2016, 14, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Yang, W.; Yan, J.; Zhou, K.; Wan, B.; Shi, P.; Chen, Y.; He, S.; Li, D. Loss of profilin 2 contributes to enhanced epithelial-mesenchymal transition and metastasis of colorectal cancer. Int. J. Oncol. 2018, 53, 1118–1128. [Google Scholar] [CrossRef] [Green Version]
- Schönichen, A.; Geyer, M. Fifteen formins for an actin filament: A molecular view on the regulation of human formins. Biochim. Biophys. Acta 2010, 1803, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Young, K.G.; Copeland, J.W. Formins in cell signaling. Biochim. Biophys. Acta 2010, 1803, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baarlink, C.; Wang, H.; Grosse, R. Nuclear Actin Network Assembly by Formins Regulates the SRF Coactivator MAL. Science 2013, 340, 864–867. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of transforming growth factor β (TGFβ) signaling regulation by actin remodeling induced by extracellular matrix (ECM) stiffness. Activation of Rho GTPase by mechanical cues promotes F-actin assembly and actin cytoskeleton contractility, which activates TGFβ signaling by: (a) Inducing TGFβ ligand release and activation. The ECM-integrin-actin cytoskeleton linkages allow integrin to shift toward an active configuration that favors TGFβ release from LTBP/latent TGFβ complex. Active TGFβ initiates signaling via TGFβ receptors, which ultimately drives the phosphorylation-dependent formation of SMAD2/3-4 complex. This complex translocates to the nucleus and facilitates the transcription of TGFβ-dependent genes; (b) Accumulating at the front of the nucleus of the lamin-binding protein Nesprin-2, which induces SMAD nuclear localization; (c) Inhibiting the formation of both cytosolic and nuclear LEMD3-SMAD2/3 complexes, resulting in the relief of LEMD3 negative regulation; (d) Activating YAP/TAZ pathway, which regulates both SMAD2/3 shuttling and SMAD-dependent transcriptional activity; (e) Controlling myocardin-related transcription factor A (MRTF-A) localization to mediate the SMAD-dependent transcriptional activity.
Figure 1.
Schematic representation of transforming growth factor β (TGFβ) signaling regulation by actin remodeling induced by extracellular matrix (ECM) stiffness. Activation of Rho GTPase by mechanical cues promotes F-actin assembly and actin cytoskeleton contractility, which activates TGFβ signaling by: (a) Inducing TGFβ ligand release and activation. The ECM-integrin-actin cytoskeleton linkages allow integrin to shift toward an active configuration that favors TGFβ release from LTBP/latent TGFβ complex. Active TGFβ initiates signaling via TGFβ receptors, which ultimately drives the phosphorylation-dependent formation of SMAD2/3-4 complex. This complex translocates to the nucleus and facilitates the transcription of TGFβ-dependent genes; (b) Accumulating at the front of the nucleus of the lamin-binding protein Nesprin-2, which induces SMAD nuclear localization; (c) Inhibiting the formation of both cytosolic and nuclear LEMD3-SMAD2/3 complexes, resulting in the relief of LEMD3 negative regulation; (d) Activating YAP/TAZ pathway, which regulates both SMAD2/3 shuttling and SMAD-dependent transcriptional activity; (e) Controlling myocardin-related transcription factor A (MRTF-A) localization to mediate the SMAD-dependent transcriptional activity.

Figure 2.
Actin regulatory proteins controlling TGFβ type I receptors (TβRI) and II trafficking. (a) The internalized TβRI into clathrin-coated vesicles is linked to the actin cytoskeleton via fascin actin-bundling protein 1 (FSCN1) which promotes the trafficking of internalized receptors from clathrin-coated vesicles to early endosomes and thus TGFβ signaling. (b) VASP promotes TGFβ activity by sustaining the complex between Rab11 and TβRII that favors TβRII recycling to the plasma membrane. (c) Myosins sustain TGFβ signaling by sustaining the cell-surface expression of TβRII receptor and inhibiting its degradation. (d) Arf GAP with GTP-binding protein-like domain, Ankyrin repeat, and PH domain 2 isoform 2 (AGAP2) sustains TGFβ signaling, by directly or indirectly interacting with TβRII and sustaining the recycling of the receptor to the plasma membrane.
Figure 2.
Actin regulatory proteins controlling TGFβ type I receptors (TβRI) and II trafficking. (a) The internalized TβRI into clathrin-coated vesicles is linked to the actin cytoskeleton via fascin actin-bundling protein 1 (FSCN1) which promotes the trafficking of internalized receptors from clathrin-coated vesicles to early endosomes and thus TGFβ signaling. (b) VASP promotes TGFβ activity by sustaining the complex between Rab11 and TβRII that favors TβRII recycling to the plasma membrane. (c) Myosins sustain TGFβ signaling by sustaining the cell-surface expression of TβRII receptor and inhibiting its degradation. (d) Arf GAP with GTP-binding protein-like domain, Ankyrin repeat, and PH domain 2 isoform 2 (AGAP2) sustains TGFβ signaling, by directly or indirectly interacting with TβRII and sustaining the recycling of the receptor to the plasma membrane.

Figure 3.
Actin cytoskeleton dynamics control the expression levels of Ski and SnoN, negative regulators of TGFβ signaling. TGFβ signaling controls expression levels of the negative regulators Ski and SnoN, by inducing both their upregulation and their ubiquitin (Ub)-mediated degradation. In normal hepatocytes, actin cytoskeleton dynamics induce Ski protein degradation and SnoN protein stabilization, and might impact TGFβ signaling outcome. This mechanism is lost in hepatoma cells [151,152].
Figure 3.
Actin cytoskeleton dynamics control the expression levels of Ski and SnoN, negative regulators of TGFβ signaling. TGFβ signaling controls expression levels of the negative regulators Ski and SnoN, by inducing both their upregulation and their ubiquitin (Ub)-mediated degradation. In normal hepatocytes, actin cytoskeleton dynamics induce Ski protein degradation and SnoN protein stabilization, and might impact TGFβ signaling outcome. This mechanism is lost in hepatoma cells [151,152].


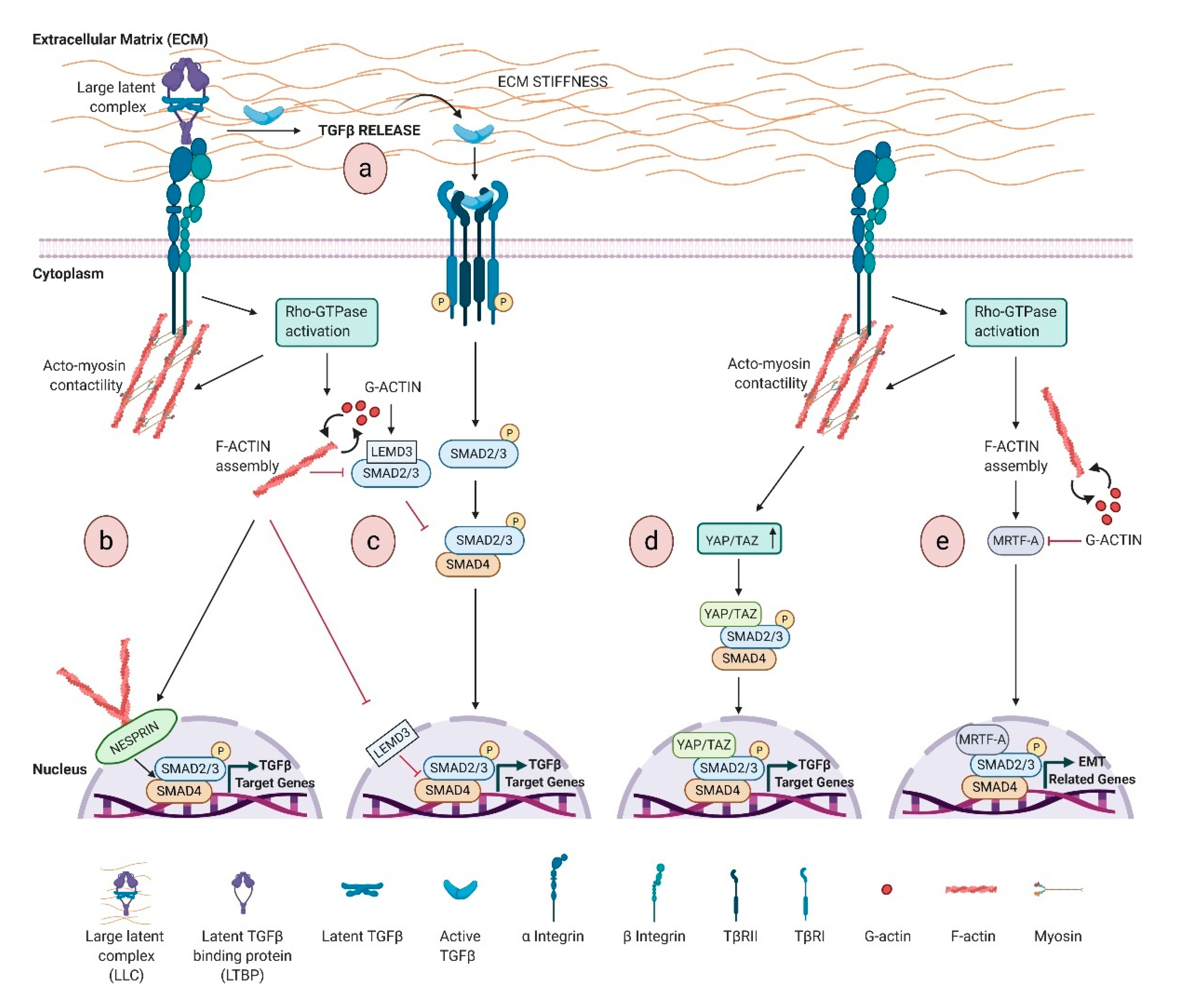{kind=link}
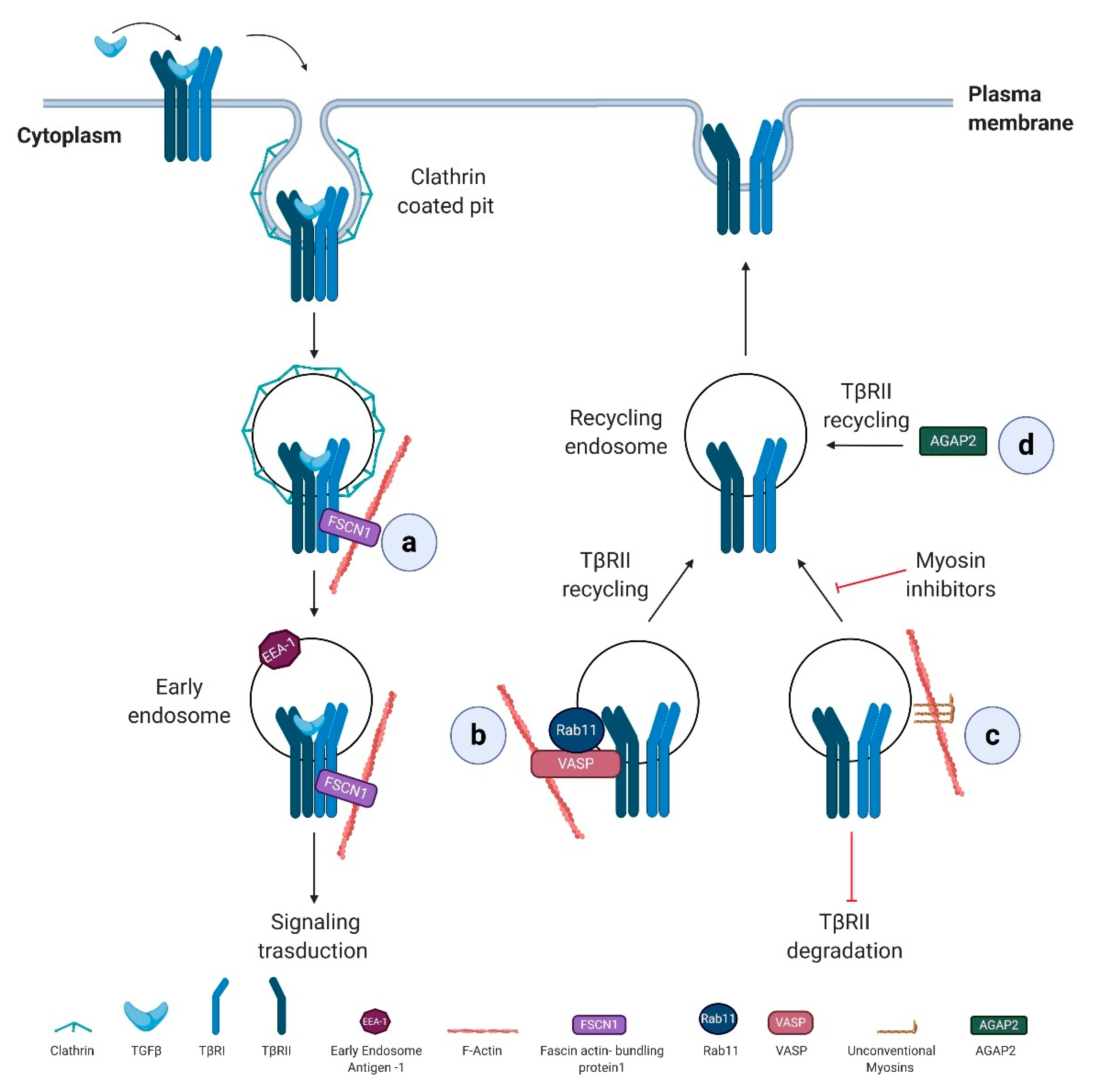{kind=link}
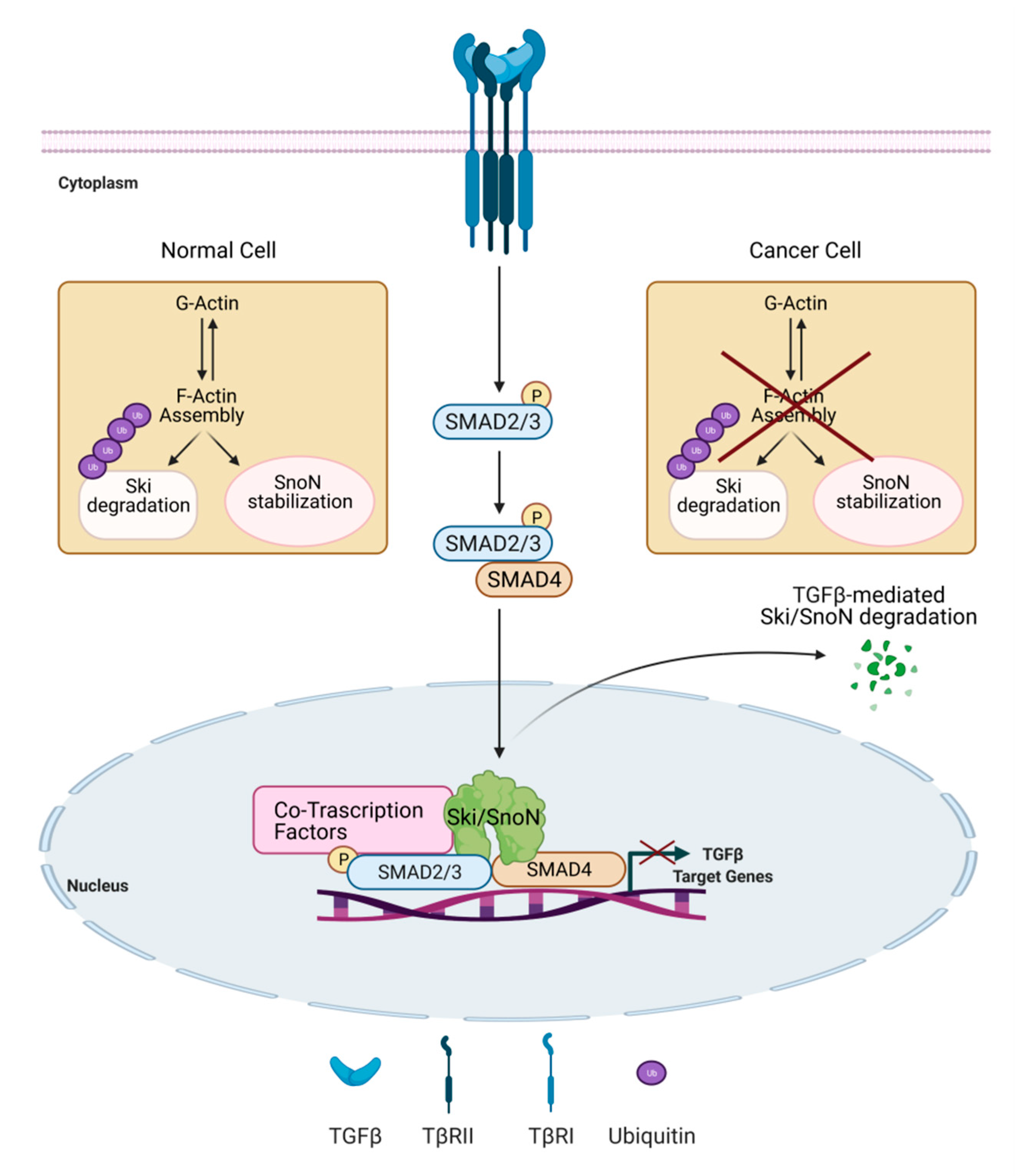{kind=link}
Table 1.
Actin regulatory proteins affecting TGFβ signaling.
| Actin Regulatory Protein | Role in TGFβ Signaling | Function | References |
|---|---|---|---|
| RhoA | SMAD2/3 phosphorylation/activation | Modulates SMAD Signaling during TGFβ-induced Smooth Muscle Differentiation | [153] |
| RhoB | SMAD-mediated transcriptional activity | Antagonizes TGFβ transcriptional program | [154] |
| Transcription of TβRII | Antagonizes TGFβ-mediated anti-proliferative and transcriptional responses in keratinocytes and pancreatic carcinoma cells | [155] | |
| Rac1 | SMAD2 phosphorylation/activation | Antagonizes TGFβ1-mediated growth inhibition and sustains TGFβ1-induced cell migration in pancreatic ductal adenocarcinoma cells | [156] |
| Rac1b | SMAD2/3 phosphorylation/activation | Inhibits TGFβ1-induced cell motility in pancreatic ductal epithelial cells | [157] |
| Synthesis and secretion of TGFβ1 | Suppresses cell motility | [158] | |
| TGFβ1-mediated p38MAPK and MEK-ERK activation | Antagonizes TGFβ1-dependent EMT | [159] | |
| WAVE3 | Transcription of EMT-related genes | Regulates TGFβ-mediated EMT and growth and metastasis of triple-negative breast cancer cells | [160] |
| hMENA/hMENAΔv6 | SMAD2/3 phosphorylation/activation | Regulates TGFβ-mediated EMT in PDAC cells | [38] |
| Zyxin | Regulation of TGFβ-induced integrin α5β1 expression | Regulates cancer cell motility and EMT during lung cancer development and progression | [161] |
| Regulation of EMT-related protein | Mediates cooperation between Hippo and TGFβ signaling pathways | [162] | |
| Cofilin | Drives Cell-Invasive and Metastatic Responses to TGF-β in Prostate Cancer | [163] | |
| Regulation of EMT-related morphology and protein expression | Sustains TGFβ-mediated EMT in gastric cancer cells | [164] | |
| Sustains migration and invasion during EMT of CRC cells | [165] | ||
| Filamin | SMAD localization | Sustains SMAD-dependent TGFβ signaling | [166] |
| Profilin 2 | Epigenetic regulation of SMAD2 and SMAD3 | Promotes lung cancer growth and metastasis | [167] |
| Formins | SMAD-mediated transcriptional activity | Sustains TGFβ-mediated EMT | [168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Melchionna, R.; Trono, P.; Tocci, A.; Nisticò, P. Actin Cytoskeleton and Regulation of TGFβ Signaling: Exploring Their Links. Biomolecules 2021, 11, 336. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020336
AMA Style
Melchionna R, Trono P, Tocci A, Nisticò P. Actin Cytoskeleton and Regulation of TGFβ Signaling: Exploring Their Links. Biomolecules. 2021; 11(2):336. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020336
Chicago/Turabian StyleMelchionna, Roberta, Paola Trono, Annalisa Tocci, and Paola Nisticò. 2021. "Actin Cytoskeleton and Regulation of TGFβ Signaling: Exploring Their Links" Biomolecules 11, no. 2: 336. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11020336
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.