
Effects of Hypoxic Preconditioning and Vascular Endothelial Growth Factor on the Survival of Isolated Primary Retinal Ganglion Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Rats
2.3. Primary RGC Harvest and Culture
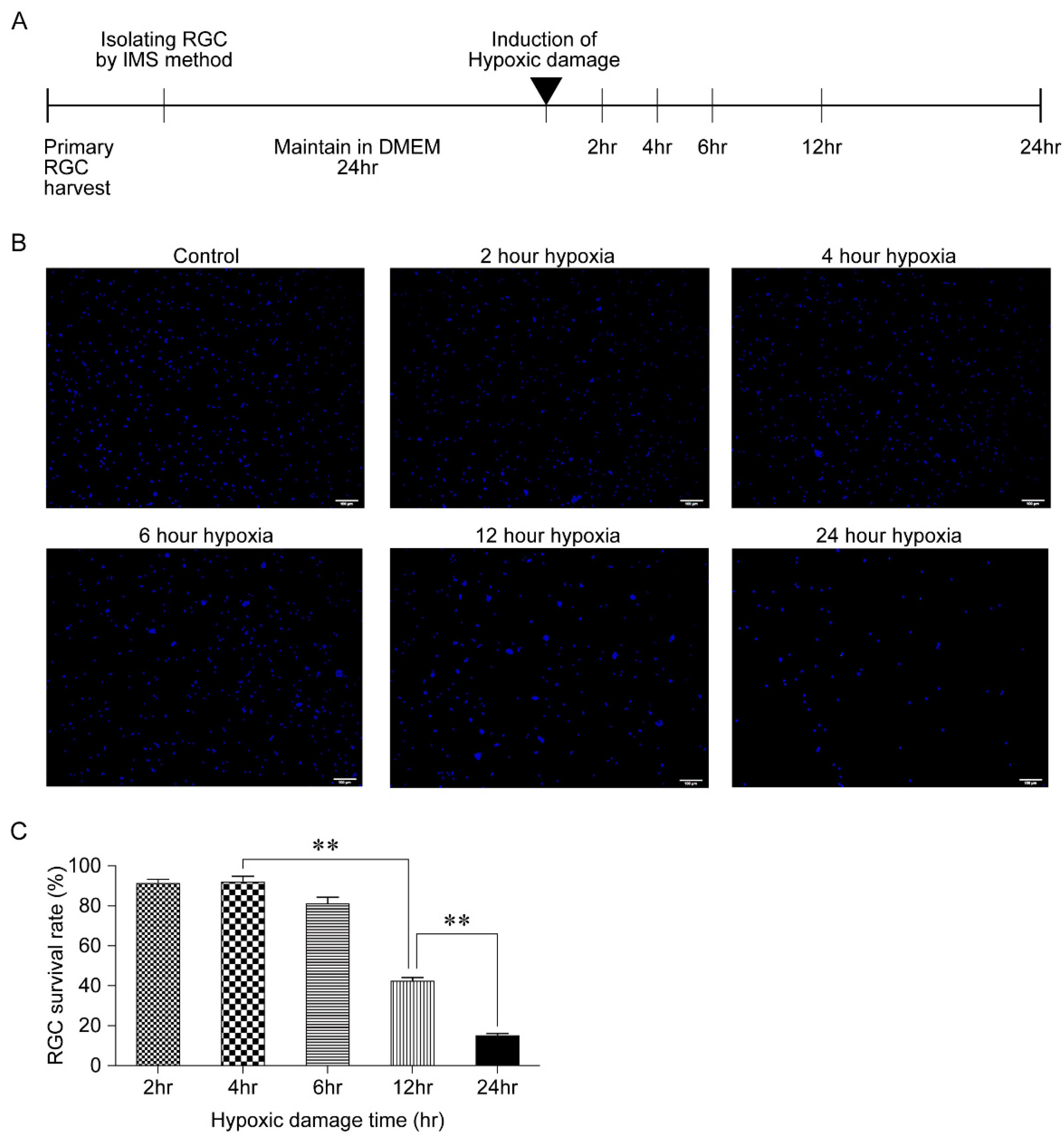
2.4. Generation of Hypoxic Damage
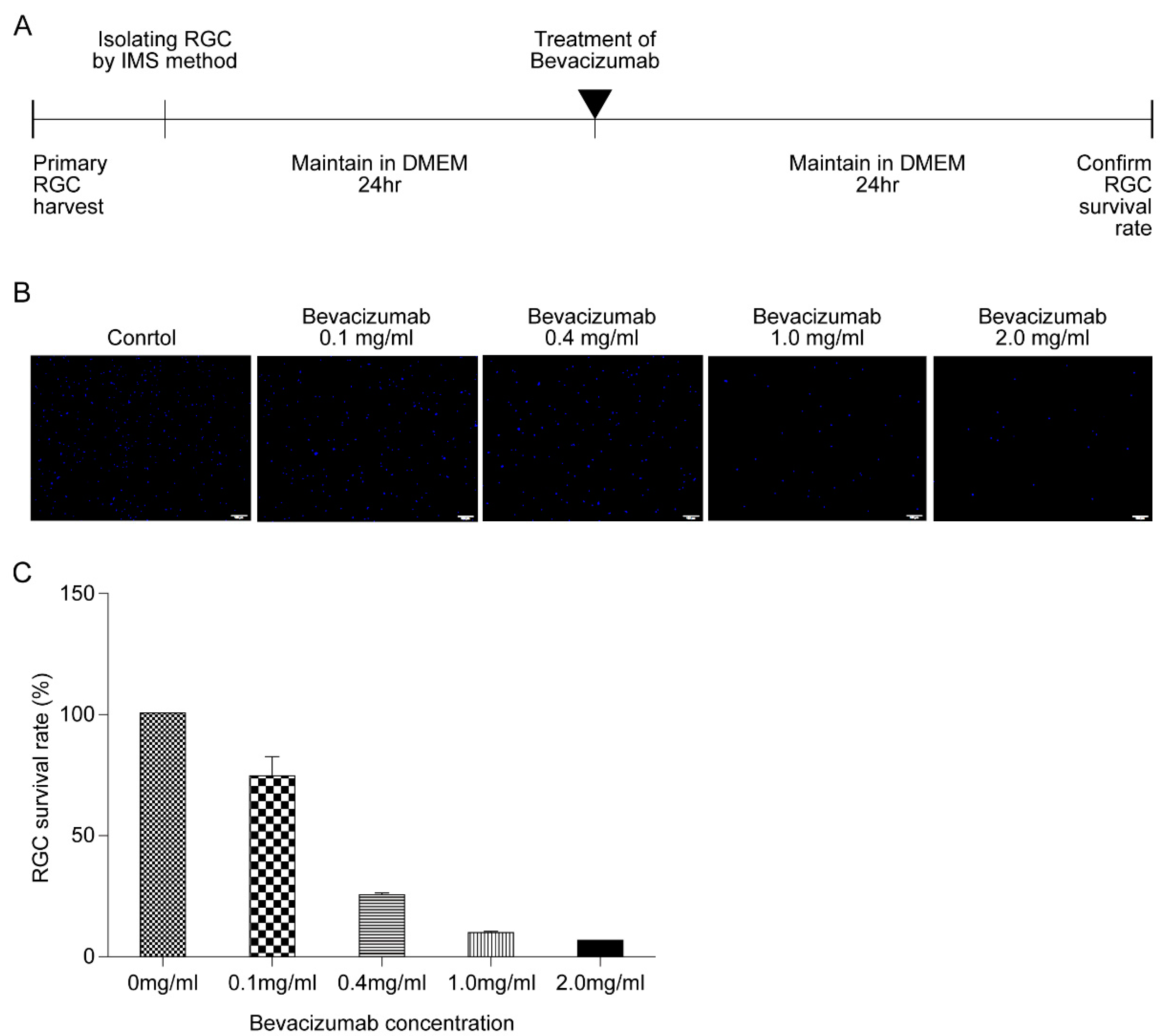
2.5. Bevacizumab Treatment
2.6. Cell Counting
2.7. Flow Cytometry
2.8. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Western Blotting
2.11. Statistics
3. Results
3.1. Outcome of Hypoxic Damage to RGCs Based on Exposure Time
3.2. Effect of HPC on RGC Survival
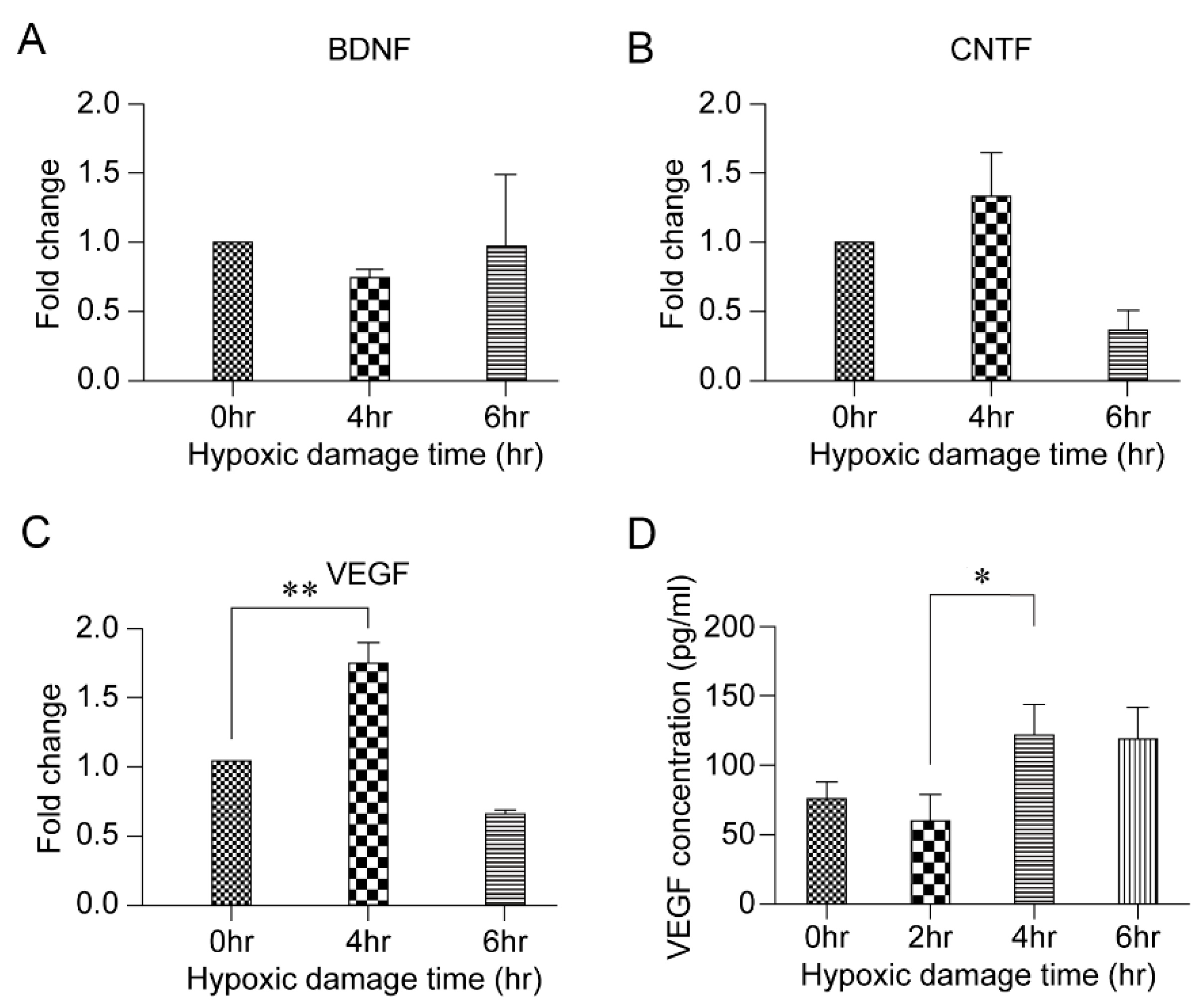
3.3. HPC and VEGF
3.4. Effect of HPC and Anti-VEGF Antibody on RGC Survival
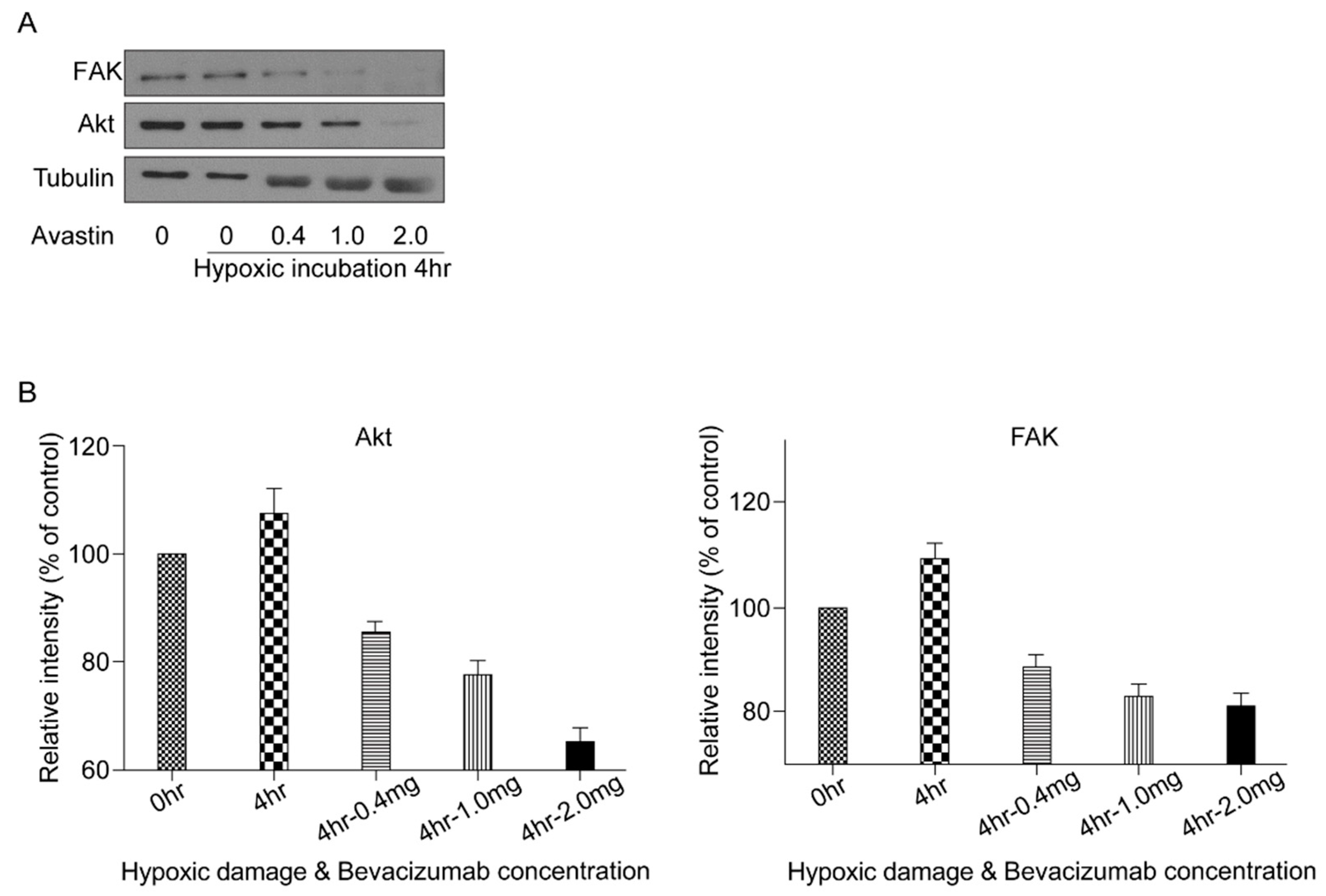
3.5. VEGF Survival Pathway Following HPC
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Quigley, H.A.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Comparison of glaucomatous progression between untreated patients with normal-tension glaucoma and patients with therapeutically reduced intraocular pressures. Collaborative normal-tension glaucoma study group. Am. J. Ophthalmol. 1998, 126, 487–497.
- Kass, M.A.; Heuer, D.K.; Higginbotham, E.J.; Johnson, C.A.; Keltner, J.L.; Miller, J.P.; Parrish, R.K., 2nd; Wilson, M.R.; Gordon, M.O. The ocular hypertension treatment study: A randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch. Ophthalmol. 2002, 120, 701–713; discussion 829–730. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, L.; Schmidt, J.F.; Gidday, J.M. Glaucoma-induced degeneration of retinal ganglion cells prevented by hypoxic preconditioning: A model of glaucoma tolerance. Mol. Med. 2012, 18, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Wangsa-Wirawan, N.D.; Linsenmeier, R.A. Retinal oxygen: Fundamental and clinical aspects. Arch. Ophthalmol. 2003, 121, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Johnstone, A.; Khoja, Z.; Rampakakis, E.; Lachapelle, P.; Wintermark, P. Sildenafil improves functional and structural outcome of retinal injury following term neonatal hypoxia-ischemia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4306–4314. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, S.; Anand, V.; Roy, S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J. Neuroimmune Pharmacol. 2014, 9, 142–160. [Google Scholar] [CrossRef] [Green Version]
- Prass, K.; Scharff, A.; Ruscher, K.; Löwl, D.; Muselmann, C.; Victorov, I.; Kapinya, K.; Dirnagl, U.; Meisel, A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke 2003, 34, 1981–1986. [Google Scholar] [CrossRef] [Green Version]
- Witmer, A.N.; Vrensen, G.F.; Van Noorden, C.J.; Schlingemann, R.O. Vascular endothelial growth factors and angiogenesis in eye disease. Prog. Retin. Eye Res. 2003, 22, 1–29. [Google Scholar] [CrossRef]
- Ng, E.W.; Adamis, A.P. Targeting angiogenesis, the underlying disorder in neovascular age-related macular degeneration. Can. J. Ophthalmol. 2005, 40, 352–368. [Google Scholar] [CrossRef]
- Hong, S.; Iizuka, Y.; Kim, C.Y.; Seong, G.J. Isolation of primary mouse retinal ganglion cells using immunopanning-magnetic separation. Mol. Vis. 2012, 18, 2922–2930. [Google Scholar] [PubMed]
- Choi, W.; Byun, Y.J.; Jung, E.; Noh, H.; Hajrasouliha, A.R.; Sadrai, Z.; Chang, E.; Lee, J.H.; Lee, H.K. Chemokine decoy receptor d6 mimicking trap (d6mt) prevents allosensitization and immune rejection in murine corneal allograft model. J. Leukoc. Biol. 2015, 97, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Kaur, C.; Sivakumar, V.; Foulds, W.S. Early response of neurons and glial cells to hypoxia in the retina. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1126–1141. [Google Scholar] [CrossRef]
- Adachi, K.; Kashii, S.; Masai, H.; Ueda, M.; Morizane, C.; Kaneda, K.; Kume, T.; Akaike, A.; Honda, Y. Mechanism of the pathogenesis of glutamate neurotoxicity in retinal ischemia. Graefes. Arch. Clin. Exp. Ophthalmol. 1998, 236, 766–774. [Google Scholar] [CrossRef]
- Barenberg, P.; Strahlendorf, H.; Strahlendorf, J. Hypoxia induces an excitotoxic-type of dark cell degeneration in cerebellar purkinje neurons. Neurosci. Res. 2001, 40, 245–254. [Google Scholar] [CrossRef]
- Joo, C.K.; Choi, J.S.; Ko, H.W.; Park, K.Y.; Sohn, S.; Chun, M.H.; Oh, Y.J.; Gwag, B.J. Necrosis and apoptosis after retinal ischemia: Involvement of nmda-mediated excitotoxicity and p53. Investig. Ophthalmol. Vis. Sci. 1999, 40, 713–720. [Google Scholar]
- Li, S.; Hafeez, A.; Noorulla, F.; Geng, X.; Shao, G.; Ren, C.; Lu, G.; Zhao, H.; Ding, Y.; Ji, X. Preconditioning in neuroprotection: From hypoxia to ischemia. Prog. Neurobiol. 2017, 157, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Xiong, L.J.; Tong, Y.; Mao, M. The neuroprotective roles of bdnf in hypoxic ischemic brain injury. Biomed. Rep. 2013, 1, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.L.; Gao, G.Q.; Ma, N.; Ye, L.L.; Zhang, L.W.; Gao, X.; Zhang, Z.B. Cntf protects neurons from hypoxic injury through the activation of stat3ptyr705. Int. J. Mol. Med. 2016, 38, 1915–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agostinone, J.; Alarcon-Martinez, L.; Gamlin, C.; Yu, W.Q.; Wong, R.O.L.; Di Polo, A. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain 2018, 141, 1963–1980. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hong, J.; Zhou, F.; Tang, S.; Wu, X. Regulatory mechanism of melatonin on the retinal ganglion cell photoreaction in mice. Exp. Ther. Med. 2017, 14, 1491–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garthwaite, J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991, 14, 60–67. [Google Scholar] [CrossRef]
- Januschowski, K.; Müller, S.; Krupp, C.; Spitzer, M.S.; Hurst, J.; Schultheiss, M.; Bartz-Schmidt, K.U.; Szurman, P.; Schnichels, S. Glutamate and hypoxia as a stress model for the isolated perfused vertebrate retina. J. Vis. Exp. 2015, 97, 52270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Yu, H.; Yan, N.; Lai, K.; Xiang, M. Hypoxia-inducible factor-1α target genes contribute to retinal neuroprotection. Front. Cell Neurosci. 2017, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Esser, S.; Wolburg, K.; Wolburg, H.; Breier, G.; Kurzchalia, T.; Risau, W. Vascular endothelial growth factor induces endothelial fenestrations in vitro. J. Cell Biol. 1998, 140, 947–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 8438–8445. [Google Scholar] [CrossRef]
- Stone, J.; Itin, A.; Alon, T.; Pe’er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (vegf) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747. [Google Scholar] [CrossRef]
- Stone, J.; Chan-Ling, T.; Pe’er, J.; Itin, A.; Gnessin, H.; Keshet, E. Roles of vascular endothelial growth factor and astrocyte degeneration in the genesis of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 1996, 37, 290–299. [Google Scholar]
- Wang, Y.; Mao, X.O.; Xie, L.; Banwait, S.; Marti, H.H.; Greenberg, D.A.; Jin, K. Vascular endothelial growth factor overexpression delays neurodegeneration and prolongs survival in amyotrophic lateral sclerosis mice. J. Neurosci. 2007, 27, 304–307. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.; Powner, M.B.; Gandhi, P.; Clarkin, C.; Gutmann, D.H.; Johnson, R.S.; Ferrara, N.; Fruttiger, M. Astrocyte-derived vascular endothelial growth factor stabilizes vessels in the developing retinal vasculature. PLoS ONE 2010, 5, e11863. [Google Scholar] [CrossRef] [Green Version]
- Saint-Geniez, M.; Maharaj, A.S.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous vegf is required for visual function: Evidence for a survival role on müller cells and photoreceptors. PLoS ONE 2008, 3, e3554. [Google Scholar] [CrossRef] [Green Version]
- Froger, N.; Matonti, F.; Roubeix, C.; Forster, V.; Ivkovic, I.; Brunel, N.; Baudouin, C.; Sahel, J.A.; Picaud, S. Vegf is an autocrine/paracrine neuroprotective factor for injured retinal ganglion neurons. Sci. Rep. 2020, 10, 12409. [Google Scholar] [CrossRef]
- Kilic, U.; Kilic, E.; Järve, A.; Guo, Z.; Spudich, A.; Bieber, K.; Barzena, U.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. Human vascular endothelial growth factor protects axotomized retinal ganglion cells in vivo by activating erk-1/2 and akt pathways. J. Neurosci. 2006, 26, 12439–12446. [Google Scholar] [CrossRef]
- Foxton, R.H.; Finkelstein, A.; Vijay, S.; Dahlmann-Noor, A.; Khaw, P.T.; Morgan, J.E.; Shima, D.T.; Ng, Y.S. Vegf-a is necessary and sufficient for retinal neuroprotection in models of experimental glaucoma. Am. J. Pathol. 2013, 182, 1379–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, R.L. What is the evidence for systemic effects of intravitreal anti-vegf agents, and should we be concerned? Br. J. Ophthalmol. 2014, 98 (Suppl. 1), i7–i10. [Google Scholar] [CrossRef]
- Thaler, S.; Fiedorowicz, M.; Choragiewicz, T.J.; Bolz, S.; Tura, A.; Henke-Fahle, S.; Yoeruek, E.; Zrenner, E.; Bartz-Schmidt, K.U.; Ziemssen, F.; et al. Toxicity testing of the vegf inhibitors bevacizumab, ranibizumab and pegaptanib in rats both with and without prior retinal ganglion cell damage. Acta Ophthalmol. 2010, 88, e170–e176. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, V.S.; Verma, N. Safety and efficacy of intravitreal anti-vegf injections for age-related macular degeneration. Curr. Opin. Ophthalmol. 2009, 20, 223–225. [Google Scholar] [CrossRef]
- Bus, P.; Gerrits, T.; Heemskerk, S.A.C.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J.; Scharpfenecker, M. Endoglin mediates vascular endothelial growth factor-a-induced endothelial cell activation by regulating akt signaling. Am. J. Pathol. 2018, 188, 2924–2935. [Google Scholar] [CrossRef] [Green Version]
- Park, H.Y.; Kim, J.H.; Park, C.K. Neuronal cell death in the inner retina and the influence of vascular endothelial growth factor inhibition in a diabetic rat model. Am. J. Pathol. 2014, 184, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Wary, K.K.; Kohler, E.E.; Chatterjee, I. Focal adhesion kinase regulation of neovascularization. Microvasc. Res. 2012, 83, 64–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitra, S.K.; Schlaepfer, D.D. Integrin-regulated fak-src signaling in normal and cancer cells. Curr. Opin. Cell Biol. 2006, 18, 516–523. [Google Scholar] [CrossRef]
- Santos, A.R.; Corredor, R.G.; Obeso, B.A.; Trakhtenberg, E.F.; Wang, Y.; Ponmattam, J.; Dvoriantchikova, G.; Ivanov, D.; Shestopalov, V.I.; Goldberg, J.L.; et al. Β1 integrin-focal adhesion kinase (fak) signaling modulates retinal ganglion cell (rgc) survival. PLoS ONE 2012, 7, e48332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, H.L.; Mercurio, A.M. Enhancing integrin function by vegf/neuropilin signaling: Implications for tumor biology. Cell Adh. Migr. 2012, 6, 554–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Sequence |
|---|---|
| BDNF | F: 5′–GCG GCA GAT AAA AAG ACT GC–3′ R: 5′–GCC AGC CAA TTC TCT TTT TG–3′ |
| CNTF | F: 5′–CAC CCC AAC TGA AGG TGA CT–3′ R: 5′–ACC TTC AAG CCC CAT AGC TT–3′ |
| VEGF | F: 5′–GGC TCT GAA ACC ATG AAC TTT CT–3′ R: 5′–GCA GTA GCT GCG CTG GTA GAC–3′ |
| Akt | F: 5′–GTG GCA AGA TGT GTA TGA G–3′ R: 5′–CTG GCT GAG TAG GAG AAC–3′ |
| FAK | F: 5′–AAA ATG TGA CGG GCC TAG TG–3′ R: 5′–TAC TCC TGC TGA AGG CTG GT–3′ |
| β-Actin | F: 5′–CAC CCG CGA GTA CAA CCT T–3′ R: 5′–CCC ATA CCC ACC ATC ACA CC–3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, H.W.; Choi, W.; Hwang, A.R.; Lee, S.Y.; Seong, G.J.; Kim, C.Y. Effects of Hypoxic Preconditioning and Vascular Endothelial Growth Factor on the Survival of Isolated Primary Retinal Ganglion Cells. Biomolecules 2021, 11, 391. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030391
Bae HW, Choi W, Hwang AR, Lee SY, Seong GJ, Kim CY. Effects of Hypoxic Preconditioning and Vascular Endothelial Growth Factor on the Survival of Isolated Primary Retinal Ganglion Cells. Biomolecules. 2021; 11(3):391. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030391
Chicago/Turabian StyleBae, Hyoung Won, Wungrak Choi, Ah Reum Hwang, Sang Yeop Lee, Gong Je Seong, and Chan Yun Kim. 2021. "Effects of Hypoxic Preconditioning and Vascular Endothelial Growth Factor on the Survival of Isolated Primary Retinal Ganglion Cells" Biomolecules 11, no. 3: 391. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11030391