
Epigenetic Insights and Potential Modifiers as Therapeutic Targets in β–Thalassemia

 ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Epigenetics and β–thalassemia
2.1. DNA Methylation
2.2. Histone Modification
3. Other Epigenetic Modifiers
3.1. IGSF4
3.2. LARP2
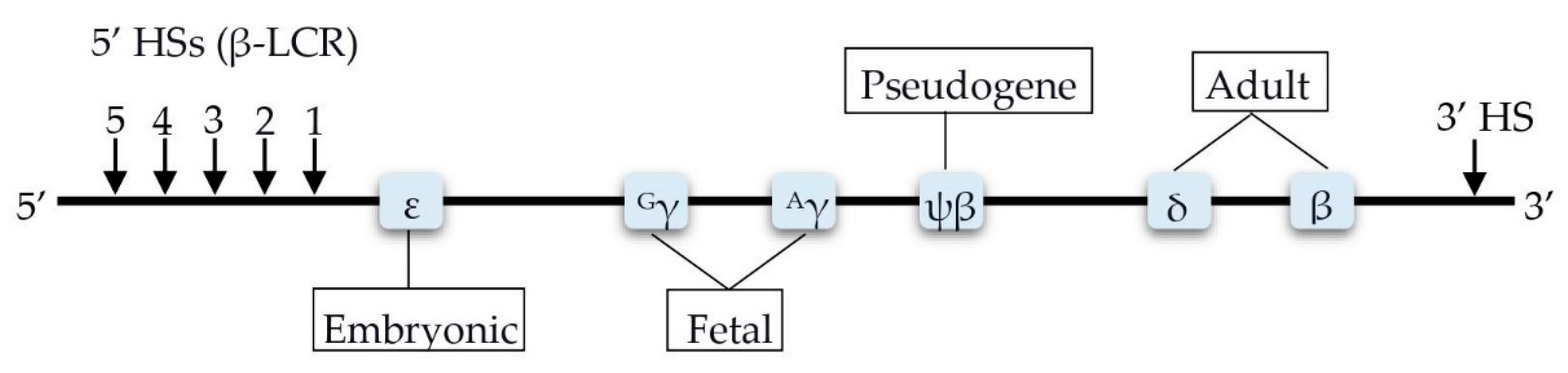
4. Factors Involved in the Transcription Control of the HBB Locus
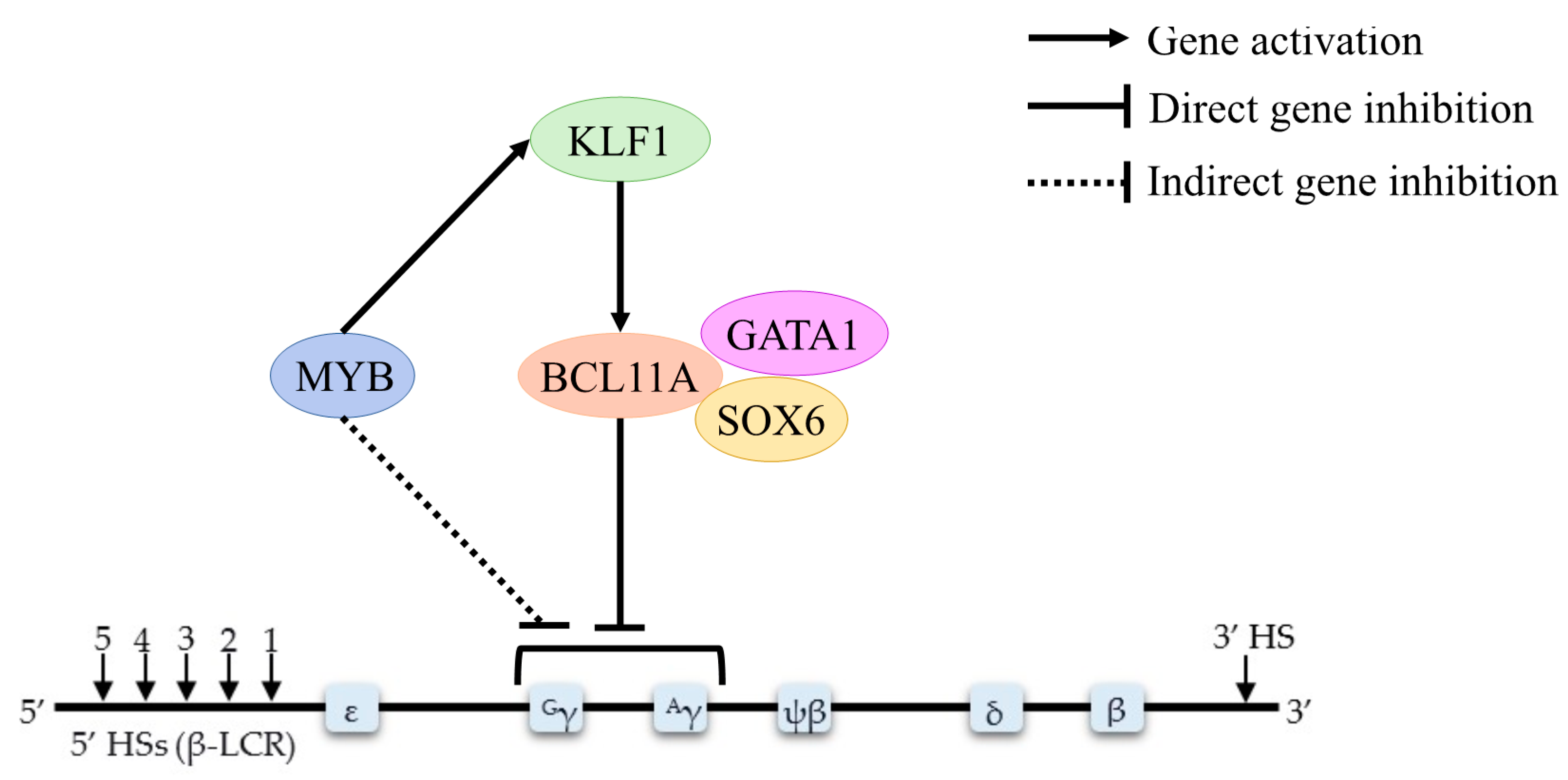
4.1. BCL11A
4.2. HBS1L-MYB
4.3. KLF1
4.4. GATA1
4.5. FLT1
4.6. BACH1
5. Genetic Findings
5.1. HBG2-Xmn1
5.2. α–Thalassemia Coinheritance
5.3. ARG2
5.4. NOS2A
5.5. MAP3K5
6. Putative Targets of Therapeutic Interventions
6.1. NRF2
6.2. PRDX1
6.3. PRDX2
6.4. TRX1
6.5. SOD1
7. Possible Targets for Gene Therapy
SOX6
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sabath, D.E. Molecular diagnosis of thalassemias and hemoglobinopathies: An ACLPS critical review. Am. J. Clin. Pathol. 2017, 148, 6–15. [Google Scholar] [CrossRef]
- Yaacob, N.S.C.; Islam, M.A.; Alsaleh, H.; Ibrahim, I.K.; Hassan, R. Alpha-hemoglobin-stabilizing protein (AHSP): A modulatory factor in β–thalassemia. Int. J. Hematol. 2020, 111, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.R.; Galanello, R.; Pennell, D.J.; Cunningham, M.J.; Vichinsky, E. Thalassemia. Hematology 2004, 2004, 14–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, A.; Galanello, R. Beta-thalassemia. Genet. Med. 2010, 12, 61–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivieri, N.F.; Pakbaz, Z.; Vichinsky, E. Hb E/beta-thalassaemia: A common & clinically diverse disorder. Indian J. Med. Res. 2011, 134, 522. [Google Scholar]
- Maryami, F.; Azarkeivan, A.; Fallah, M.S.; Zeinali, S. A large cohort study of genotype and phenotype correlations of beta-thalassemia in Iranian population. Int. J. Hematol. Oncol. Stem Cell Res. 2015, 9, 198–202. [Google Scholar]
- Mettananda, S.; Higgs, D.R. Molecular basis and genetic modifiers of thalassemia. Hematol./Oncol. Clin. 2018, 32, 177–191. [Google Scholar] [CrossRef]
- Kiefer, C.M.; Hou, C.; Little, J.A.; Dean, A. Epigenetics of β–globin gene regulation. Mutat. Res. 2008, 647, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Thein, S.L. The emerging role of fetal hemoglobin induction in non-transfusion-dependent thalassemia. Blood Rev. 2012, 26, 35–39. [Google Scholar] [CrossRef]
- Levings, P.P.; Bungert, J. The human β-globin locus control region. Eur. J. Biochem. 2002, 269, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadhouders, R.; Aktuna, S.; Thongjuea, S.; Aghajanirefah, A.; Pourfarzad, F.; van IJcken, W.; Lenhard, B.; Rooks, H.; Best, S.; Menzel, S. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J. Clin. Investig. 2014, 124, 1699–1710. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Liu, K.; Sun, C.-W.; Pawlik, K.M.; Townes, T.M. KLF1 regulates BCL11A expression and γ-to β–globin gene switching. Nat. Genet. 2010, 42, 742–744. [Google Scholar] [CrossRef]
- Ahmadvand, M.; Noruzinia, M.; Fard, A.D.; Zohour, M.M.; Tabatabaiefar, M.A.; Soleimani, M.; Kaviani, S.; Abroun, S.; Beiranvand, S.; Saki, N. The role of epigenetics in the induction of fetal hemoglobin: A combination therapy approach. Int. J. Hematol. Oncol. Stem Cell Res. 2014, 8, 9–14. [Google Scholar] [PubMed]
- Bao, X.; Zuo, Y.; Chen, D.; Zhao, C. DNA methylation patterns of β–globin cluster in β–thalassemia patients. Clin. Epigenet. 2020, 12, 187. [Google Scholar] [CrossRef] [PubMed]
- Fard, A.D.; Kaviani, S.; Noruzinia, M.; Soleimani, M.; Abroun, S.; Chegeni, R.; Hajifathali, A.; Zonoubi, Z.; Ahmadvand, M.; Mohammadi, M.M.; et al. Evaluation of H3 histone methylation and colony formation in erythroid progenitors treated with thalidomide and sodium butyrate. Lab. Hematol. 2013, 19, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Mettananda, S.; Yasara, N.; Fisher, C.A.; Taylor, S.; Gibbons, R.; Higgs, D. Synergistic silencing of α-globin and induction of γ-globin by histone deacetylase inhibitor, vorinostat as a potential therapy for β–thalassaemia. Sci. Rep. 2019, 9, 11649. [Google Scholar] [CrossRef] [PubMed]
- Voskou, S.; Phylactides, M.; Afantitis, A.; Melagraki, G.; Tsoumanis, A.; Koutentis, P.A.; Mitsidi, T.; Mirallai, S.I.; Kleanthous, M. MS-275 Chemical Analogues Promote Hemoglobin Production and Erythroid Differentiation of K562 Cells. Hemoglobin 2019, 43, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Breton, A.; Theodorou, A.; Aktuna, S.; Sonzogni, L.; Darling, D.; Chan, L.; Menzel, S.; van der Spek, P.J.; Swagemakers, S.M.; Grosveld, F. ASH 1L (a histone methyltransferase protein) is a novel candidate globin gene regulator revealed by genetic study of an English family with beta-thalassaemia unlinked to the beta-globin locus. Br. J. Haematol. 2016, 175, 525–530. [Google Scholar] [CrossRef]
- Mettananda, S.; Fisher, C.A.; Sloane-Stanley, J.A.; Taylor, S.; Oppermann, U.; Gibbons, R.J.; Higgs, D.R. Selective silencing of α-globin by the histone demethylase inhibitor IOX1: A potentially new pathway for treatment of β–thalassemia. Haematologica 2017, 102, e80. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Chen, T.; Ijaz, H.; Cho, E.H.; Steinberg, M.H. SIRT1 activates the expression of fetal hemoglobin genes. Am. J. Hematol. 2017, 92, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, T.; Nie, Y.; Hu, H.; Liang, Z. Hypermethylation of IGSF4 gene for noninvasive prenatal diagnosis of thalassemia. Med. Sci. Monit. 2012, 18, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohd Yassim, H.; Abdullah, W.Z.; Johan, M.F. DNA Methylation of IGSF4 gene as an Epigenetic Modifier in HbE/β–Thalassaemia. J. Biomed. Clin. Sci. 2017, 2, 17–19. [Google Scholar]
- Mohd Yassim, H.; Sudin, A.; Nasir, A.; Abdullah, W.Z.; Johan, M.F. Methylation Status Of LARP2 and IGSF4 Gene Promoter Region In Hb E/Β–Thalassemia And Β–Thalassemia Major Patients. Asian J. Med. Biomed. 2018, 10 (Suppl. 2). Available online: https://journal.unisza.edu.my/ajmb/index.php/ajmb/article/view/151 (accessed on 29 December 2020).
- Gao, T.; Nie, Y.; Guo, J. Hypermethylation of the gene LARP2 for noninvasive prenatal diagnosis of β–thalassemia based on DNA methylation profile. Mol. Biol. Rep. 2012, 39, 6591–6598. [Google Scholar] [CrossRef]
- Haiyuni, M.Y.; Aziee, S.; Nasir, A.; Abdullah, W.Z.; Johan, M.F. LARP2 DNA methylation in transfusion-dependent haemoglobin E-beta (HBE/β) and β–thalassaemia major patients. Malays. J. Med. Health Sci. 2019, 15, 46–53. [Google Scholar]
- Li, J.; Lai, Y.; Shi, L. BCL11A down-regulation induces γ-globin in human β–thalassemia major erythroid cells. Hemoglobin 2018, 42, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Xu, J.; Ragoczy, T.; Ippolito, G.C.; Walkley, C.R.; Maika, S.D.; Fujiwara, Y.; Ito, M.; Groudine, M.; Bender, M. Developmental and species-divergent globin switching are driven by BCL11A. Nature 2009, 460, 1093–1097. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Peng, C.; Sankaran, V.G.; Shao, Z.; Esrick, E.B.; Chong, B.G.; Ippolito, G.C.; Fujiwara, Y.; Ebert, B.L.; Tucker, P.W. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 2011, 334, 993–996. [Google Scholar] [CrossRef] [Green Version]
- Roosjen, M.; McColl, B.; Kao, B.; Gearing, L.J.; Blewitt, M.E.; Vadolas, J. Transcriptional regulators Myb and BCL11A interplay with DNA methyltransferase 1 in developmental silencing of embryonic and fetal beta-like globin genes. FASEB J. 2014, 28, 1610–1620. [Google Scholar] [CrossRef] [Green Version]
- Lessard, S.; Beaudoin, M.; Benkirane, K.; Lettre, G. Comparison of DNA methylation profiles in human fetal and adult red blood cell progenitors. Genome Med. 2015, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rujito, L.; Basalamah, M.; Siswandari, W.; Setyono, J.; Wulandari, G.; Mulatsih, S.; Sofro, A.S.M.; Sadewa, A.H.; Sutaryo, S. Modifying effect of XmnI, BCL11A, and HBS1L-MYB on clinical appearances: A study on β–thalassemia and hemoglobin E/β–thalassemia patients in Indonesia. Hematol. Oncol. Stem Cell Ther. 2016, 9, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasing, W.; Mekki, C.; Traisathit, P.; Pissard, S.; Pornprasert, S. Genotyping of BCL11A and HBS1L-MYB Single Nucleotide Polymorphisms in β–thalassemia/HbE and Homozygous HbE Subjects with Low and High Levels of HbF. Walailak J. Sci. Technol. 2018, 15, 627–636. [Google Scholar] [CrossRef]
- Nuinoon, M.; Makarasara, W.; Mushiroda, T.; Setianingsih, I.; Wahidiyat, P.A.; Sripichai, O.; Kumasaka, N.; Takahashi, A.; Svasti, S.; Munkongdee, T. A genome-wide association identified the common genetic variants influence disease severity in β 0-thalassemia/hemoglobin E. Hum. Genet. 2010, 127, 303–314. [Google Scholar] [CrossRef]
- Fornari, T.A.; Lanaro, C.; Albuquerque, D.M.; Ferreira, R.; Costa, F.F. Featured Article: Modulation of fetal hemoglobin in hereditary persistence of fetal hemoglobin deletion type-2, compared to Sicilian δ β–thalassemia, by BCL11A and SOX6-targeting microRNAs. Exp. Biol. Med. 2017, 242, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Mahdavi, M.R.; Pourfarzad, F.; Kosaryan, M.; Akbari, M.T. In Vitro Hb Production in B-thalassemia Patients Is Not a Predictor of Clinical Responsiveness to Hydroxyurea. Iran. J. Public Health 2017, 46, 948. [Google Scholar]
- Chu, N.-L.; Wu, Z.-K.; Zhang, X.-H.; Fang, S.-P.; Wang, W.-J.; Cheng, Y.-L. Molecular mechanism of Yisui Shengxue Granule, a complex Chinese medicine, on thalassemia patients suffering from hemolysis and anemia of erythrocytes. Evid.-Based Complement. Altern. Med. 2014, 2014, 213782. [Google Scholar] [CrossRef]
- Thein, S.L. Molecular basis of β thalassemia and potential therapeutic targets. Blood Cells Mol. Dis. 2018, 70, 54–65. [Google Scholar] [CrossRef]
- Fanis, P.; Kousiappa, I.; Phylactides, M.; Kyrri, A.; Hadjigavriel, M.; Christou, S.; Sitarou, M.; Kleanthous, M. A novel mutation in the erythroid transcription factor KLF1 is likely responsible for ameliorating β–thalassemia major. Hum. Mutat. 2019, 40, 1768–1780. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; He, Y.; Zhou, J.-Y.; Xie, X.-M.; Li, J.; Li, R.; Liao, C.; Li, D.-Z. KLF1 gene mutations in Chinese adults with increased fetal hemoglobin. Hemoglobin 2013, 37, 501–506. [Google Scholar] [CrossRef]
- Gallienne, A.E.; Dréau, H.M.; Schuh, A.; Old, J.M.; Henderson, S. Ten novel mutations in the erythroid transcription factor KLF1 gene associated with increased fetal hemoglobin levels in adults. Haematologica 2012, 97, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Zhang, X.; Yu, L.; Cai, R.; Ma, X.; Zheng, C.; Zhou, Y.; Liu, Q.; Wei, X.; Lin, L. KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of β–thalassemia. Blood 2014, 124, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, P.; Colah, R.; Ghosh, K.; Nadkarni, A. Differential role of Kruppel like factor 1 (KLF1) gene in red blood cell disorders. Genomics 2019, 111, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
- Borgio, J.F.; AbdulAzeez, S.; Al-Muslami, A.M.; Naserullah, Z.A.; Al-Jarrash, S.; Al-Suliman, A.M.; Al-Madan, M.S.; Al-Ali, A.K. KLF1 gene and borderline hemoglobin A2 in Saudi population. Arch. Med. Sci. 2018, 14, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinjamur, D.S.; Alhashem, Y.N.; Mohamad, S.F.; Amin, P.; Williams, D.C., Jr.; Lloyd, J.A. Krüppel-like transcription factor KLF1 is required for optimal γ-and β–globin expression in human fetal erythroblasts. PLoS ONE 2016, 11, e0146802. [Google Scholar] [CrossRef] [PubMed]
- Khamphikham, P.; Sripichai, O.; Munkongdee, T.; Fucharoen, S.; Tongsima, S.; Smith, D.R. Genetic variation of Krüppel-like factor 1 (KLF1) and fetal hemoglobin (HbF) levels in β 0-thalassemia/HbE disease. Int. J. Hematol. 2018, 107, 297–310. [Google Scholar] [CrossRef]
- Gutiérrez, L.; Caballero, N.; Fernández-Calleja, L.; Karkoulia, E.; Strouboulis, J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life 2020, 72, 89–105. [Google Scholar] [CrossRef]
- Goren, A.; Simchen, G.; Fibach, E.; Szabo, P.E.; Tanimoto, K.; Chakalova, L.; Pfeifer, G.P.; Fraser, P.J.; Engel, J.D.; Cedar, H. Fine tuning of globin gene expression by DNA methylation. PLoS ONE 2006, 1, e46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wu, Z.-K. Molecular pharmacological basis of the YiSui ShenXu Granule in β–thalassemia therapy. J. Ethnopharmacol. 2008, 120, 437–441. [Google Scholar] [CrossRef]
- Kolliopoulou, A.; Siamoglou, S.; John, A.; Sgourou, A.; Kourakli, A.; Symeonidis, A.; Vlachaki, E.; Chalkia, P.; Theodoridou, S.; Ali, B.R. Role of genomic biomarkers in increasing fetal hemoglobin levels upon hydroxyurea therapy and in β–thalassemia intermedia: A validation cohort study. Hemoglobin 2019, 43, 27–33. [Google Scholar] [CrossRef]
- Lee, T.Y.; Muniandy, L.; Teh, L.K.; Abdullah, M.; George, E.; Sathar, J.; Lai, M.I. Correlation of BACH1 and Hemoglobin E/Beta-Thalassemia Globin Expression. Turk. J. Hematol. 2016, 33, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Sivalingam, M.; Looi, M.; Zakaria, S.Z.S.; Hamidah, N.; Alias, H.; Latiff, Z.A.; Ibrahim, H.; Jamal, R. Molecular study and genotype/phenotype correlation of β thalassemia in Malaysia. Int. J. Lab. Hematol. 2012, 34, 377–382. [Google Scholar] [CrossRef]
- Kalantri, S.A.; Ray, R.; Choudhuri, S.; Roy, S.; Bhattacharyya, M. Key Determinants of Phenotypic Heterogeneity of Hb E/β Thalassemia: A Comparative Study from Eastern India. Indian J. Hematol. Blood Transfus. 2019, 36, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zulkifli, M.M.; Yusof, W.; Azman, N.F.; Ab Hamid, S.A.; Adnan, W.N.A.W.; Othman, A.; Draman, N.; Zilfalil, B.A.; Hassan, R.; Abdullah, W.Z. Factors Affecting Health Related Quality of Life and Its Association with Xmn1 Gene Modifier among Transfusion-Dependent β–Thalassemia and HbE/β–Thalassemia Adolescent in East Coast Malaysia. Asian J. Med. Biomed. 2018, 19. Available online: https://journal.unisza.edu.my/ajmb/index.php/ajmb/article/view/119 (accessed on 29 December 2020).
- Prasing, W.; Odawara, T.; Traisathit, P.; Yamashiro, Y.; Hattori, Y.; Pornprasert, S. Analysis of the Xmn1-Gγ polymorphism in β–thalassemia/hemoglobin E (HbE) and homozygous HbE patients with low and high levels of HbF. Int. J. Lab. Hematol. 2015, 37, 25–28. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Nag, A.; Ghosh, K.; Ray, R.; Roy, K.; Bandyopadhyay, A.; Bhattacharyya, M. Genetic determinants related to pharmacological induction of foetal haemoglobin in transfusion-dependent HbE-β thalassaemia. Ann. Hematol. 2019, 98, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Ray, R.; Roy, K.; Bandyopadhyay, A.; Ghosh, K.; Bhattacharyya, M. Alpha Globin Gene Mutation: A Major Determinant of Hydroxyurea Response in Transfusion-Dependent HbE-β–Thalassaemia. Acta Haematol. 2019, 142, 132–141. [Google Scholar] [CrossRef]
- Sharma, V.; Saxena, R. Effect of [alpha]-gene numbers on phenotype of HbE/[beta] thalassemia patients. Ann. Hematol. 2009, 88, 1035. [Google Scholar] [CrossRef]
- Sripichai, O.; Munkongdee, T.; Kumkhaek, C.; Svasti, S.; Winichagoon, P.; Fucharoen, S. Coinheritance of the different copy numbers of α-globin gene modifies severity of β–thalassemia/Hb E disease. Ann. Hematol. 2008, 87, 375–379. [Google Scholar] [CrossRef]
- Keikhaei, B.; Yousefi, H.; Bahadoram, M. Clinical and haematological effects of hydroxyurea in β–thalassemia intermedia patients. J. Clin. Diagn. Res. JCDR 2015, 9, OM01. [Google Scholar] [CrossRef]
- Chalikiopoulou, C.; Tavianatou, A.-G.; Sgourou, A.; Kourakli, A.; Kelepouri, D.; Chrysanthakopoulou, M.; Kanelaki, V.-K.; Mourdoukoutas, E.; Siamoglou, S.; John, A. Genomic variants in the ASS1 gene, involved in the nitric oxide biosynthesis and signaling pathway, predict hydroxyurea treatment efficacy in compound sickle cell disease/β–thalassemia patients. Pharmacogenomics 2016, 17, 393–403. [Google Scholar] [CrossRef]
- Tafrali, C.; Paizi, A.; Borg, J.; Radmilovic, M.; Bartsakoulia, M.; Giannopoulou, E.; Giannakopoulou, O.; Stojiljkovic-Petrovic, M.; Zukic, B.; Poulas, K. Genomic variation in the MAP3K5 gene is associated with β–thalassemia disease severity and hydroxyurea treatment efficacy. Pharmacogenomics 2013, 14, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.J.; Duarte, T.L.; Arezes, J.; Garcia-Santos, D.; Hamdi, A.; Pasricha, S.-R.; Armitage, A.E.; Mehta, H.; Wideman, S.; Santos, A.G. Nrf2 controls iron homoeostasis in haemochromatosis and thalassaemia via Bmp6 and hepcidin. Nat. Metab. 2019, 1, 519–531. [Google Scholar] [CrossRef]
- Somparn, N.; Prawan, A.; Senggunprai, L.; Kukongviriyapan, U.; Jetsrisuparb, A.; Lee, M.-H.; Kim, D.-H.; Kukongviriyapan, V.; Surh, Y.-J. Cellular adaptation mediated through Nrf2-induced glutamate cysteine ligase up-regulation against oxidative stress caused by iron overload in β–thalassemia/HbE patients. Free Radic. Res. 2019, 53, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Romanello, K.S.; Teixeira, K.K.; Silva, J.P.M.; Nagamatsu, S.T.; Bezerra, M.A.C.; Domingos, I.F.; Martins, D.A.; Araujo, A.S.; Lanaro, C.; Breyer, C.A. Global analysis of erythroid cells redox status reveals the involvement of Prdx1 and Prdx2 in the severity of beta thalassemia. PLoS ONE 2018, 13, e0208316. [Google Scholar] [CrossRef]
- De Franceschi, L.; Bertoldi, M.; De Falco, L.; Franco, S.S.; Ronzoni, L.; Turrini, F.; Colancecco, A.; Camaschella, C.; Cappellini, M.D.; Iolascon, A. Oxidative stress modulates heme synthesis and induces peroxiredoxin-2 as a novel cytoprotective response in β–thalassemic erythropoiesis. Haematologica 2011, 96, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.-H.; Kim, S.-U.; Kwon, T.-H.; Lee, D.-S.; Ha, H.-L.; Park, D.-S.; Woo, E.-J.; Lee, S.-H.; Kim, J.-M.; Chae, H.-B. Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem. Biophys. Res. Commun. 2012, 426, 427–432. [Google Scholar] [CrossRef]
- Ozturk, Z.; Genc, G.; Kupesiz, A.; Kurtoglu, E.; Gumuslu, S. Thalassemia major patients using iron chelators showed a reduced plasma thioredoxin level and reduced thioredoxin reductase activity, despite elevated oxidative stress. Free Radic. Res. 2015, 49, 309–316. [Google Scholar] [CrossRef]
- Shariati, L.; Rohani, F.; Heidari Hafshejani, N.; Kouhpayeh, S.; Boshtam, M.; Mirian, M.; Rahimmanesh, I.; Hejazi, Z.; Modarres, M.; Pieper, I.L. Disruption of SOX6 gene using CRISPR/Cas9 technology for gamma-globin reactivation: An approach towards gene therapy of β–thalassemia. J. Cell. Biochem. 2018, 119, 9357–9363. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, Y.; Luo, J.; Luo, L.; Liu, R.; Liu, Z.; Zhao, W. SOX6 Downregulation Induces γ-Globin in Human β–Thalassemia Major Erythroid Cells. BioMed Res. Int. 2017, 2017, 9496058. [Google Scholar] [CrossRef] [Green Version]
- Modares Sadeghi, M.; Shariati, L.; Hejazi, Z.; Shahbazi, M.; Tabatabaiefar, M.A.; Khanahmad, H. Inducing indel mutation in the SOX6 gene by zinc finger nuclease for gamma reactivation: An approach towards gene therapy of beta thalassemia. J. Cell. Biochem. 2018, 119, 2512–2519. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Epigenetic Modifiers Involved | Study Type | Findings | References |
|---|---|---|---|
| DNA methylation | |||
| β–globin cluster | Experimental study | Hypomethylation in the CpG sites around the LCR HS4-HS3, γ– and β–globin promoter | [15] |
| Histone Modification | |||
| H3K27 | Experimental study | Thalidomide treatment markedly reduced the H3K27 methylation | [16] |
| γ–globin | Experimental study | Vorinostat induced γ–globin production and simultaneously suppressed α–globin expression | [17] |
| MS-275 | Experimental study | MS-275 analogues (MD48) highly induced HbF production | [18] |
| ASH1L | Experimental study | ASH1L expression increased, thus ASH1L binding on the β– and α–globin promoters was also increased | [19] |
| IOX1 | Experimental study | IOX1 down-regulated α– and α–like globin expression without affecting β–like globin expression | [20] |
| SIRT1 | Experimental study | SIRT1 knockdown decreased, and its overexpression increased the γ–globin gene | [21] |
| Other epigenetic modifiers | |||
| IGSF4 | Cohort study | IGSF4 promoter was fully and partially methylated in β–thalassemia major and HbE/β–thalassemia, respectively | [23] |
| Case-control study | High methylation of IGSF4 in β–thalassemia patients | [22] | |
| LARP2 | Cohort study | Partial methylation of LARP2 in HbE/β–thalassemia and β–thalassemia major patients | [24] |
| Cohort study | Up-regulation of LARP2 expression in β–thalassemia major | [26] | |
| Cohort study | Hypermethylation of LARP2 promoter region | [25] | |
| Factors involved in the transcription control of the HBB locus | |||
| BCL11A | Experimental study | Down-regulation of BCL11A via lentiviral RNA interference (RNAi) reactivated HbF expression | [27] |
| Experimental study | Double knockdowns of BCL11A and DNMT1 enhanced 90% expression of γ–globin | [30] | |
| Experimental study | Erythroblasts from bone-marrow had significant expression of BCL11A compared to fetal erythroblasts | [31] | |
| Cohort study | rs11886868 and rs766432 increase HbF level | [33,34] | |
| Cohort study | Up-regulation of 12 microRNAs targeting BCL11A gene, namely miR-21, miR-23b, miR-29a, miR-29b, miR-29c, miR-146a, miR-146b-5p, miR-148a, miR-148b, miR-128, miR-181a, and miR-590-5p may explain the down-regulation of BCL11A | [35] | |
| Experimental study | Hydroxyurea treatment significantly decreased the BCL11A expression | [36] | |
| Cohort study | A complex Chinese medicine, yisui shengxue granule, was demonstrated to down-regulate the BCL11A gene expression | [37] | |
| HBS1L-MYB | Experimental study | Double knockdowns of MYB and DNMT1 significantly induced ε–globin | [30] |
| Cohort study | rs9399137 had no significant effect in modifying HbF level or clinical appearance in both β–thalassemia and HbE/β–thalassemia | [32] | |
| Cohort study | rs9399137 frequencies were high in homozygous HbE subjects with high HbF levels | [33] | |
| Cohort study | rs9376092 was significantly associated with the HbE/β0-thalassemia severity | [34] | |
| KLF1 | Experimental study | KLF1 controls the globin gene switching by directly influence BCL11A level and γ–globin/β–globin expression ratios | [13] |
| Cohort study | KLF1 mutation significantly associated with high HbF levels | [40] | |
| Cohort study | 11 KLF1 mutations were observed in high HbF hemoglobinopathies patients but the mutations were not functionally defective KLF1 mutations | [41] | |
| Cohort study | KLF1 mutations were significantly higher in the patients from the endemic thalassemia region than in the non-endemic thalassemia region | [42] | |
| Cohort study | KLF1 mutation (NM_006563.4:c.968C>T) was suggested to ameliorate severe β–thalassemia genotype, especially homozygous IVS1-110 | [39] | |
| Cohort study | KLF1 and BCL11A were inversely correlated with γ–globin gene expression in patients with KLF1 gene mutations | [43] | |
| Cohort study | KLF1 gene variations were not significantly related to borderline HbA2 β–thalassemia carriers | [44] | |
| Experimental study | The amount of KLF1 expression is weakly positively correlated with BCL11A mRNA | [45] | |
| Experimental study | KLF1 knockdown decreases BCL11A expression and elevates HbF levels | [46] | |
| GATA1 | Experimental study | GATA1 was suggested to favor binding to the hypomethylated sites in fetal erythroblasts | [31] |
| Experimental study | No significant footprint was observed in adult erythroid cells and lymphocytes when the γA promoter is methylated | [48] | |
| Experimental study | Yisui shengxue granule decreased GATA1 and GATA2 expressions | [49] | |
| FLT1 | Cohort study | FLT1 gene SNP (rs2182008 (G>A)) was strongly associated with the elevation of HbF levels | [50] |
| BACH1 | Cohort study | BACH1, where it was significantly correlated with age, α–, β– and γ–globin gene expression levels and heme oxygenase-1 protein | [51] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakaria, N.A.; Islam, M.A.; Abdullah, W.Z.; Bahar, R.; Mohamed Yusoff, A.A.; Abdul Wahab, R.; Shamsuddin, S.; Johan, M.F. Epigenetic Insights and Potential Modifiers as Therapeutic Targets in β–Thalassemia. Biomolecules 2021, 11, 755. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050755
Zakaria NA, Islam MA, Abdullah WZ, Bahar R, Mohamed Yusoff AA, Abdul Wahab R, Shamsuddin S, Johan MF. Epigenetic Insights and Potential Modifiers as Therapeutic Targets in β–Thalassemia. Biomolecules. 2021; 11(5):755. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050755
Chicago/Turabian StyleZakaria, Nur Atikah, Md Asiful Islam, Wan Zaidah Abdullah, Rosnah Bahar, Abdul Aziz Mohamed Yusoff, Ridhwan Abdul Wahab, Shaharum Shamsuddin, and Muhammad Farid Johan. 2021. "Epigenetic Insights and Potential Modifiers as Therapeutic Targets in β–Thalassemia" Biomolecules 11, no. 5: 755. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11050755