
Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Generation Routes for Various AGEs in the Human Body
3. Formation/Metabolism of MGO
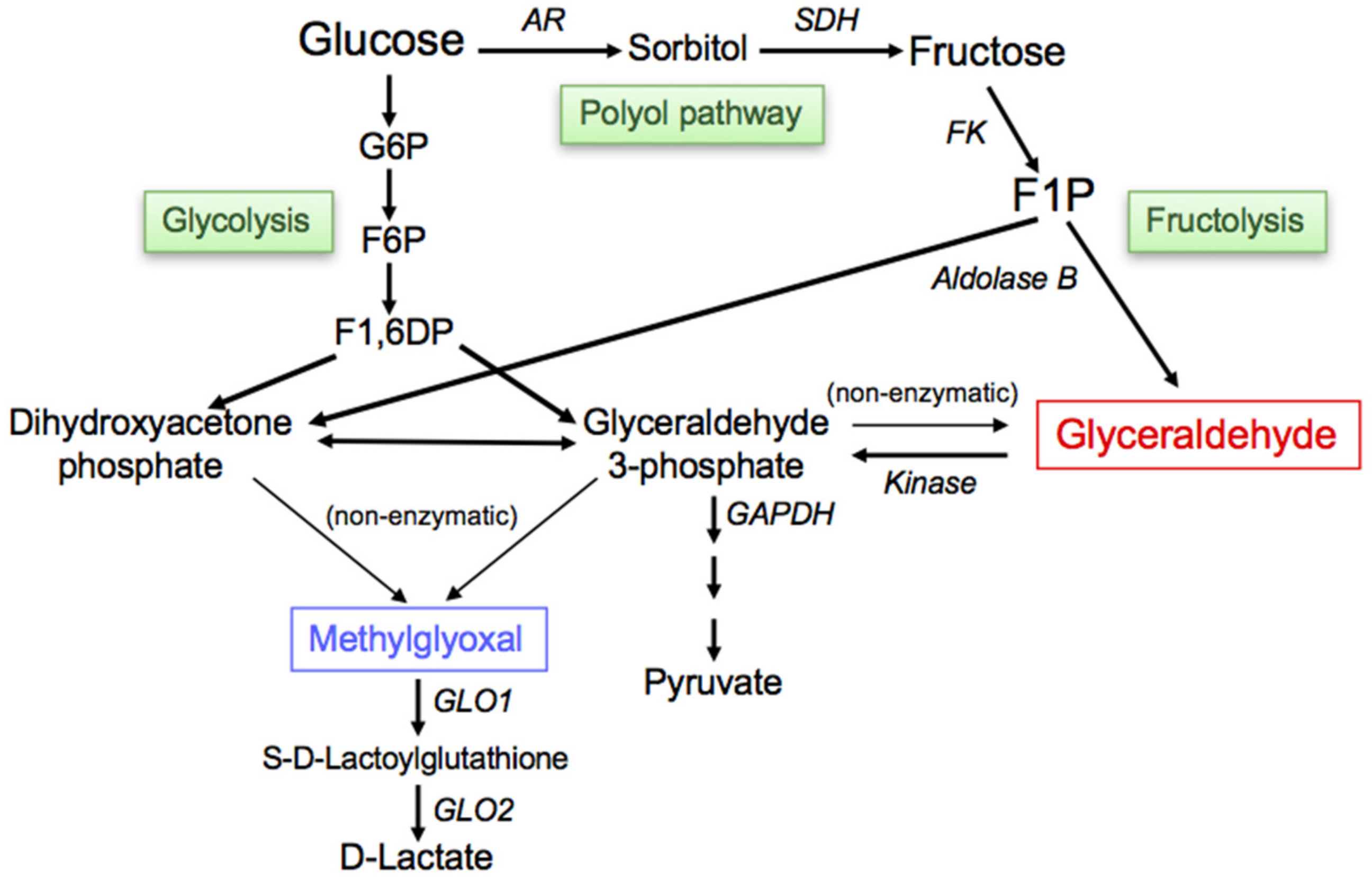
4. The Generation/Accumulation of GA, a Precursor of TAGE
4.1. Fructolysis
4.2. Glycolysis
4.3. Polyol Pathway
5. RAGE
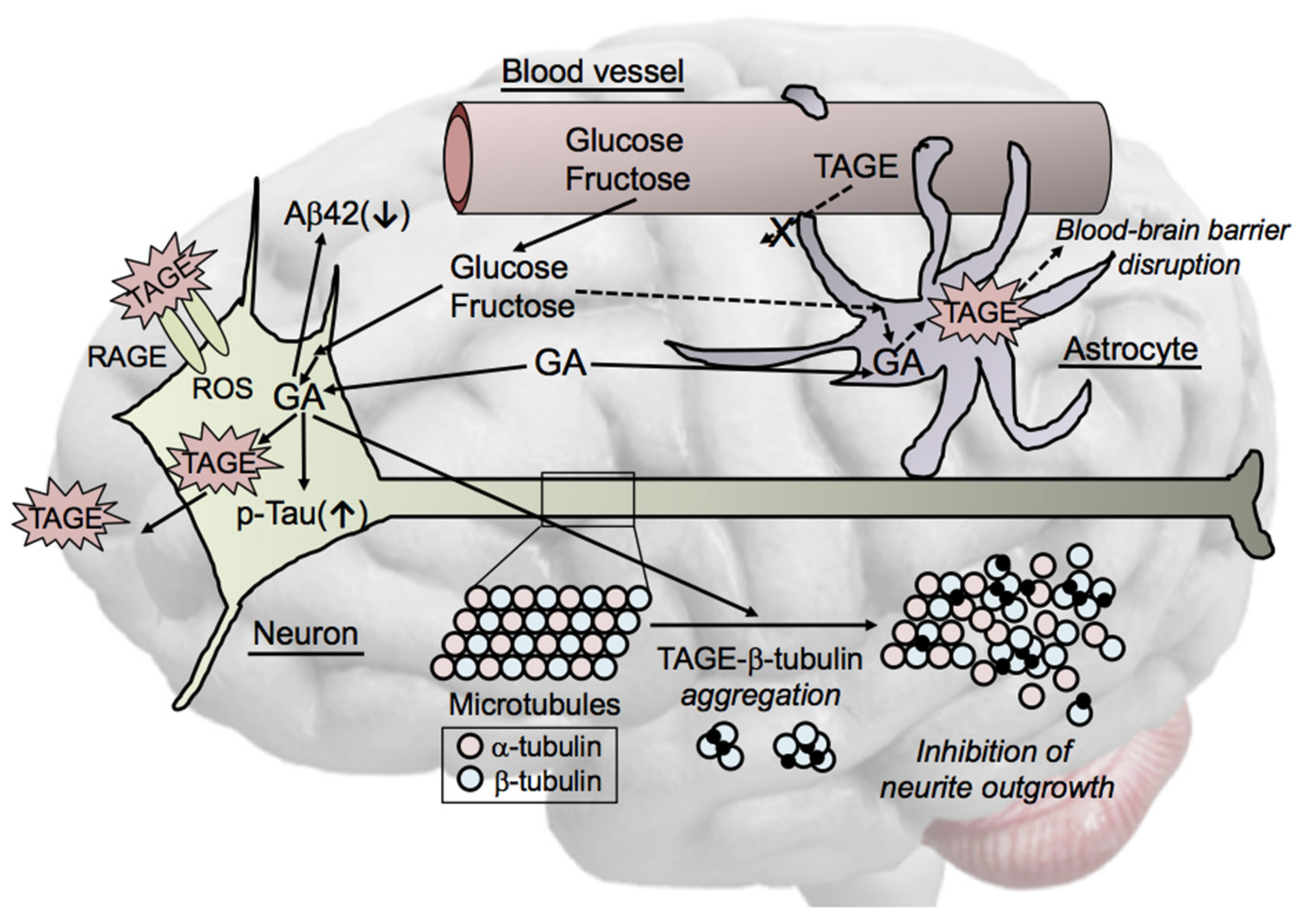
6. Cytotoxicity of TAGE in the Brain
6.1. Localization of TAGE in AD Brains
6.2. Effects of Extracellular TAGE on Neuronal Cells
6.3. Intracellular TAGE and Neuronal Cell Death
- (i)
- The intracellular accumulation of TAGE was induced in GA-treated SH-SY5Y cells, which, ultimately, led to cell death. The production of TAGE in SH-SY5Y cells was dose-dependently increased by GA [24].
- (ii)
- GAPDH activity was decreased in neuronal cells treated with GA [24]. GAPDH activity was reduced in AD patients [57], and GAPDH played a role in apoptosis in neurodegenerative disorders [58]. We previously reported that TAGE inactivated GAPDH in a neuronal culture system [11]. These findings are indicative of a feed-forward mechanism, with extracellular TAGE-induced GAPDH inactivation further stimulating the generation of intracellular TAGE.
- (iii)
- Aβ42 levels in cerebrospinal fluid (CSF) were significantly lower in AD patients than in age-matched healthy elderly controls, whereas total tau and p-tauT181 levels in CSF were significantly higher in AD patients than in the controls [59,60]. Furthermore, the levels of other AD biomarkers, such as vascular endothelial growth factor (VEGF) [61] and transforming growth factor-β1 (TGF-β1) [62], were higher in the CSF of AD patients. We previously reported that the intracellular generation of TAGE decreased Aβ42 levels and increased total tau and p-tauT181 levels in culture media, and also elevated the intracellular levels of AD biomarkers in SH-SY5Y cells [24].
6.4. Mechanisms by Which Intracellular TAGE Cause Cell Damage in Neuronal Cells
6.5. Cell Signaling of TAGE in Neurons
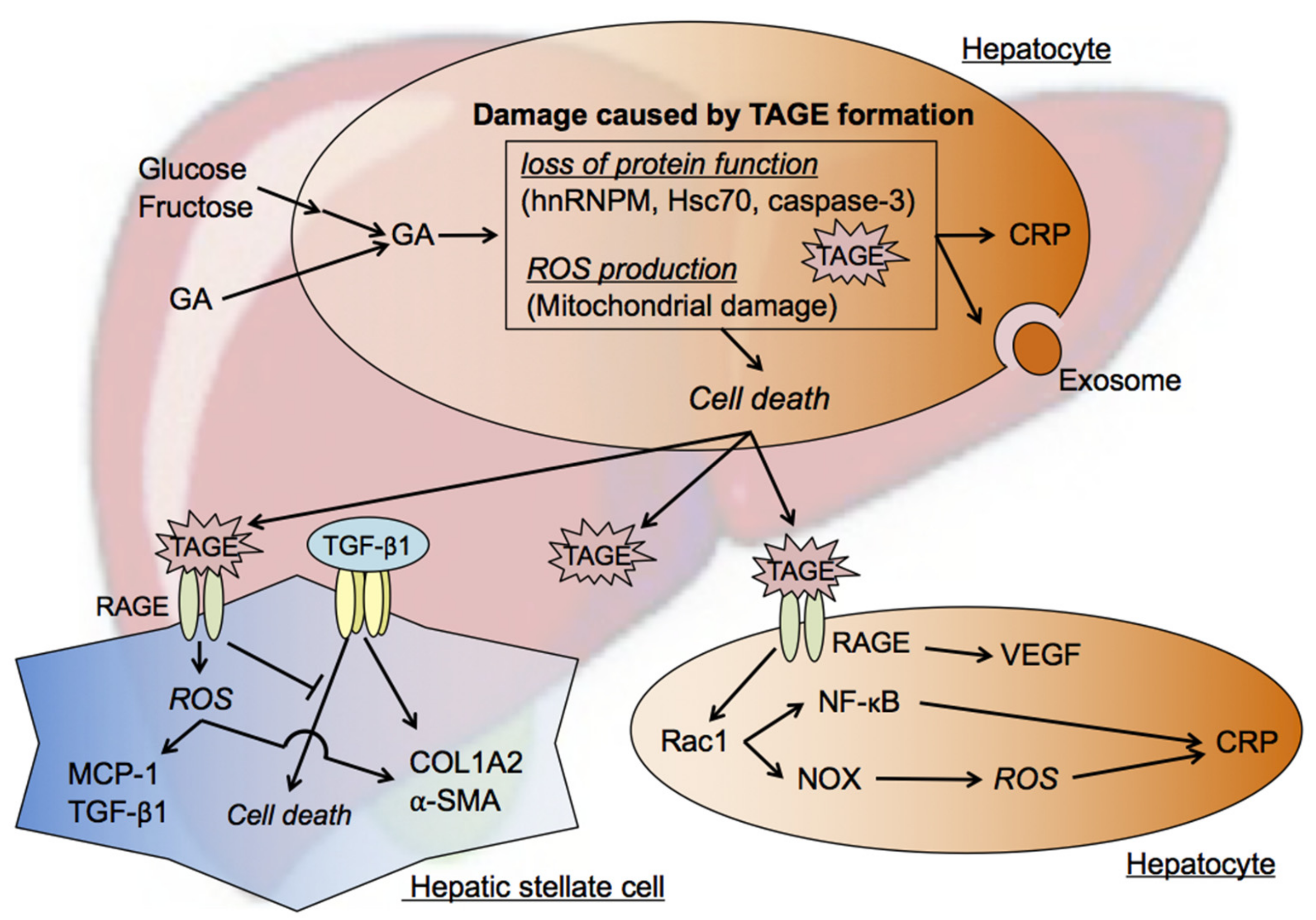
7. Cytotoxicity of TAGE in the Liver
7.1. Intracellular TAGE and Hepatocyte Cell Death
- (i)
- The accumulation of TAGE and abnormal heat shock cognate 70 (Hsc70) protein levels were observed in GA-treated human hepatocellular carcinoma (HCC) Hep3B cells without changes in Hsc70 mRNA expression levels [26]. Elevated C-reactive protein (CRP) mRNA levels were reduced to control levels by a pretreatment with aminoguanidine (AG), an inhibitor of the formation of AGEs, which indicated that the intracellular generation and accumulation of TAGE activated inflammatory responses [26].
- (ii)
- Previous studies examined Hep3B cells incubated with GA- or high fructose-containing media and identified heterogeneous nuclear ribonucleoprotein M (hnRNPM) as a TAGE-modified protein [76,77]. hnRNPM, an RNA-binding protein, contributes to many processes in nucleic acid metabolism, such as alternative splicing, mRNA stabilization, and transcriptional and translational regulation. The expression levels of genes associated with extracellular exosome-containing extracellular spaces were altered (up- or down-regulated) by the knockdown of hnRNPM [77].
- (iii)
- Caspase-3 plays a crucial role in apoptosis and has been identified as a target of TAGE-induced modifications in GA-treated HCC HepG2 cells. The cleavage and activation of caspase-3, resulting in protease activity, was previously reported during apoptosis [79]. However, a relationship was observed between increases in TAGE-modified caspase-3 levels and the loss of enzymatic activity, and TAGE-induced modifications also inhibited the cleavage of poly (ADP-ribose) polymerase, which is downstream of caspase-3 in the apoptotic cascade. Furthermore, necrotic-type cell death appeared to be promoted by TAGE-modified caspase-3 [27,78].
7.2. Mechanisms by Which Intracellular TAGE Induce Cell Death in Hepatocytes
7.3. Effects of Extracellular TAGE on Hepatocytes
7.4. Effects of Extracellular TAGE on HSC
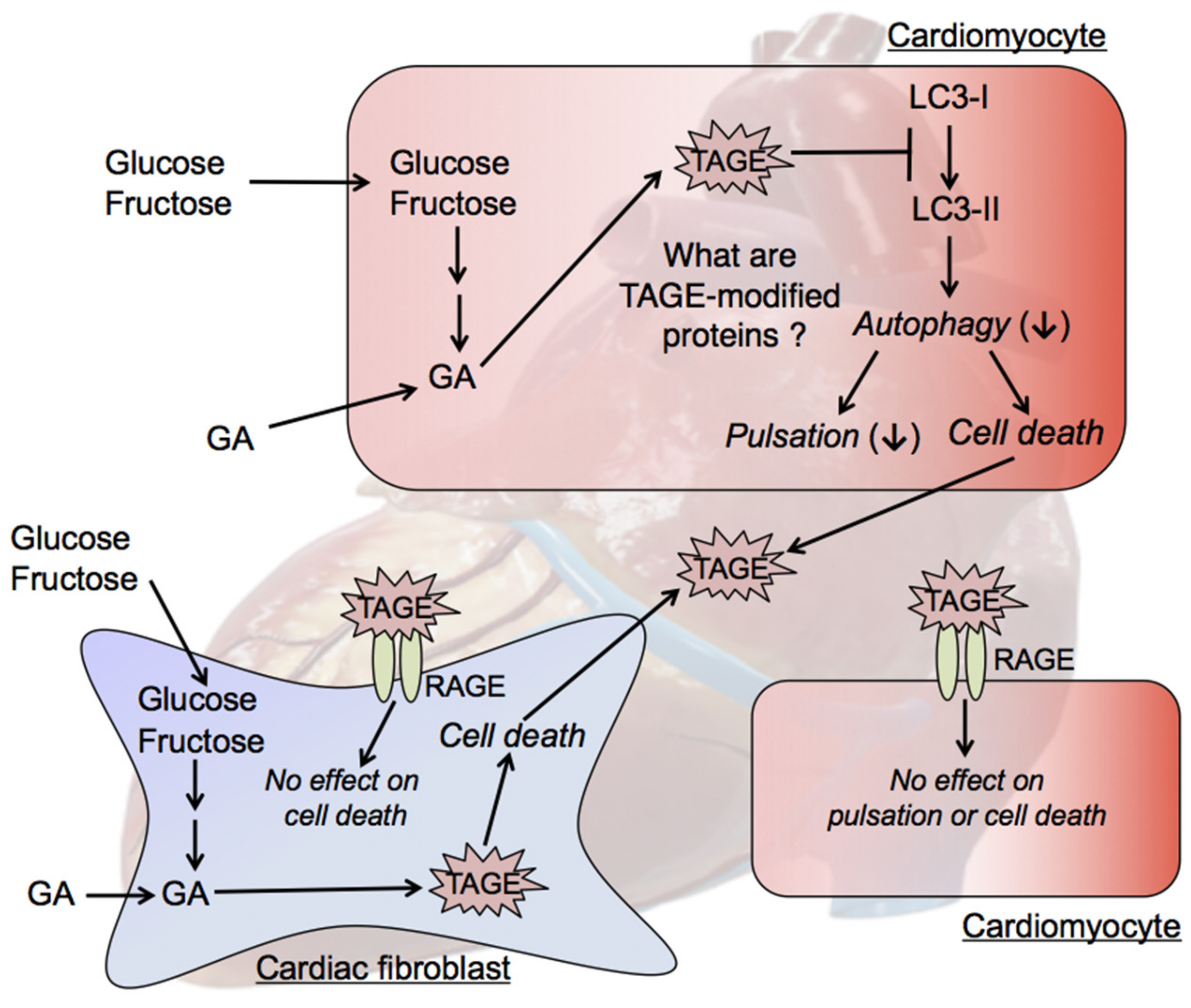
8. Cytotoxicity of TAGE in the Heart
8.1. Postprandial Hyperglycemia and TAGE Generation
8.2. Intracellular TAGE and Cardiomyocyte Cell Damage
8.3. Mechanisms by Which Intracellular TAGE Cause Cell Damage in Cardiomyocytes
8.4. Intracellular TAGE and Cardiac Fibroblast Cell Death
8.5. Effects of Extracellular TAGE on Cardiomyocytes and Cardiac Fibroblasts
9. Effects of TAGE on Cancer Progression
10. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Saeedi, P.; Petersohn, I.; Salpea, P.; Malanda, B.; Karuranga, S.; Unwin, N.; Colagiuri, S.; Guariguata, L.; Motala, A.A.; Ogurtsova, K.; et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res. Clin. Pract. 2019, 157, 107843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Yu, M.; Fang, L.; Hu, R.Y. Association between sugar-sweetened beverages and type 2 diabetes: A meta-analysis. J. Diabetes Investig. 2015, 6, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Imamura, F.; O’Connor, L.; Ye, Z.; Mursu, J.; Hayashino, Y.; Bhupathiraju, S.N.; Forouhi, N.G. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: Systematic review, meta-analysis, and estimation of population attributable fraction. Br. J. Sports Med. 2016, 50, 496–504. [Google Scholar] [CrossRef]
- Malik, V.S. Sugar sweetened beverages and cardiometabolic health. Curr. Opin. Cardiol. 2017, 32, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Hoffmann, G.; Lampousi, A.M.; Knüppel, S.; Iqbal, K.; Schwedhelm, C.; Bechthold, A.; Schlesinger, S.; Boeing, H. Food groups and risk of type 2 diabetes mellitus: A systematic review and meta-analysis of prospective studies. Eur. J. Epidemiol. 2017, 32, 363–375. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Makita, Z. Alternative routes for the formation of immunochemically distinct advanced glycation end-products in vivo. Curr. Mol. Med. 2001, 1, 305–315. [Google Scholar] [CrossRef]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bügel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem. Toxicol. 2013, 60, 1037. [Google Scholar] [CrossRef]
- Hollenbach, M. The Role of glyoxalase-I (Glo-I), advanced glycation endproducts (AGEs), and their receptor (RAGE) in chronic liver disease and hepatocellular carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and advanced glycation end-products in the development of diabetic complications and targets for intervention. Int. J. Mol. Sci. 2017, 18, 984. [Google Scholar] [CrossRef] [Green Version]
- Jud, P.; Sourij, H. Therapeutic options to reduce advanced glycation end products in patients with diabetes mellitus: A review. Diabetes Res. Clin. Pract. 2019, 148, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Bucala, R.; Suzuki, T.; Ohkubo, T.; Yamazaki, M.; Koike, T.; Kameda, Y.; Makita, Z. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J. Neuropathol. Exp. Neurol. 2000, 59, 1094–1105. [Google Scholar] [CrossRef] [Green Version]
- Choei, H.; Sasaki, N.; Takeuchi, M.; Yoshida, T.; Ukai, W.; Yamagishi, S.; Kikuchi, S.; Saito, T. Glyceraldehyde-derived advanced glycation end products in Alzheimer’s disease. Acta Neuropathol. 2004, 108, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Involvement of toxic AGEs (TAGE) in the pathogenesis of diabetic vascular complications and Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 845–858. [Google Scholar] [CrossRef]
- Hyogo, H.; Yamagishi, S.; Iwamoto, K.; Arihiro, K.; Takeuchi, M.; Sato, T.; Ochi, H.; Nonaka, M.; Nabeshima, Y.; Inoue, M.; et al. Elevated levels of serum advanced glycation end-products in patients with nonalcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2007, 22, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Ishitobi, T.; Nabeshima, Y.; Arihiro, K.; Chayama, K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: Clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 2010, 45, 750–757. [Google Scholar] [CrossRef]
- Kan, H.; Yamagishi, S.; Ojima, A.; Fukami, K.; Ueda, S.; Takeuchi, M.; Hyogo, H.; Aikata, H.; Chayama, K. Elevation of serum levels of advanced glycation end products in patients with non-B or non-C hepatocellular carcinoma. J. Clin. Lab. Anal. 2015, 29, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Sakasai-Sakai, A.; Takata, T.; Tsutsumi, M. Toxic AGE (TAGE) theory for the pathophysiology of the onset/progression of NAFLD and ALD. Nutrients 2017, 9, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, Y.; Daida, H.; Morimoto, T.; Kasai, T.; Miyauchi, K.; Yamagishi, S.; Takeuchi, M.; Hiro, T.; Kimura, T.; Nakagawa, Y.; et al. Relationship between advanced glycation end products and plaque progression in patients with acute coronary syndrome: The JAPAN-ACS sub-study. Cardiovasc. Diabetol. 2013, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Kawai, Y.; Kitayama, M.; Akao, H.; Motoyama, A.; Wakasa, M.; Saito, R.; Aoki, H.; Fujibayashi, K.; Tsuchiya, T.; et al. Diurnal glycemic fluctuation is associated with severity of coronary artery disease in prediabetic patients: Possible role of nitrotyrosine and glyceraldehyde-derived advanced glycation end products. J. Cardiol. 2017, 69, 625–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomosugi, N.; Yamamoto, S.; Takeuchi, M.; Yonekura, H.; Ishigaki, Y.; Numata, N.; Katsuda, S.; Sakai, Y. Effect of collagen tripeptide on atherosclerosis in healthy humans. J. Atheroscler. Thromb. 2017, 24, 530–538. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Yamagishi, S.; Adachi, H.; Matsui, T.; Kurita-Nakamura, Y.; Takeuchi, M.; Inoue, H.; Imaizumi, T. Circulating advanced glycation end products (AGEs) and soluble form of receptor for AGEs (sRAGE) are independent determinants of serum monocyte chemoattractant protein-1 (MCP-1) levels in patients with type 2 diabetes. Diabetes Metab. Res. Rev. 2008, 24, 109–114. [Google Scholar] [CrossRef]
- Tsunosue, M.; Mashiko, N.; Ohta, Y.; Matsuo, Y.; Ueda, K.; Ninomiya, M.; Tanaka, S.; Hoshiko, M.; Yoshiyama, Y.; Takeuchi, M.; et al. An α-glucosidase inhibitor, acarbose treatment decreases serum levels of glyceraldehyde-derived advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin. Exp. Med. 2010, 10, 139–141. [Google Scholar] [CrossRef]
- Takeuchi, M.; Takino, J.; Yamagishi, S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: A novel therapeutic strategy. Curr. Drug Targets 2010, 11, 1468–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koriyama, Y.; Furukawa, A.; Muramatsu, M.; Takino, J.; Takeuchi, M. Glyceraldehyde caused Alzheimer’s disease-like alterations in diagnostic marker levels in SH-SY5Y human neuroblastoma cells. Sci. Rep. 2015, 5, 13313. [Google Scholar] [CrossRef] [Green Version]
- Nasu, R.; Furukawa, A.; Suzuki, K.; Takeuchi, M.; Koriyama, Y. The effect of glyceraldehyde-derived advanced glycation end-products on β-tubulin-inhibited neurite outgrowth in SH-SY5Y human neuroblastoma cells. Nutrients 2020, 12, 2958. [Google Scholar] [CrossRef]
- Takino, J.; Kobayashi, Y.; Takeuchi, M. The formation of intracellular glyceraldehyde-derived advanced glycation end-products and cytotoxicity. J. Gastroenterol. 2010, 45, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. Impact of intracellular glyceraldehyde-derived advanced glycation end-products on human hepatocyte cell death. Sci. Rep. 2017, 7, 14282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakasai-Sakai, A.; Takata, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products promote the production of reactive oxygen species in HepG2 cells. Int. J. Mol. Sci. 2020, 21, 4861. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Ueda, T.; Sakasai-Sakai, A.; Takeuchi, M. Generation of glyceraldehyde-derived advanced glycation end-products in pancreatic cancer cells and the potential of tumor promotion. World J. Gastroenterol. 2017, 23, 4910–4919. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Sakasai-Sakai, A.; Ueda, T.; Takeuchi, M. Intracellular toxic advanced glycation end-products in cardiomyocytes may cause cardiovascular disease. Sci. Rep. 2019, 9, 2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, T.; Sakasai-Sakai, A.; Takeuchi, M. Impact of intracellular toxic advanced glycation end-products (TAGE) on murine myoblast cell death. Diabetol. Metab. Syndr. 2020, 12, 54. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M. Serum levels of toxic AGEs (TAGE) may be a promising novel biomarker for the onset/progression of lifestyle-related diseases. Diagnostics 2016, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Advanced protein glycosylation in diabetes and aging. Ann. Rev. Med. 1995, 46, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Iwaki, M.; Takino, J.; Shirai, H.; Kawakami, M.; Bucala, R.; Yamagishi, S. Immunological detection of fructose-derived advanced glycation end-products. Lab. Investig. 2010, 90, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, M.; Yamagishi, S. TAGE (toxic AGEs) hypothesis in various chronic diseases. Med. Hypotheses 2004, 63, 449–452. [Google Scholar] [CrossRef]
- Usui, T.; Hayase, F. Isolation and identification of the 3-hydroxy-5-hydroxymethyl-pyridinium compound as a novel advanced glycation end product on glyceraldehyde-related Maillard reaction. Biosci. Biotechnol. Biochem. 2003, 67, 930–932. [Google Scholar] [CrossRef] [PubMed]
- Tessier, F.J.; Monnier, V.M.; Sayre, L.M.; Kornfield, J.A. Triosidines: Novel Maillard reaction products and cross-links from the reaction of triose sugars with lysine and arginine residues. Biochem. J. 2003, 369, 705–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellier, J.; Nokin, M.J.; Lardé, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcène, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef]
- Yumnam, S.; Subedi, L.; Kim, S.Y. Glyoxalase system in the progression of skin aging and skin malignancies. Int. J. Mol. Sci. 2021, 22, 310. [Google Scholar] [CrossRef]
- Takeuchi, M.; Yamagishi, S. Alternative routes for the formation of glyceraldehyde-derived AGEs (TAGE) in vivo. Med. Hypotheses 2004, 63, 453–455. [Google Scholar] [CrossRef] [PubMed]
- Bais, R.; James, H.M.; Rofe, A.M.; Conyers, R.A. The purification and properties of human liver ketohexokinase. A role for ketohexokinase and fructose-bisphosphate aldolase in the metabolic production of oxalate from xylitol. Biochem. J. 1985, 230, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oates, P.J. Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 2002, 50, 325–392. [Google Scholar]
- Fuentes, E.; Rojas, A.; Palomo, I. Role of multiligand/RAGE axis in platelet activation. Thromb. Res. 2014, 133, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Leerach, N.; Harashima, A.; Munesue, S.; Kimura, K.; Oshima, Y.; Goto, H.; Yamamoto, H.; Higashida, H.; Yamamoto, Y. Glycation reaction and the role of the receptor for advanced glycation end-products in immunity and social behavior. Glycoconj. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Takino, J.; Sakasai-Sakai, A.; Takata, T.; Ueda, T.; Tsutsumi, M.; Hyogo, H.; Yamagishi, S. Involvement of the TAGE-RAGE system in non-alcoholic steatohepatitis: Novel treatment strategies. World J. Hepatol. 2014, 6, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab. Res. Rev. 2001, 17, 436–443. [Google Scholar] [CrossRef]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am. J. Physiol. Cell. Physiol. 2008, 294, C145–C152. [Google Scholar] [CrossRef] [Green Version]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrove, R.G.; Abedin, M.J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Yonekura, H.; Watanabe, T.; Sakurai, S.; Li, H.; Harashima, A.; Myint, K.M.; Osawa, M.; Takeuchi, A.; Takeuchi, M.; et al. Short-chain aldehyde-derived ligands for RAGE and their actions on endothelial cells. Diabetes Res. Clin. Pract. 2007, 77 (Suppl. 1), S30–S40. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen species-mediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkoe, D.J. Normal and abnormal biology of the beta-amyloid precursor protein. Annu. Rev. Neurosci. 1994, 17, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Katre, D.P. Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab. Syndr. 2016, 10 (Suppl. 1), S144–S149. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Peters, S.A.E.; Woodward, M.; Arango, S.M.; Batty, G.D.; Beckett, N.; Beiser, A.; Borenstein, A.R.; Crane, P.K.; Haan, M.; et al. Type 2 diabetes as a risk factor for dementia in women compared with men: A pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care 2016, 39, 300–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledesma, M.D.; Bonay, P.; Colaco, C.; Avila, J. Analysis of microtubule-associated protein tau glycation in paired helical filaments. J. Biol. Chem. 1994, 269, 21614–21619. [Google Scholar] [CrossRef]
- Mazzola, J.L.; Sirover, M.A. Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. J. Neurochem. 2001, 76, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Mazzola, J.L.; Sirover, M.A. Subcellular alteration of glyceraldehyde-3-phosphate dehydrogenase in Alzheimer’s disease fibroblasts. J. Neurosci. Res. 2003, 71, 279–285. [Google Scholar] [CrossRef]
- Blennow, K.; Hampel, H. CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003, 2, 605–613. [Google Scholar] [CrossRef]
- Kang, J.H.; Ryoo, N.Y.; Shin, D.W.; Trojanowski, J.Q.; Shaw, L.M. Role of cerebrospinal fluid biomarkers in clinical trials for Alzheimer’s disease modifying therapies. Korean J. Physiol. Pharmacol. 2014, 18, 447–456. [Google Scholar] [CrossRef] [Green Version]
- Tarkowski, E.; Issa, R.; Sjögren, M.; Wallin, A.; Blennow, K.; Tarkowski, A.; Kumar, P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol. Aging 2002, 23, 237–243. [Google Scholar] [CrossRef]
- Zetterberg, H.; Andreasen, N.; Blennow, K. Increased cerebrospinal fluid levels of transforming growth factor-β1 in Alzheimer’s disease. Neurosci. Lett. 2004, 367, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Ihara, Y.; Nukina, N.; Miura, R.; Ogawara, M. Phosphorylated tau protein is integrated into paired helical filaments in Alzheimer’s disease. J. Biochem. 1986, 99, 1807–1810. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Grundke-Iqbal, I.; Smith, A.J.; George, L.; Tung, Y.C.; Zaidi, T. Identification and localization of a tau peptide to paired helical filaments of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1989, 86, 5646–5650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornalley, P.J. Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell. Mol. Biol. 1998, 44, 1013–1023. [Google Scholar]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Lue, L.F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogers, J.; Schmidt, A.M.; Stern, D.M.; Yan, S.D. Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer’s disease: Identification of a cellular activation mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef]
- Li, J.J.; Dickson, D.; Hof, P.R.; Vlassara, H. Receptors for advanced glycosylation endproducts in human brain: Role in brain homeostasis. Mol. Med. 1998, 4, 46–60. [Google Scholar] [CrossRef]
- Chou, S.M.; Han, C.Y.; Wang, H.S.; Vlassara, H.; Bucala, R. A receptor for advanced glycosylation endproducts (AGEs) is colocalized with neurofilament-bound AGEs and SOD1 in motoneurons of ALS: Immunohistochemical study. J. Neurol. Sci. 1999, 169, 87–92. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef]
- Yin, Q.Q.; Dong, C.F.; Dong, S.Q.; Dong, X.L.; Hong, Y.; Hou, X.Y.; Luo, D.Z.; Pei, J.J.; Liu, X.P. AGEs induce cell death via oxidative and endoplasmic reticulum stresses in both human SH-SY5Y neuroblastoma cells and rat cortical neurons. Cell Mol. Neurobiol. 2012, 32, 1299–1309. [Google Scholar] [CrossRef]
- Arancio, O.; Zhang, H.P.; Chen, X.; Lin, C.; Trinchese, F.; Puzzo, D.; Liu, S.; Hegde, A.; Yan, S.F.; Stern, A. RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J. 2004, 23, 4096–4105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, D.; Lue, L.F.; Paul, G.; Patel, A.; Sabbagh, M.N. Receptor for advanced glycation endproduct modulators: A new therapeutic target in Alzheimer’s disease. Expert Opin. Investig. Drugs 2015, 24, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of HCC by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef]
- Takino, J.; Nagamine, K.; Takeuchi, M.; Hori, T. In vitro identification of nonalcoholic fatty liver disease-related protein hnRNPM. World J. Gastroenterol. 2015, 21, 1784–1793. [Google Scholar] [CrossRef]
- Takino, J.; Nagamine, K.; Suzuki, M.; Sakasai-Sakai, A.; Takeuchi, M.; Hori, T. Gene expression changes associated with the loss of heterogeneous nuclear ribonucleoprotein M function. Am. J. Mol. Biol. 2017, 7, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Sakasai-Sakai, A.; Takata, T.; Takino, J.; Takeuchi, M. The relevance of toxic AGEs (TAGE) cytotoxicity to NASH pathogenesis: A mini-review. Nutrients 2019, 11, 462. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, J.; Pang, X.; Wang, S.; Wu, D.; Zhang, X.; Feng, L. Angiotensin II induces C-reactive protein expression via AT1-ROS-MAPK-NF-κB signal pathway in hepatocytes. Cell. Physiol. Biochem. 2013, 32, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.Y.; Sun, L.; Chen, X.P.; Zhang, D.S. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Videla, L.A.; Tapia, G.; Rodrigo, R.; Pettinelli, P.; Haim, D.; Santibãez, C.; Araya, A.V.; Smok, G.; Csendes, A.; Gutierrez, L.; et al. Liver NF-κB and AP-1 DNA binding in obese patients. Obesity 2009, 17, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Jiang, J.X.; Sasaki, Y.; Chao, T.I.; Kiss, Z.; Chen, X.; Tian, J.; Katsuyama, M.; Yabe-Nishimura, C.; Xi, Y.; et al. Hepatocyte nicotinamide adenine dinucleotide phosphate reduced oxidase 4 regulates stress signaling, fibrosis, and insulin sensitivity during development of steatohepatitis in mice. Gastroenterology 2015, 149, 468–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Fukami, K.; Okuda, S.; Imaizumi, T. Olmesartan blocks advanced glycation end products (AGEs)-induced angiogenesis in vitro by suppressing receptor for AGEs (RAGE) expression. Microvasc. Res. 2008, 75, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Niiya, Y.; Abumiya, T.; Yamagishi, S.; Takino, J.; Takeuchi, M. Advanced glycation end products increase permeability of brain microvascular endothelial cells through reactive oxygen species-induced vascular endothelial growth factor expression. J. Stroke Cerebrovasc. Dis. 2012, 21, 293–298. [Google Scholar] [CrossRef]
- Inagaki, Y.; Yamagishi, S.; Okamoto, T.; Takeuchi, M.; Amano, S. Pigment epithelium-derived factor prevents advanced glycation end products-induced monocyte chemoattractant protein-1 production in microvascular endothelial cells by suppressing intracellular reactive oxygen species generation. Diabetologia 2003, 46, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Niiya, Y.; Abumiya, T.; Shichinohe, H.; Kuroda, S.; Kikuchi, S.; Ieko, M.; Yamagishi, S.; Takeuchi, M.; Sato, T.; Iwasaki, Y. Susceptibility of brain microvascular endothelial cells to advanced glycation end products-induced tissue factor upregulation is associated with intracellular reactive oxygen species. Brain Res. 2006, 1108, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T.; Nakamura, K.; Inoue, H.; Takeuchi, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Olmesartan blocks inflammatory reactions in endothelial cells evoked by advanced glycation end products by suppressing generation of reactive oxygen species. Ophthalmic. Res. 2008, 40, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Baiocchini, A.; Montaldo, C.; Conigliaro, A.; Grimaldi, A.; Correani, V.; Mura, F.; Ciccosanti, F.; Rotiroti, N.; Brenna, A.; Montalbano, M.; et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS ONE 2016, 11, e0151736. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yuan, W.; He, P.; Lei, J.; Wang, B. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Coulouarn, C.; Clément, B. Stellate cells and the development of liver cancer: Therapeutic potential of targeting the stroma. J. Hepatol. 2014, 60, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Fang, C.; Yin, R.; Qiao, B.; Shang, R.; Wang, J.; Song, W.; He, Y.; Chen, Y. Agrin para-secreted by PDGF-activated human hepatic stellate cells promotes hepatocarcinogenesis in vitro and in vivo. Oncotarget 2017, 8, 105340–105355. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Sato, T.; Nagamine, K.; Sakasai-Sakai, A.; Takeuchi, M.; Hori, T. Suppression of hepatic stellate cell death by toxic advanced glycation end-products. Biol. Pharm. Bull. 2021, 44, 112–117. [Google Scholar] [CrossRef] [PubMed]
- DECODE Study Group. Glucose tolerance and cardiovascular mortality: Comparison of fasting and 2-hour diagnostic criteria. Arch. Intern. Med. 2001, 161, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Levitan, E.B.; Song, Y.; Ford, E.S.; Liu, S. Is nondiabetic hyperglycemia a risk factor for cardiovascular disease? A meta-analysis of prospective studies. Arch. Intern. Med. 2004, 164, 2147–2155. [Google Scholar] [CrossRef] [Green Version]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Hanefeld, M.; Fischer, S.; Julius, U.; Schulze, J.; Schwanebeck, U.; Schmechel, H.; Ziegelasch, H.J.; Lindner, J. Risk factors for myocardial infarction and death in newly detected NIDDM: The Diabetes Intervention Study, 11-year follow-up. Diabetologia 1996, 39, 1577–1583. [Google Scholar] [CrossRef]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Ueda, S.; Imaizumi, T. Role of postprandial hyperglycaemia in cardiovascular disease in diabetes. Int. J. Clin. Pract. 2007, 61, 83–87. [Google Scholar] [CrossRef]
- Inagaki, N.; Harashima, S.; Maruyama, N.; Kawaguchi, Y.; Goda, M.; Iijima, H. Efficacy and safety of canagliflozin in combination with insulin: A double-blind, randomized, placebo-controlled study in Japanese patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2016, 15, 89. [Google Scholar] [CrossRef] [Green Version]
- Lei, Y.; Yang, G.; Hu, L.; Piao, L.; Inoue, A.; Jiang, H.; Sasaki, T.; Zhao, G.; Yisireyili, M.; Yu, C. Increased dipeptidyl peptidase-4 accelerates diet-related vascular aging and atherosclerosis in ApoE-deficient mice under chronic stress. Int. J. Cardiol. 2017, 243, 413–420. [Google Scholar] [CrossRef]
- Grasser, E.K.; Dulloo, A.; Montani, J. Cardiovascular responses to the ingestion of sugary drinks using a randomised cross-over study design: Dose glucose attenuate the blood pressure-elevating effect of fructose? Br. J. Nutr. 2014, 112, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Rippe, J.M.; Angelopoulos, T.J. Fructose-containing sugars and cardiovascular disease. Adv. Nutr. 2015, 6, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelopoulos, T.J.; Lowndes, J.; Sinnett, S.; Rippe, J.M. Fructose containing sugars at normal levels of consumption do not effect adversely components of the metabolic syndrome and risk factors for cardiovascular disease. Nutrients 2016, 8, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaei, B.; Abdi, H.; Serahati, S.; Barzin, M.; Niroomand, M.; Azizi, F.; Hosseinpanah, F. Cardiovascular risk in different obesity phenotypes over a decade follow-up: Tehran lipid and glucose study. Atherosclerosis 2017, 258, 65–71. [Google Scholar] [CrossRef]
- Brahma, M.K.; Pepin, M.E.; Wende, A.R. My sweetheart is broken: Role of glucose in diabetic cardiomyopathy. Diabetes Metab. J. 2017, 41, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, Y.; Takeuchi, M.; Miura, K.; Mine, T.; Matsui, T.; Yamagishi, S. Glyceraldehyde-derived advanced glycation end products (AGEs). A novel biomarker of postprandial hyperglycaemia in diabetic rats. Clin. Exp. Med. 2008, 8, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacethylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [Green Version]
- Drieβen, S.; Berleth, N.; Friesen, O.; Löffler, A.S.; Böhler, P.; Hieke, N.; Stuhldreier, F.; Peter, C.; Schink, K.O.; Schultz, S.W.; et al. Deubiquitinase inhibition by WP1130 leads to ULK1 aggregation and blockade of autophagy. Autophagy 2015, 11, 1458–1470. [Google Scholar]
- Gu, J.; Hu, W.; Song, Z.P.; Chen, Y.G.; Zhang, D.D.; Wang, C.Q. Rapamycin inhibits cardiac hypertrophy by promoting autophagy via the MEK/ERK/Beclin-1 pathway. Front. Physiol. 2016, 7, 104. [Google Scholar] [CrossRef]
- Li, S.; Sun, X.; Wu, H.; Yu, P.; Wang, X.; Jiang, Z.; Gao, E.; Chen, J.; Li, D.; Qiu, C.; et al. TRPA1 promotes cardiac myofibroblast transdifferentiation after myocardial infarction injury via the calcineurin-NFAT-DYRK1A signaling pathway. Oxid. Med. Cell. Longev. 2019, 2019, 6408352. [Google Scholar] [CrossRef] [Green Version]
- Hara, A.; Kobayashi, H.; Asai, N.; Saito, S.; Higuchi, T.; Kato, K.; Okumura, T.; Bando, Y.K.; Takefuji, M.; Mizutani, Y.; et al. Roles of the mesenchymal stromal/stem cell marker meflin in cardiac tissue repair and the development of diastolic dysfunction. Circ. Res. 2019, 125, 414–430. [Google Scholar] [CrossRef]
- Enomoto, M.; Adachi, H.; Yamagishi, S.; Takeuchi, M.; Furuki, K.; Hino, A.; Hiratsuka, A.; Takajo, Y.; Imaizumi, T. Positive association of serum levels of advanced glycation end products with thrombogenic markers in humans. Metabolism 2006, 55, 912–917. [Google Scholar] [CrossRef]
- Tahara, N.; Yamagishi, S.; Takeuchi, M.; Honda, A.; Tahara, A.; Nitta, Y.; Kodama, N.; Mizoguchi, M.; Kaida, H.; Ishibashi, M.; et al. Positive association between serum level of glyceraldehyde-derived advanced glycation end products (AGEs) and vascular inflammation evaluated by 18F-fluorodeoxyglucose positron emission tomography (FDG-PET). Diabetes Care 2012, 35, 2618–2625. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.Y.; Takeuchi, M.; Hyogo, H.; McKeown-Eyssen, G.; Yamagishi, S.; Chayama, K.; O’Brien, P.J.; Ferrari, P.; Overvad, K.; Olsen, A.; et al. The association between glyceraldehyde-derived advanced glycation end-products and colorectal cancer risk. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1855–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takino, J.; Nagamine, K.; Hori, T.; Sakasai-Sakai, A.; Takeuchi, M. Contribution of the toxic advanced glycation end-products-receptor axis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2459–2469. [Google Scholar] [CrossRef]
- Abe, R.; Shimizu, T.; Sugawara, H.; Watanabe, H.; Nakamura, H.; Choei, H.; Sasaki, N.; Yamagishi, S.; Takeuchi, M.; Shimizu, H. Regulation of human melanoma growth and metastasis by AGE-AGE receptor interactions. J. Investig. Dermatol. 2004, 122, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Cancer malignancy is enhanced by glyceraldehyde-derived advanced glycation end-products. J. Oncol. 2010, 2010, 739852. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Metformin inhibits advanced glycation end products (AGEs)-induced growth and VEGF expression in MCF-7 breast cancer cells by suppressing AGEs receptor expression via AMP-activated protein kinase. Horm. Metab. Res. 2013, 45, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Kaida, Y.; Fukami, K.; Matsui, T.; Higashimoto, Y.; Nishino, Y.; Obara, N.; Nakayama, Y.; Ando, R.; Toyonaga, M.; Ueda, S.; et al. DNA aptamer raised against AGEs blocks the progression of experimental diabetic nephropathy. Diabetes 2013, 62, 3241–3250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojima, A.; Matsui, T.; Maeda, S.; Takeuchi, M.; Inoue, H.; Higashimoto, Y.; Yamagishi, S. DNA aptamer raised against advanced glycation end products inhibits melanoma growth in nude mice. Lab. Investig. 2014, 94, 422–429. [Google Scholar] [CrossRef] [Green Version]
- Ojima, A.; Oda, E.; Higashimoto, Y.; Matsui, T.; Yamagishi, S. DNA aptamer raised against advanced glycation end products inhibits neointimal hyperplasia in balloon-injured rat carotid arteries. Int. J. Cardiol. 2014, 171, 443–446. [Google Scholar] [CrossRef] [PubMed]
- Ojima, A.; Matsui, T.; Nakamura, N.; Higashimoto, Y.; Ueda, S.; Fukami, K.; Okuda, S.; Yamagishi, S. DNA aptamer raised against advanced glycation end products (AGEs) improves glycemic control and decreases adipocyte size in fructose-fed rats by suppressing AGE-RAGE axis. Horm. Metab. Res. 2015, 47, 253–258. [Google Scholar] [CrossRef]
- Johnson, L.L.; Johnson, J.; Ober, R.; Holland, A.; Zhang, G.; Backer, M.; Backer, J.; Ali, Z.; Tekabe, Y. Novel receptor for advanced glycation end products-blocking antibody to treat diabetic peripheral artery disease. J. Am. Heart. Assoc. 2021, 10, e016696. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Matsui, T.; Ishibashi, Y.; Sotokawauchi, A.; Fukami, K.; Higashimoto, Y.; Yamagishi, S. RAGE-aptamer attenuates the growth and liver metastasis of malignant melanoma in nude mice. Mol. Med. 2017, 23, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Yamagishi, S.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, 2686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotokawauchi, A.; Matsui, T.; Higashimoto, Y.; Nishino, Y.; Koga, Y.; Yagi, M.; Yamagishi, S. DNA aptamer raised against receptor for advanced glycation end products suppresses renal tubular damage and improves insulin resistance in diabetic mice. Diab. Vasc. Dis. Res. 2021, 18. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeuchi, M.; Sakasai-Sakai, A.; Takata, T.; Takino, J.-i.; Koriyama, Y.; Kikuchi, C.; Furukawa, A.; Nagamine, K.; Hori, T.; Matsunaga, T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules 2021, 11, 387. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030387
Takeuchi M, Sakasai-Sakai A, Takata T, Takino J-i, Koriyama Y, Kikuchi C, Furukawa A, Nagamine K, Hori T, Matsunaga T. Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage. Biomolecules. 2021; 11(3):387. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030387
Chicago/Turabian StyleTakeuchi, Masayoshi, Akiko Sakasai-Sakai, Takanobu Takata, Jun-ichi Takino, Yoshiki Koriyama, Chigusa Kikuchi, Ayako Furukawa, Kentaro Nagamine, Takamitsu Hori, and Tamihide Matsunaga. 2021. "Intracellular Toxic AGEs (TAGE) Triggers Numerous Types of Cell Damage" Biomolecules 11, no. 3: 387. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030387