
Evaluating Mechanisms of IDH1 Regulation through Site-Specific Acetylation Mimics

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Construct Preparation
2.3. Kinetic Assays
2.4. Structural Modeling
3. Results
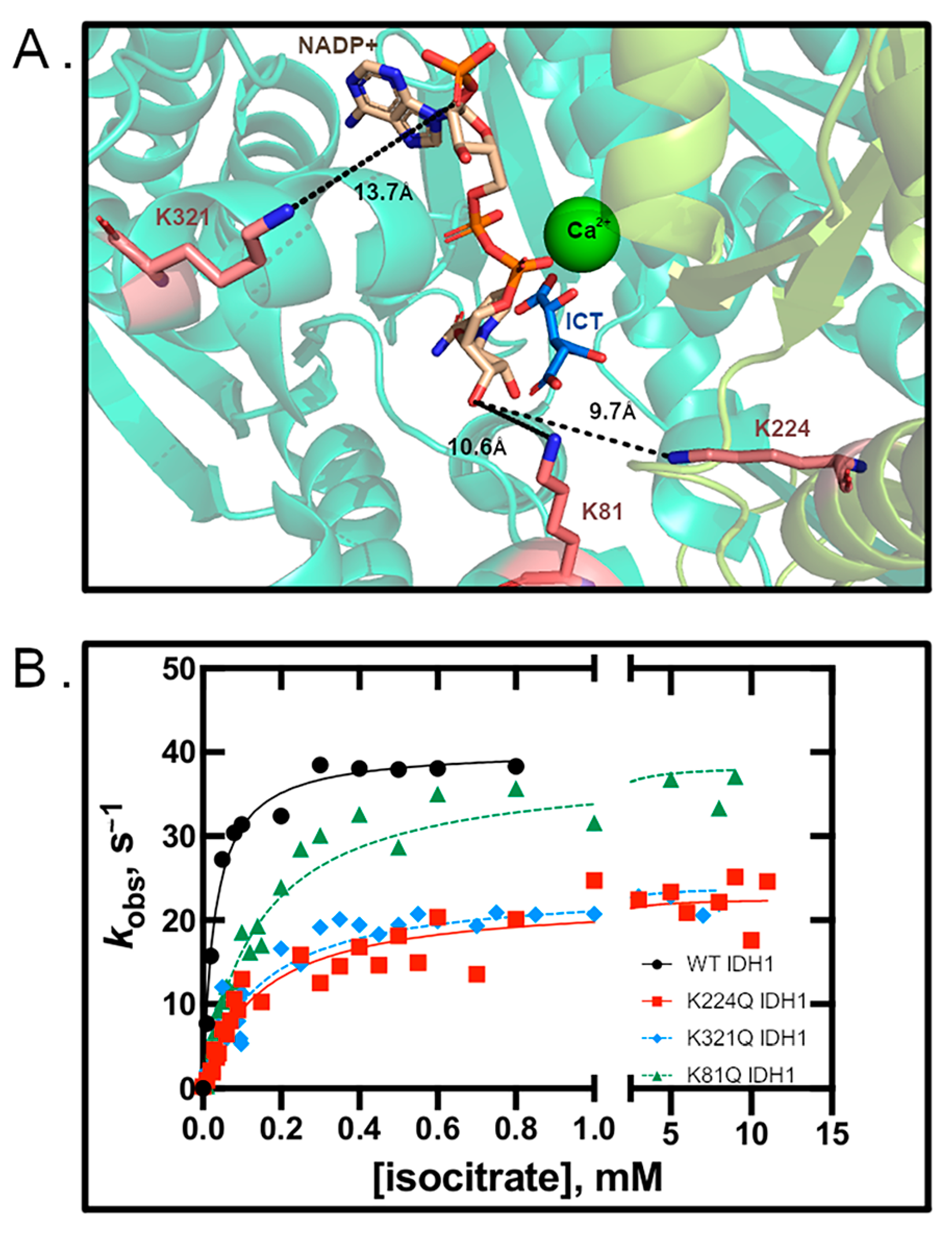
3.1. Glutamine Mutation Results in Decreased Kinetic Activity
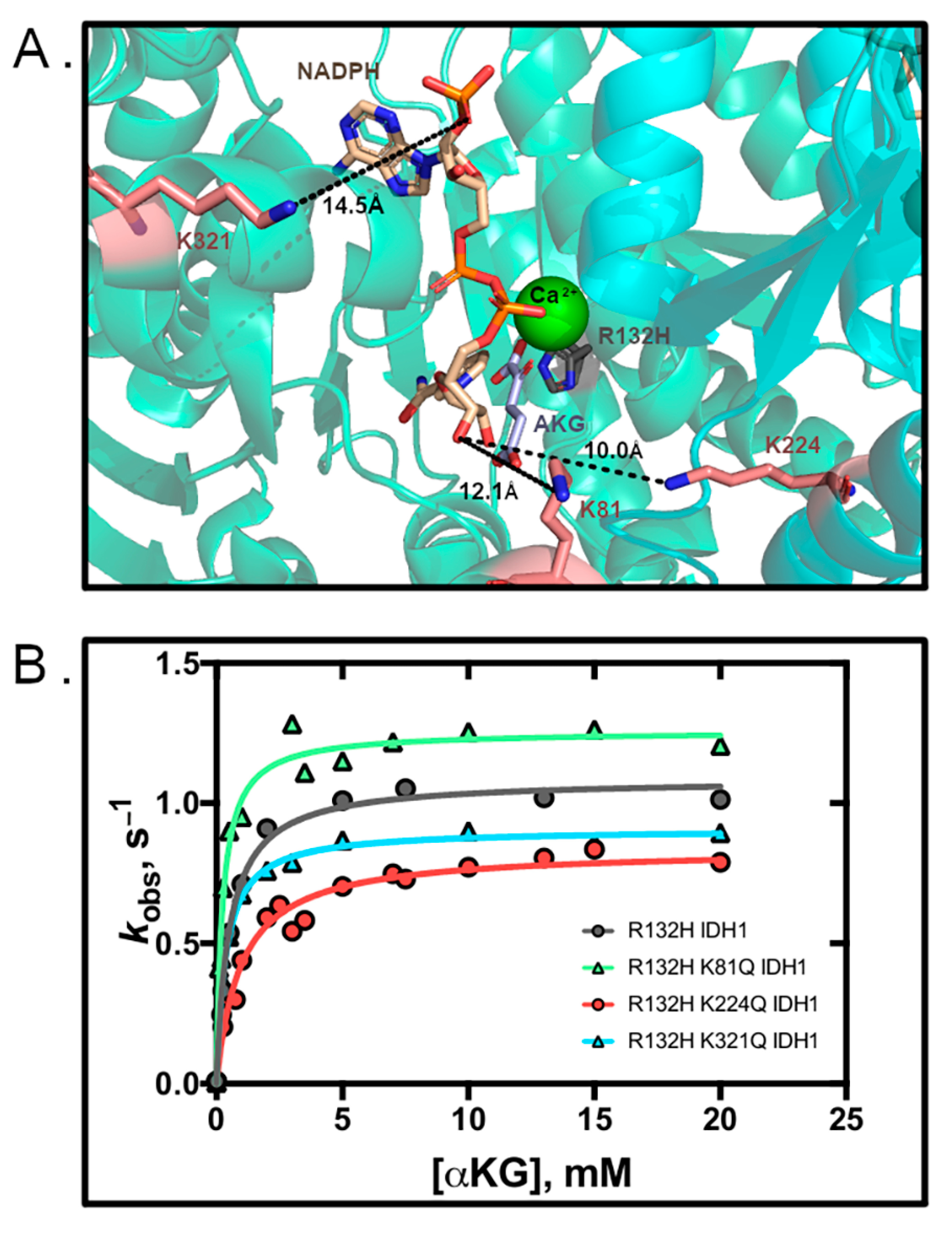
3.2. R132H IDH1 Acetylation Mimics Have Minor Kinetic Effects
3.3. Residue D79 Plays an Important Role in WT IDH1 Activity
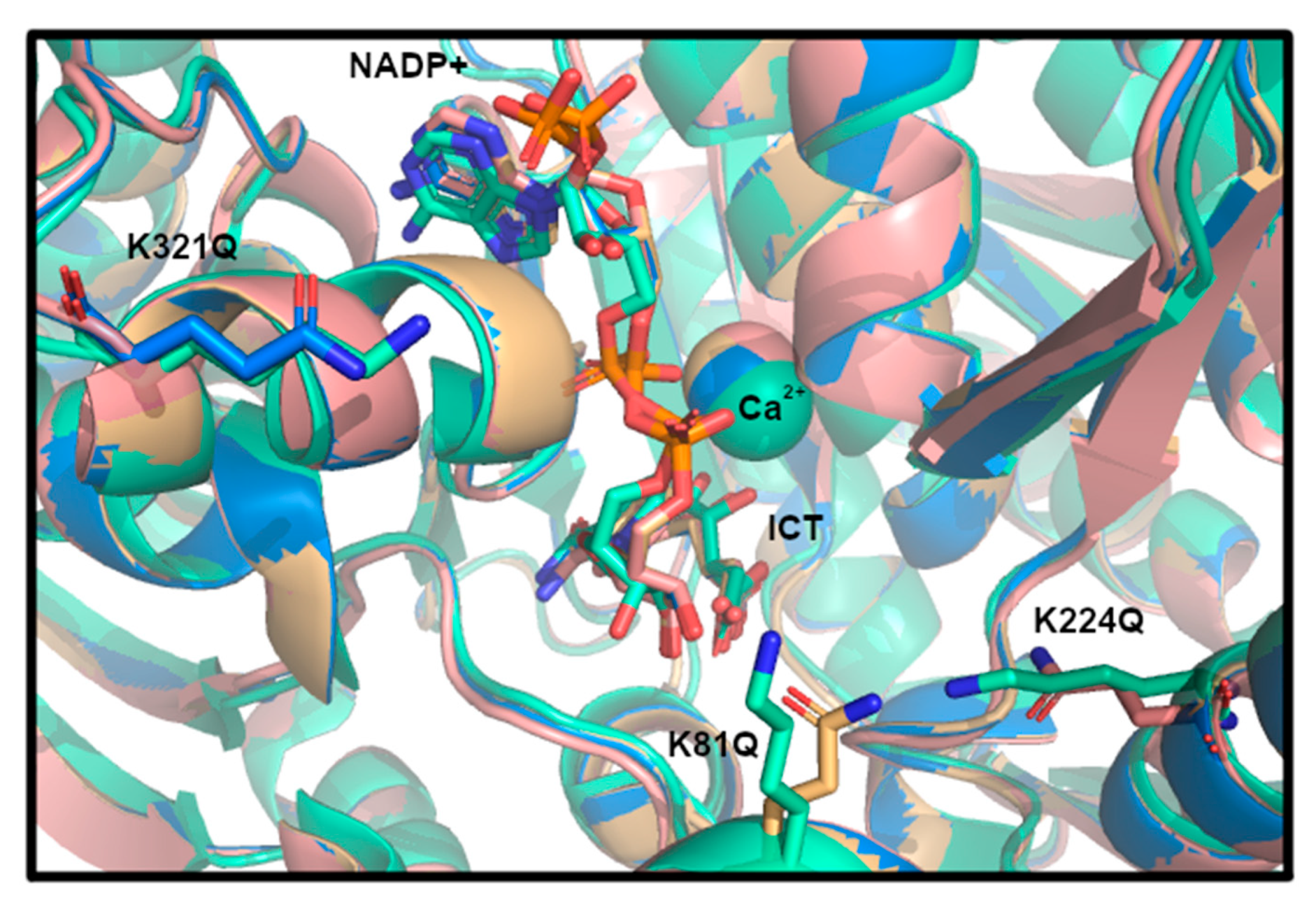
3.4. Structural Modeling of IDH1 Mutations
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Avellaneda Matteo, D.; Grunseth, A.J.; Gonzalez, E.R.; Anselmo, S.L.; Kennedy, M.A.; Moman, P.; Scott, D.A.; Hoang, A.; Sohl, C.D. Molecular mechanisms of isocitrate dehydrogenase 1 (IDH1) mutations identified in tumors: The role of size and hydrophobicity at residue 132 on catalytic efficiency. J. Biol. Chem. 2017, 292, 7971–7983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G.; et al. The IDH1 mutation-induced oncometabolite, 2-hydroxyglutarate, may affect DNA methylation and expression of PD-L1 in gliomas. Front. Mol. Neurosci. 2018, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Wu, J.; Ma, S.; Zhang, L.; Yao, J.; Hoadley, K.A.; Wilkerson, M.D.; Perou, C.M.; Guan, K.L.; Ye, D.; et al. Oncometabolite D-2-hydroxyglutarate inhibits ALKBH DNA repair enzymes and sensitizes IDH mutant cells to alkylating agents. Cell Rep. 2015, 13, 2353–2361. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Tönjes, M.; Barbus, S.; Park, Y.J.; Wang, W.; Schlotter, M.; Lindroth, A.M.; Pleier, S.V.; Bai, A.H.C.; Karra, D.; Piro, R.M.; et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nat. Med. 2013, 19, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, Y.; Chen, J.; Qiu, J.; Huang, K.; Wu, M.; Xia, C. IDH1 R132H mutation enhances cell migration by activating AKT-mTOR signaling pathway, but sensitizes cells to 5-FU treatment as NADPH and GSH are reduced. PLoS ONE 2017, 12, e0169038. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Dean, A.M.; Sohl, J.L.; Koshland, D.E.; Stroud, R.M. Regulation of an Enzyme by Phosphorylation at the Active Site. Science 1990, 249, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.P.; McAlister-Henn, L. Homologous binding sites in yeast isocitrate dehydrogenase for cofactor (NAD+) and allosteric activator (AMP). J. Biol. Chem. 2003, 278, 12864–12872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J. Natl. Cancer Inst. 2010, 102, 932–941. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Xia, S.; Wang, M.; Lin, R.; Li, Y.; Mao, H.; Aguiar, M.; Famulare, C.A.; Shih, A.H.; Brennan, C.W.; et al. Mutant and wild-type isocitrate dehydrogenase 1 share enhancing mechanisms involving distinct tyrosine kinase cascades in cancer. Cancer Discov. 2019, 9, 756–777. [Google Scholar] [CrossRef] [Green Version]
- Baeza, J.; Smallegan, M.J.; Denu, J.M. Site-specific reactivity of nonenzymatic lysine acetylation. ACS Chem. Biol. 2015, 10, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef] [Green Version]
- Lombard, D.B.; Alt, F.W.; Cheng, H.L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A.; et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starai, V.J.; Celic, I.; Cole, R.N.; Boeke, J.D.; Escalante-Semerena, J.C. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 2002, 298, 2390–2392. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, K.; Wang, J.; Diagouraga, B.; Khochbin, S. The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2011, 2011, 970382. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Drazic, A.; Myklebust, L.M.; Ree, R.; Arnesen, T. The world of protein acetylation. Biochim. Biophys. Acta 2016, 1864, 1372–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Dittenhafer-Reed, K.E.; Denu, J.M. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J. Biol. Chem. 2012, 287, 14078–14086. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Liu, L.; Nakamura, A.; Someya, S.; Miyakawa, T.; Tanokura, M. Studies on the regulatory mechanism of isocitrate dehydrogenase 2 using acetylation mimics. Sci. Rep. 2017, 7, 9785. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rardin, M.J.; Newman, J.C.; Held, J.M.; Cusack, M.P.; Sorensen, D.J.; Li, B.; Schilling, B.; Mooney, S.D.; Kahn, C.R.; Verdin, E.; et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA 2013, 110, 6601–6606. [Google Scholar] [CrossRef] [Green Version]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, T. The BLAST Sequence Analysis Tool; National Center for Biotechnology Information: Bethesda, MD, USA, 2002. [Google Scholar]
- Wang, B.; Ye, Y.; Yang, X.; Liu, B.; Wang, Z.; Chen, S.; Jiang, K.; Zhang, W.; Jiang, H.; Mustonen, H.; et al. SIRT2-dependent IDH1 deacetylation inhibits colorectal cancer and liver metastases. EMBO Rep. 2020, e48183. [Google Scholar] [CrossRef]
- Avellaneda Matteo, D.; Wells, G.A.; Luna, L.A.; Grunseth, A.J.; Zagnitko, O.; Scott, D.A.; Hoang, A.; Luthra, A.; Swairjo, M.A.; Schiffer, J.M.; et al. Inhibitor potency varies widely among tumor-relevant human isocitrate dehydrogenase 1 mutants. Biochem. J. 2018, 475, 3221–3238. [Google Scholar] [CrossRef]
- Rendina, A.R.; Pietrak, B.; Smallwood, A.; Zhao, H.; Qi, H.; Quinn, C.; Adams, N.D.; Concha, N.; Duraiswami, C.; Thrall, S.H.; et al. Mutant IDH1 enhances the production of 2-hydroxyglutarate due to its kinetic mechanism. Biochemistry 2013, 52, 4563–4577. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 2.0; Schrödinger, LLC: Portland, OR, USA, 2017. [Google Scholar]
- Schrodinger, LLC. The AxPyMOL Molecular Graphics Plugin for Microsoft PowerPoint, Version 2.0; Schrödinger, LLC: Portland, OR, USA, 2010. [Google Scholar]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Cryst. Sect. D 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Morizono, H.; Yu, X.; Tong, L.; Allewell, N.M.; Tuchman, M. Human ornithine transcarbamylase: Crystallographic insights into substrate recognition and conformational changes. Biochem. J. 2001, 354, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Guan, K.-L. Mechanistic insights into the regulation of metabolic enzymes by acetylation. J. Cell Biol. 2012, 198, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Lin, Y.; Yao, J.; Huang, W.; Lei, Q.; Xiong, Y.; Zhao, S.; Guan, K.-L. Lysine 88 acetylation negatively regulates ornithine carbamoyltransferase activity in response to nutrient signals. J. Biol. Chem. 2009, 284, 13669–13675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-M.; You, D.; Ye, B.-C. Site-specific and kinetic characterization of enzymatic and nonenzymatic protein acetylation in bacteria. Sci. Rep. 2017, 7, 14790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Rao, M.; Wang, C.; Sakamaki, T.; Wang, J.; Di Vizio, D.; Zhang, X.; Albanese, C.; Balk, S.; Chang, C.; et al. Acetylation of Androgen Receptor Enhances Coactivator Binding and Promotes Prostate Cancer Cell Growth. Mol. Cell Biol. 2003, 23, 8563–8575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, R.H.; Keberlein, M.; Jackson, V. A Mutational Mimic Analysis of Histone H3 Post-Translational Modifications: Specific Sites Influence the Conformational State of H3/H4, Causing either Positive or Negative Supercoiling of DNA. Biochemistry 2012, 51, 8173–8188. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Genetically encoding Nε-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008, 4, 232–234. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IDH1 | kcat, s−1 | Km, mM | kcat/Km, µM−1 s−1 |
|---|---|---|---|
| WT | 40.4 ± 0.8 | 0.03001 ± 0.0008 | 1.3 ± 0.1 |
| K81Q | 38 ± 1 | 0.14 ± 0.02 | 0.27 ± 0.04 |
| K224Q | 23.0 ± 0.8 | 0.14 ± 0.02 | 0.16 ± 0.02 |
| K321Q | 24 ± 1 | 0.14 ± 0.02 | 0.17 ± 0.02 |
| D79L | 0.315 ± 0.006 | 0.028 ± 0.003 | 0.010 ± 0.001 |
| IDH1 | kcat, s−1 | Km, mM | kcat/Km, µM−1 s−1 |
|---|---|---|---|
| R132H | 1.09 ± 0.02 | 0.51 ± 0.04 | 0.0021 ± 0.0002 |
| R132H/K81Q | 1.26 ± 0.03 | 0.24 ± 0.03 | 0.0052 ± 0.0007 |
| R132H/K224Q | 0.84 ± 0.02 | 1.0 ± 0.1 | 0.0009 ± 0.0001 |
| R132H/K321Q | 0.91 ± 0.01 | 0.37 ± 0.02 | 0.0025 ± 0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weeks, J.; Strom, A.I.; Widjaja, V.; Alexander, S.; Pucher, D.K.; Sohl, C.D. Evaluating Mechanisms of IDH1 Regulation through Site-Specific Acetylation Mimics. Biomolecules 2021, 11, 740. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050740
Weeks J, Strom AI, Widjaja V, Alexander S, Pucher DK, Sohl CD. Evaluating Mechanisms of IDH1 Regulation through Site-Specific Acetylation Mimics. Biomolecules. 2021; 11(5):740. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050740
Chicago/Turabian StyleWeeks, Joi, Alexandra I. Strom, Vinnie Widjaja, Sati Alexander, Dahra K. Pucher, and Christal D. Sohl. 2021. "Evaluating Mechanisms of IDH1 Regulation through Site-Specific Acetylation Mimics" Biomolecules 11, no. 5: 740. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050740