
Parallel and Sequential Pathways of Molecular Recognition of a Tandem-Repeat Protein and Its Intrinsically Disordered Binding Partner


Abstract
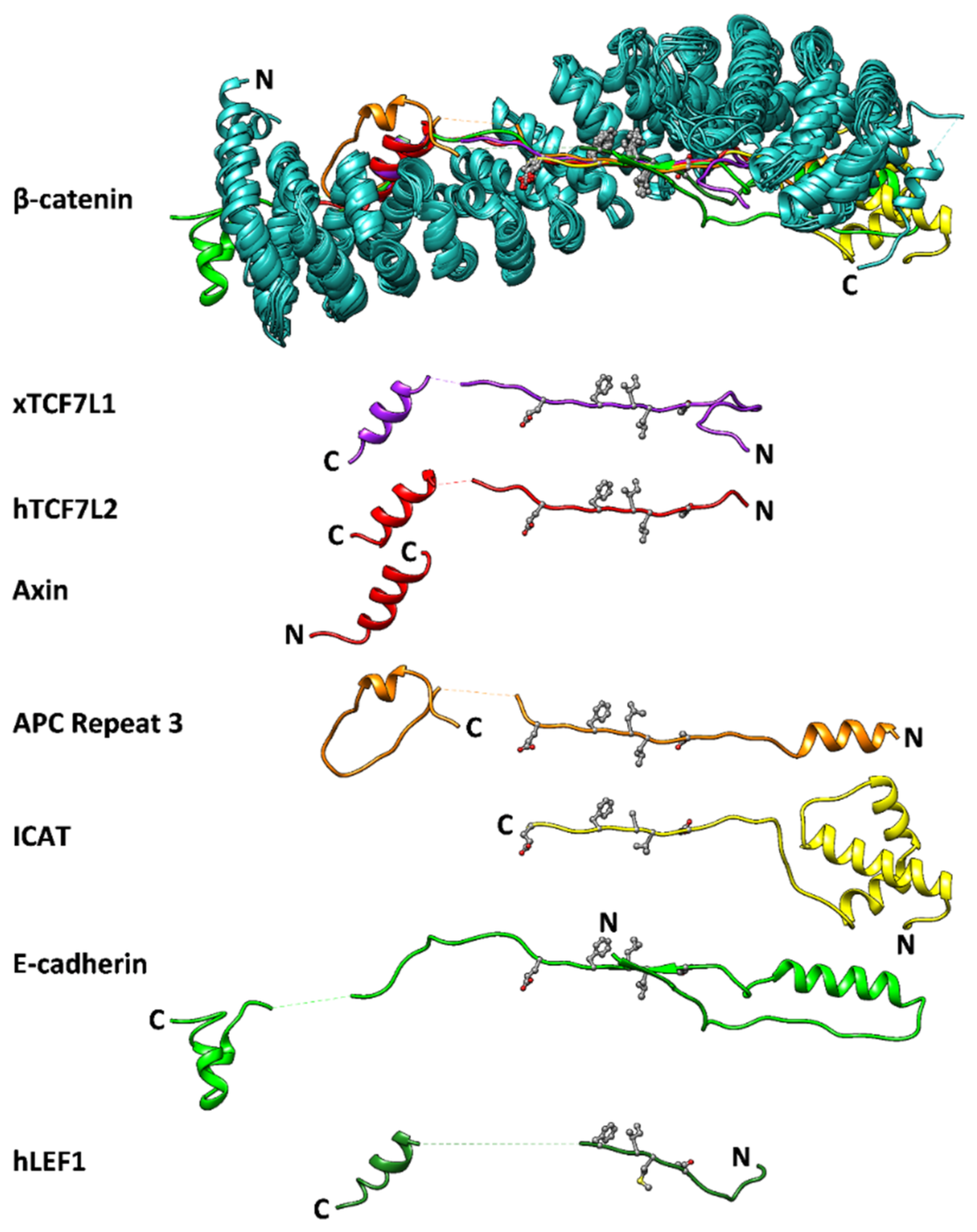
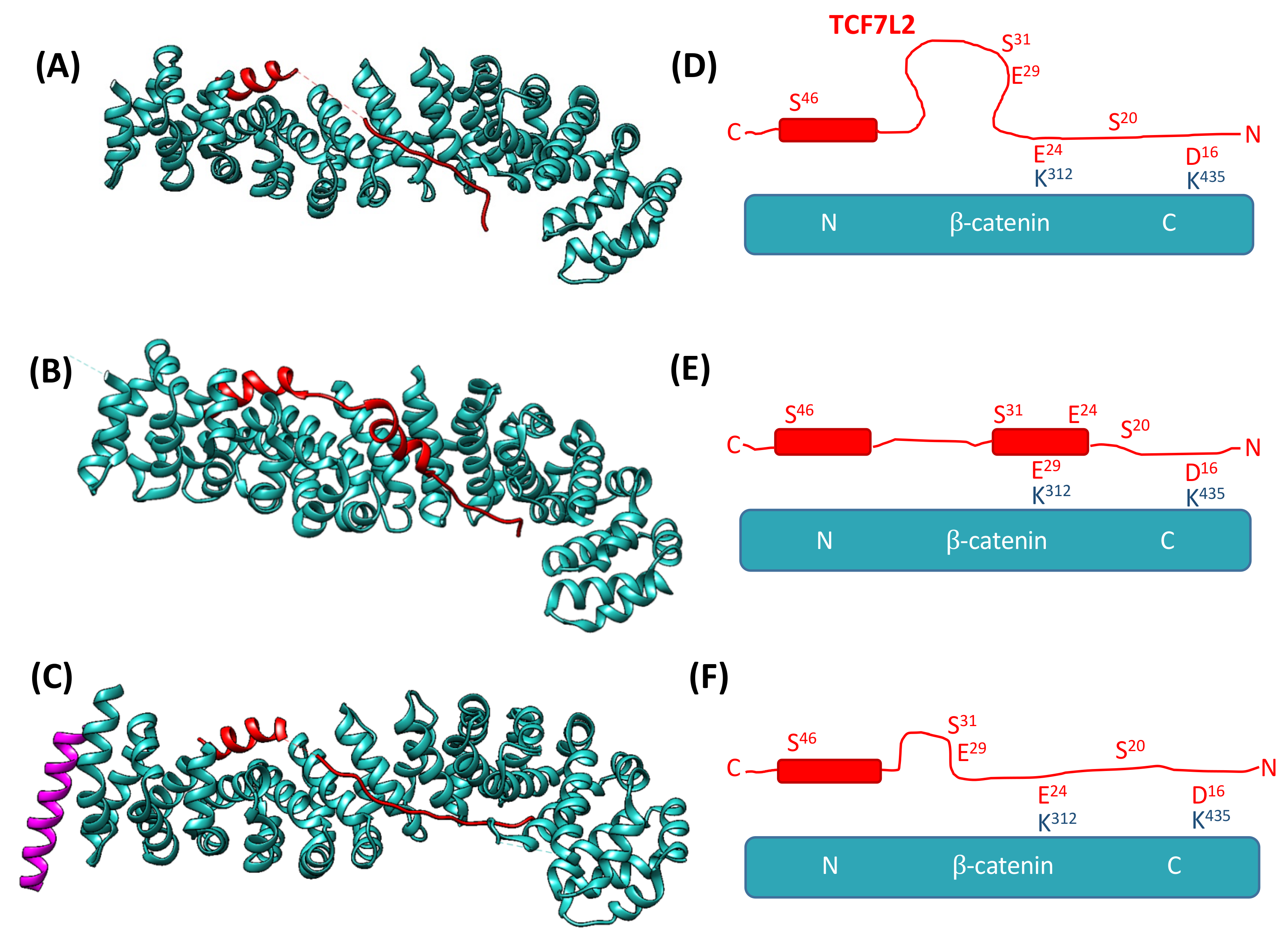
:1. Introduction
2. Materials and Methods
2.1. Molecular Biology and Protein Expression and Purification
2.2. Fluorescent Labelling of TCF7L2
2.3. Isothermal Titration Calorimetry (ITC)
2.4. Kinetic Experiments
3. Results
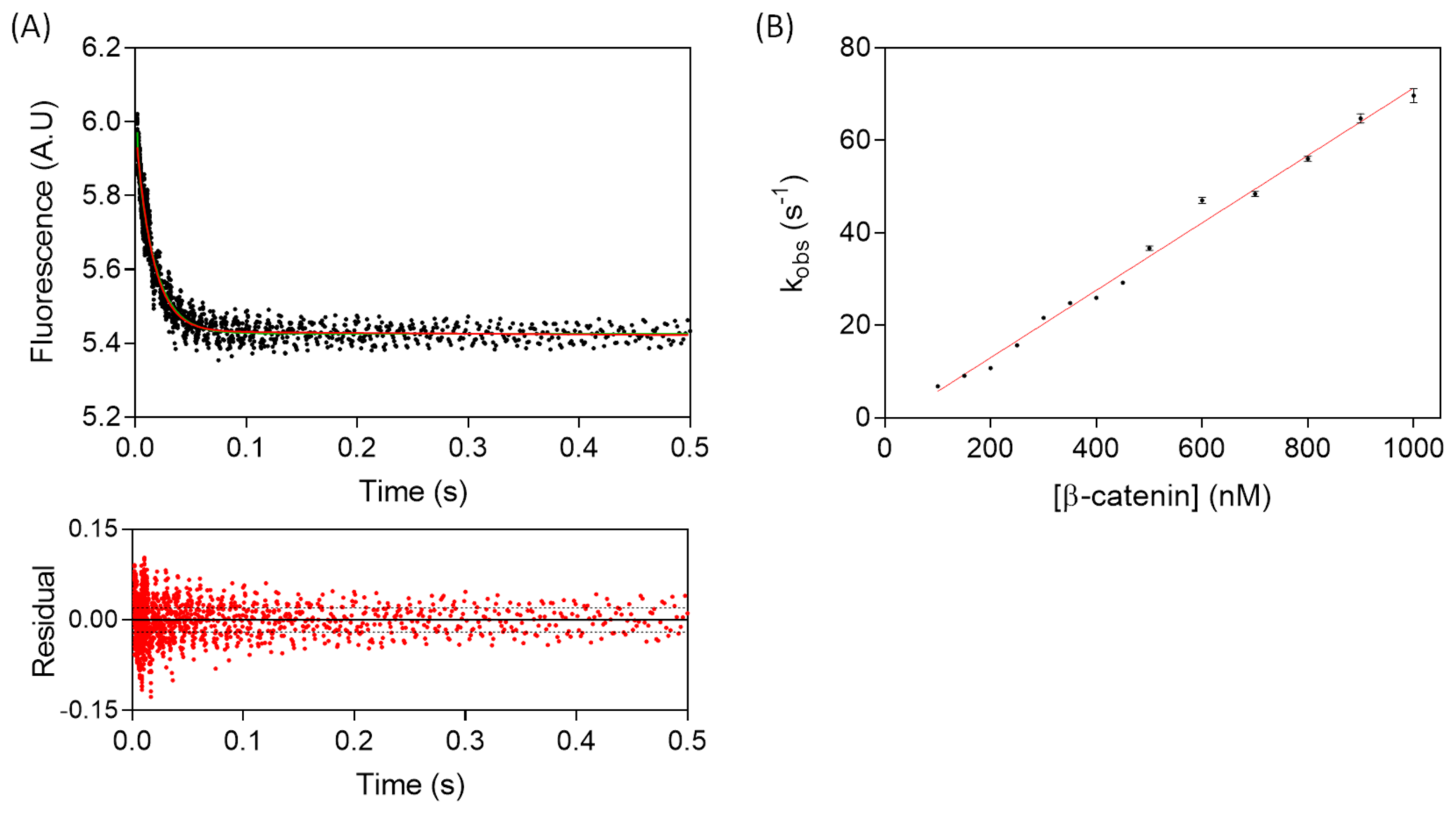
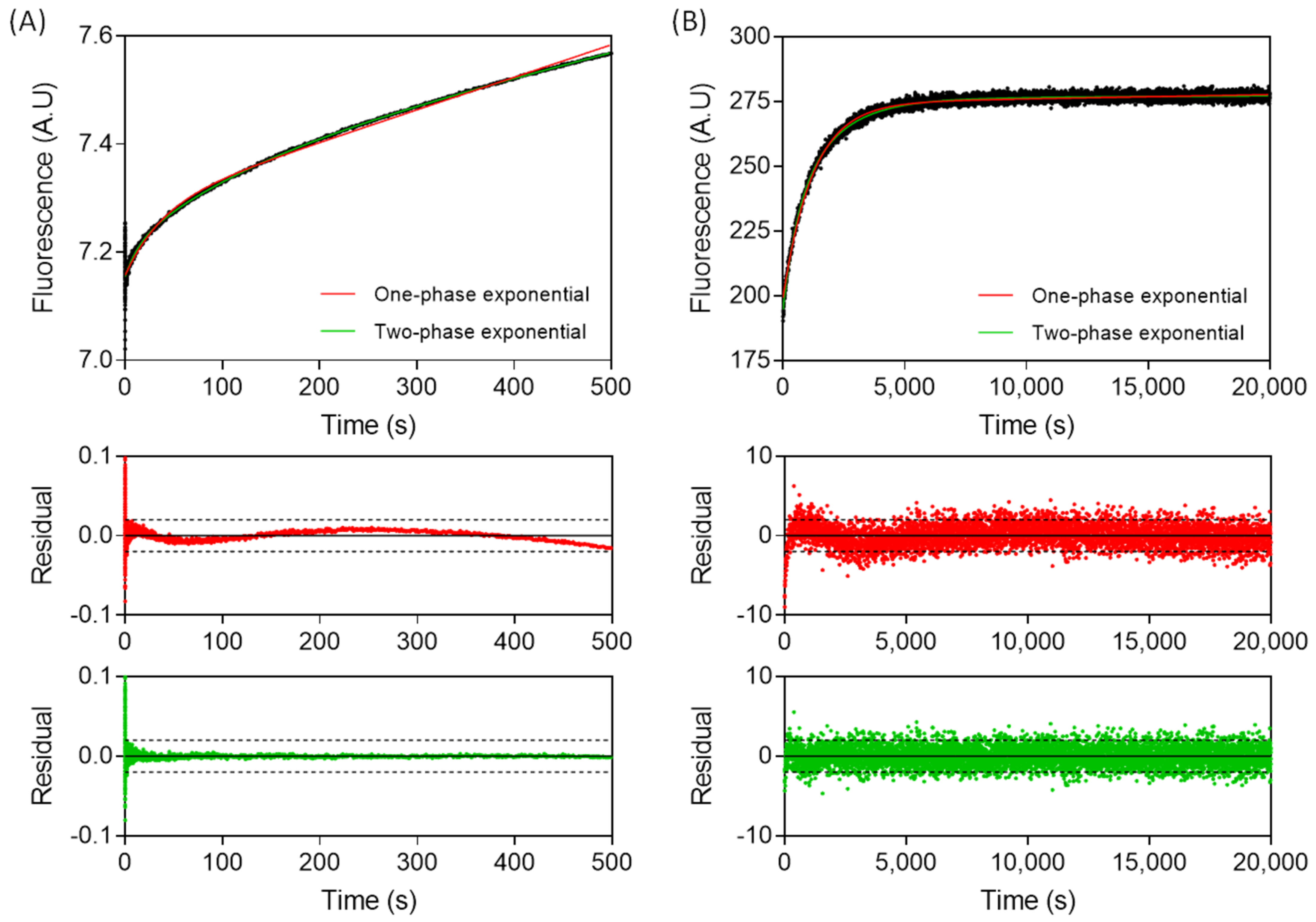
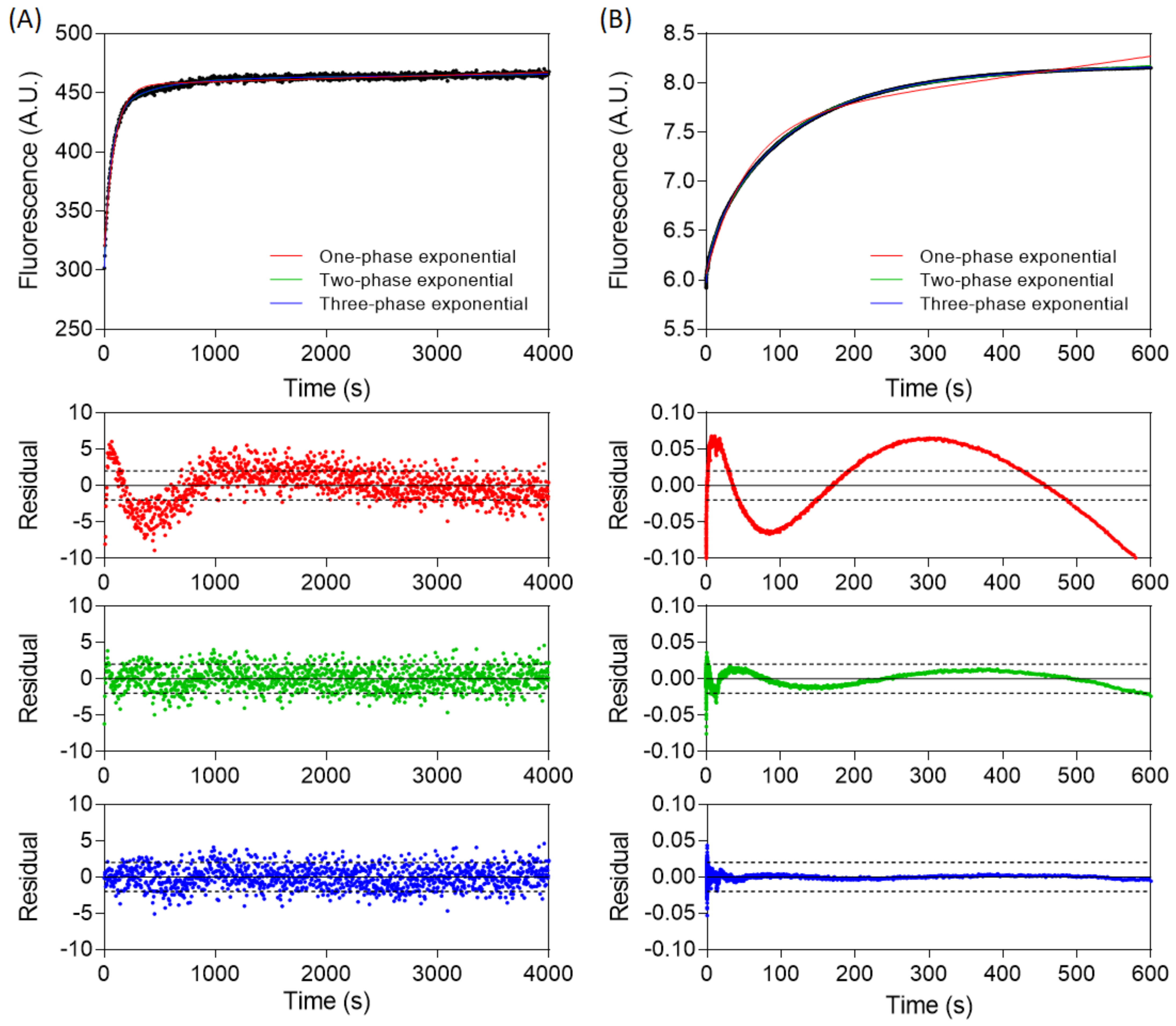
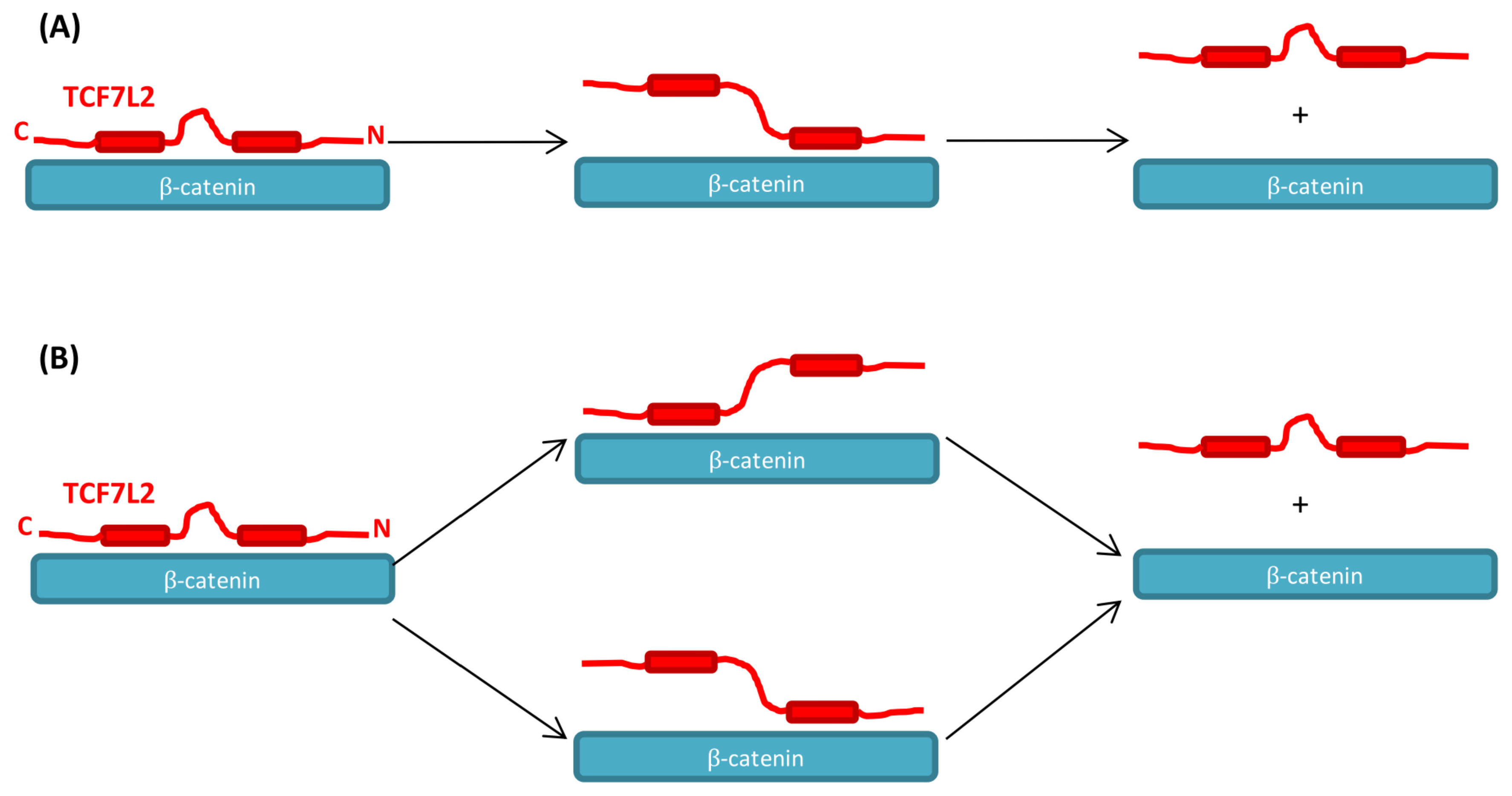
3.1. The Association of TCF7L2 and β-Catenin Is Monophasic, and the Dissociation of the Complex Is Biphasic
3.2. Alanine Scanning of TCF7L2 Reveals Three Distinct Interface Regions
3.3. Multi-Site Mutations in the Variable Region of TCF7L2 Do Not Perturb the Kinetics
3.4. TCF7L2 Mutants I19A and L21A Display More Complex Dissociation Kinetics
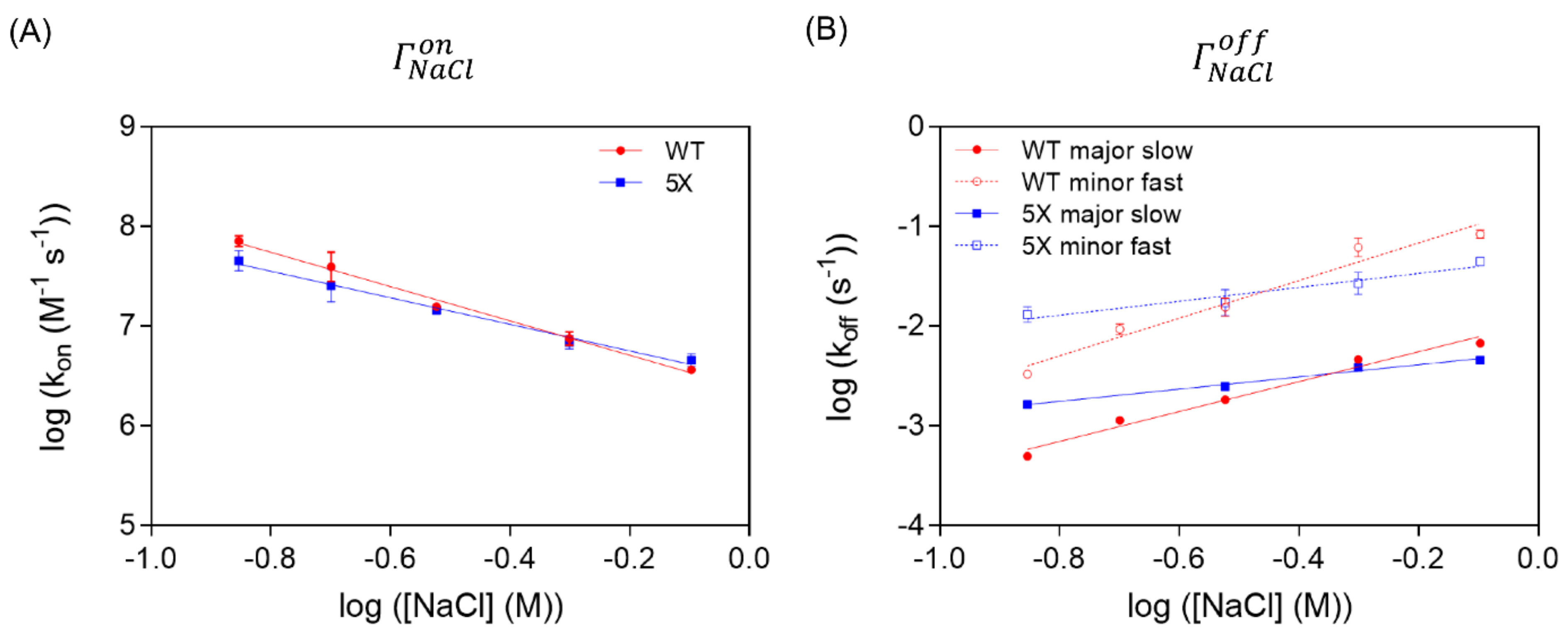
3.5. The Variable Region of TCF7L2 Is Very Sensitive to Ionic Strength
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Van Der Wal, T.; Van Amerongen, R. Walking the tight wire between cell adhesion and WNT signalling: A balancing act for β-catenin. Open Biol 2020, 10, 200267. [Google Scholar] [CrossRef] [PubMed]
- Arce, L.; Yokoyama, N.N.; Waterman, M.L. Diversity of LEF/TCF action in development and disease. Oncogene 2006, 25, 7492–7504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammons, M.; Bienz, M. Multiprotein complexes governing Wnt signal transduction. Curr. Opin. Cell Biol. 2018, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, C.; Hausmann, G.; Basler, K. β-Catenin hits Chromatin: Regulation of Wnt Target Gene Activation. Nat. Rev. Mol. Cell Biol. 2009, 10, 276–286. [Google Scholar] [CrossRef]
- Willert, K.; Jones, K.A. Wnt Signaling: Is the Party in the Nucleus? Genes Dev. 2006, 20, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, K.N.; Bonello, T.T.; Zhang, S.; Williams, C.E.; Roberts, D.M.; McKay, D.J.; Peifer, M. Supramolecular assembly of the beta-catenin destruction complex and the effect of Wnt signaling on its localization, molecular size, and activity in vivo. PLoS Genet. 2018, 14, e1007339. [Google Scholar] [CrossRef] [Green Version]
- Kimelman, D.; Xu, W. β-catenin Destruction Complex: Insights and Questions from a Structural Perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Xu, G.; Schulman, B.A.; Jeffrey, P.D.; Harper, J.W.; Pavletich, N.P. Structure of a β-TrCP1-Skp1-β-catenin complex: Destruction motif binding and lysine specificity of the SCFβ-TrCP1 ubiquitin ligase. Mol. Cell 2003, 11, 1445–1456. [Google Scholar] [CrossRef]
- Harris, T.J.C.; Tepass, U. Adherens Junctions: From Molecules to Morphogenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 502–514. [Google Scholar] [CrossRef]
- Huber, A.H.; Weis, W.I. The structure of beta-catenin/E-cadherin Complex and the Molecular Basis of Diverse Ligand Recognition by beta-catenin. Cell 2001, 105, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Pokutta, S.; Weis, W.I. Structure and Mechanism of Cadherins and Catenins in Cell-Cell Contacts. Annu. Rev. Cell Dev. Biol. 2007, 23, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Bugter, J.M.; Fenderico, N.; Maurice, M.M. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat. Rev. Cancer 2021, 21, 5–21. [Google Scholar] [CrossRef]
- Yoshida, R.; Kimura, N.; Harada, Y.; Ohuchi, N. The Loss of E-cadherin, α- and β-catenin Expression is Associated with Metastasis and Poor Prognosis in Invasive Breast Cancer. Int. J. Oncol. 2001, 18, 513–520. [Google Scholar] [CrossRef]
- Conacci-Sorrell, M.; Zhurinsky, J.; Ben-Ze’ev, A. The Cadherin-catenin Adhesion System in Signaling and Cancer. J. Clin. Invest. 2002, 109, 987–991. [Google Scholar] [CrossRef]
- Herzig, M.; Savarese, F.; Novatchkova, M.; Semb, H.; Christofori, G. Tumor Progression Induced by the Loss of E-cadherin Independent of β-catenin/Tcf-mediated Wnt Signaling. Oncogene 2007, 26, 2290–2298. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. The Many Ways of Wnt in Cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef]
- Huber, A.H.; Nelson, W.J.; Weis, W.I. Three-dimensional Structure of the Armadillo Repeat Region of β-catenin. Cell 1997, 90, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Takemaru, K.-I.; Liu, J.; Berndt, J.D.; Zheng, J.J.; Moon, R.T.; Xu, W. Crystal Structure of a Full-length β-catenin. Structure 2008, 16, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Eklof Spink, K.; Fridman, S.G.; Weis, W.I. Molecular Mechanisms of β-catenin Recognition by Adenomatous Polyposis Coli Revealed by the Structure of an APC-β-catenin Complex. EMBO J. 2001, 20, 6203–6212. [Google Scholar] [CrossRef]
- Graham, T.A.; Weaver, C.; Mao, F.; Kimelman, D.; Xu, W. Crystal Structure of a β-catenin/Tcf Complex. Cell 2000, 103, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Ha, N.C.; Tonozuka, T.; Stamos, J.L.; Choi, H.J.; Weis, W.I. Mechanism of phosphorylation-dependent binding of APC to β-catenin and its role in β-catenin degradation. Mol. Cell 2004, 15, 511–521. [Google Scholar] [CrossRef]
- Poy, F.; Lepourcelet, M.; Shivdasani, R.A.; Eck, M.J. Structure of a Human Tcf4-β-catenin Complex. Nat. Struct. Biol. 2001, 8, 1053–1057. [Google Scholar] [CrossRef]
- Xing, Y.; Clements, W.K.; Le Trong, I.; Hinds, T.R.; Stenkamp, R.; Kimelman, D.; Xu, W. Crystal Structure of a β-catenin/APC Complex Reveals a Critical Role for APC Phosphorylation in APC Function. Mol. Cell 2004, 15, 523–533. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera - A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampietro, J.; Dahlberg, C.L.; Cho, U.S.; Hinds, T.R.; Kimelman, D.; Xu, W. Crystal Structure of a β-catenin/BCL9/Tcf4 Complex. Mol. Cell 2006, 24, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Clements, W.K.; Kimelman, D.; Xu, W. Crystal Structure of a β-catenin/Axin Complex Suggests a Mechanism for the β-catenin Destruction Complex. Genes Dev. 2003, 17, 2753–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, T.A.; Clements, W.K.; Kimelman, D.; Xu, W. The Crystal Structure of the β-catenin/ICAT Complex Reveals the Inhibitory Mechanism of ICAT. Mol. Cell 2002, 10, 563–571. [Google Scholar] [CrossRef]
- Choi, H.-J.; Gross, J.C.; Pokutta, S.; Weis, W.I. Interactions of Plakoglobin and β-catenin with Desmosomal Cadherins. J. Biol. Chem. 2009, 284, 31776–31788. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Weis, W.I. Biochemical and structural characterization of β-catenin interactions with nonphosphorylated and CK2-phosphorylated Lef-1. J. Mol. Biol. 2011, 405, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt Signaling in the Nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4, a007906. [Google Scholar] [CrossRef]
- Schuijers, J.; Mokry, M.; Hatzis, P.; Cuppen, E.; Clevers, H. Wnt-induced transcriptional activation is exclusively mediated by TCF/LEF. EMBO J. 2014, 33, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Dodge, M.; Gundapaneni, D.; Michnoff, C.; Roth, M.; Lum, L. A Genome-wide RNAi Screen for Wnt/β-catenin Pathway Components Identifies Unexpected Roles for TCF Transcription Factors in Cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 9697–9702. [Google Scholar] [CrossRef] [Green Version]
- Hazra, A.; Fuchs, C.S.; Chan, A.T.; Giovannucci, E.L.; Hunter, D.J. Association of the TCF7L2 Polymorphism with Colorectal Cancer and Adenoma Risk. Cancer Causes Control 2008, 19, 975–980. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L.; Folsom, A.R.; Wolff, R.; Herrick, J.; Caan, B.J.; Potter, J.D. Transcription Factor 7-like 2 Polymorphism and Colon Cancer. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 978–982. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, J.; Rose, K.; Haghighi, E.B.; Lamprecht, C.; Rauen, G.; Freihen, V.; Kesselring, R.; Boerries, M.; Hecht, A. Loss of the nuclear Wnt pathway effector TCF7L2 promotes migration and invasion of human colorectal cancer cells. Oncogene 2020, 39, 3893–3909. [Google Scholar] [CrossRef] [Green Version]
- Vaquero, A.R.; Ferreira, N.E.; Omae, S.V.; Rodrigues, M.V.; Teixeira, S.K.; Krieger, J.E.; Pereira, A.C. Using Gene-network Landscape to Dissect Genotype Effects of TCF7L2 Genetic Variant on Diabetes and Cardiovascular Risk. Physiol. Genomics 2012, 44, 903–914. [Google Scholar] [CrossRef]
- Zhang, C.; Bao, W.; Rong, Y.; Yang, H.; Bowers, K.; Yeung, E.; Kiely, M. Genetic Variants and the Risk of Gestational Diabetes Mellitus: A Systematic Review. Hum. Reprod. Update 2013, 19, 376–390. [Google Scholar] [CrossRef] [Green Version]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Csizmok, V.; Follis, A.V.; Kriwacki, R.W.; Forman-Kay, J.D. Dynamic Protein Interaction Networks and New Structural Paradigms in Signaling. Chem. Rev. 2016, 116, 6424–6462. [Google Scholar] [CrossRef]
- Holmstrom, E.D.; Liu, Z.; Nettels, D.; Best, R.B.; Schuler, B. Disordered RNA chaperones can enhance nucleic acid folding via local charge screening. Nat. Commun. 2019, 10, 2453. [Google Scholar] [CrossRef]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev. 2014. [Google Scholar] [CrossRef] [PubMed]
- Schuler, B.; Borgia, A.; Borgia, M.B.; Heidarsson, O.; Holmstrom, E.D.; Nettels, D.; Sottini, A. Binding without folding-the biomolecular function of disordered polyelectrolyte complexes This review comes from a themed issue on Folding and binding. Curr. Opin. Struct. Biol. 2020, 60, 66–76. [Google Scholar] [CrossRef]
- Shammas, S.L. Mechanistic roles of protein disorder within transcription. Curr. Opin. Struct. Biol. 2017, 42, 155–161. [Google Scholar] [CrossRef]
- Tsafou, K.; Tiwari, P.B.; Forman-Kay, J.D.; Metallo, S.J.; Toretsky, J.A. Targeting Intrinsically Disordered Transcription Factors: Changing the Paradigm. J. Mol. Biol. 2018, 430, 2321–2341. [Google Scholar] [CrossRef]
- Uversky, V.N. Multitude of Binding Modes Attainable by Intrinsically Disordered Proteins: A Portrait Gallery of Disorder-based Complexes. Chem. Soc. Rev. 2011, 40, 1623–1634. [Google Scholar] [CrossRef]
- Mittag, T.; Kay, L.E.; Forman-Kay, J.D. Protein Dynamics and Conformational Disorder in Molecular Recognition. J. Mol. Recognit. 2009, 23, 105–116. [Google Scholar] [CrossRef]
- Borg, M.; Mittag, T.; Pawson, T.; Tyers, M.; Forman-Kay, J.D.; Chan, H.S. Polyelectrostatic Interactions of Disordered Ligands Suggest a Physical Basis for Ultrasensitivity. Proc. Natl. Acad. Sci. USA 2007, 104, 9650–9655. [Google Scholar] [CrossRef] [Green Version]
- Sigalov, A.B.; Kim, W.M.; Saline, M.; Stern, L.J. The Intrinsically Disordered Cytoplasmic Domain of the T Cell Receptor ζ Chain Binds to the Nef Protein of Simian Immunodeficiency Virus without a Disorder-to-order Transition? Biochemistry 2008, 47, 12942–12944. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.G.; Teilum, K.; Kragelund, B.B. Behaviour of Intrinsically Disordered Proteins in Protein-Protein Complexes with an Emphasis on Fuzziness. Cell. Mol. Life Sci. 2017, 74, 3175–3183. [Google Scholar] [CrossRef]
- Bachmann, A.; Wildemann, D.; Praetorius, F.; Fischer, G.; Kiefhaber, T.; Baldwin, R. Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proc. Natl. Acad. Sci. USA 2011, 108, 3952–3957. [Google Scholar] [CrossRef] [Green Version]
- Dogan, J.; Gianni, S.; Jemth, P. The Binding Mechanisms of Intrinsically Disordered Proteins. Phys. Chem. Chem. Phys. 2014, 16, 6323–6331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, E.; Paissoni, C.; Erkelens, A.M.; Tehranizadeh, Z.A.; Sorgenfrei, F.A.; Andersson, E.; Ye, W.; Camilloni, C.; Jemth, P. Mapping the transition state for a binding reaction between ancient intrinsically disordered proteins. J. Biol. Chem. 2020, 295, 17698–17712. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M.; Wong, C.T.; Clarke, J. Coupled Folding and Binding of the Disordered Protein PUMA Does Not Require Particular Residual Structure. J. Am. Chem. Soc. 2014, 136, 5197–5200. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009, 19, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Graham, T.A.; Ferkey, D.M.; Mao, F.; Kimelman, D.; Xu, W. Tcf4 can Specifically Recognize β-catenin using Alternative Conformations. Nat. Struct. Biol. 2001, 8, 1048–1052. [Google Scholar] [CrossRef]
- Daniels, D.L.; Weis, W.I. ICAT inhibits β-catenin binding to tcf/lef-family transcription factors and the general coactivator p300 using independent structural modules. Mol. Cell 2002, 10, 573–584. [Google Scholar] [CrossRef]
- Wiggers, F.; Wohl, S.; Dubovetskyi, A.; Rosenblum, G.; Zheng, W.; Hofmann, H. Diffusion of the disordered E-cadherin tail on β -catenin tail. bioRxiv 2021. [Google Scholar] [CrossRef]
- Miroux, B.; Walker, J.E. Over-production of proteins in Escherichia coli: Mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996, 260, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. The Proteomics Protocols Handbook—Chapter 52: Protein Identification and Analysis Tools on the ExPASy Server; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; ISBN 978-1-58829-343-5. [Google Scholar]
- Vivès, E.; Lebleu, B. One-pot labeling and purification of peptides and proteins with fluorescein maleimide. Tetrahedron Lett. 2003, 44, 5389–5391. [Google Scholar] [CrossRef]
- Omer, C.A.; Miller, P.J.; Diehl, R.E.; Kral, A.M. Identification of Tcf4 Residues Involved in High-Affinity β-catenin Binding. Biochem. Biophys. Res. Commun. 1999, 256, 584–590. [Google Scholar] [CrossRef]
- Knapp, S.; Zamai, M.; Volpi, D.; Nardese, V.; Avanzi, N.; Breton, J.; Plyte, S.; Flocco, M.; Marconi, M.; Isacchi, A.; et al. Thermodynamics of the high-affinity interaction of TCF4 with β-catenin. J. Mol. Biol. 2001, 306, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Gail, R.; Frank, R.; Wittinghofer, A. Systematic Peptide Array-based Delineation of the Differential β-catenin Interaction with Tcf4, E-cadherin, and Adenomatous Polyposis Coli. J. Biol. Chem. 2005, 280, 7107–7117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Kimelman, D. Mechanistic Insights from Structural Studies of β-catenin and Its Binding Partners. J. Cell Sci. 2007, 120, 3337–3344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemes, L.B.; Sánchez, I.E.; de Prat-Gay, G. Kinetic Recognition of the Retinoblastoma Tumor Suppressor by a Specific Protein Target. J. Mol. Biol. 2011, 412, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Grucza, R.A.; Bradshaw, J.M.; Mitaxov, V.; Waksman, G. Role of Electrostatic Interactions in SH2 Domain Recognition: Salt-Dependence of Tyrosyl-Phosphorylated Peptide Binding to the Tandem SH2 Domain of the Syk Kinase and the Single SH2 Domain of the Src Kinase. Biochemistry 2000, 39, 10072–10081. [Google Scholar] [CrossRef] [PubMed]
- Vindigni, A.; White, C.E.; Komives, E.A.; Di Cera, E. Energetics of Thrombin−Thrombomodulin Interaction. Biochemistry 1997, 36, 6674–6681. [Google Scholar] [CrossRef]
- Fuxreiter, M. Classifying the binding modes of disordered proteins. Int. J. Mol. Sci. 2020, 21, 8615. [Google Scholar] [CrossRef]
- Toto, A.; Malagrinò, F.; Visconti, L.; Troilo, F.; Pagano, L.; Brunori, M.; Jemth, P.; Gianni, S. Templated folding of intrinsically disordered proteins. J. Biol. Chem. 2020, 295, 6586–6593. [Google Scholar] [CrossRef] [Green Version]
- Rudeen, A.J.; Douglas, J.T.; Xing, M.; McDonald, W.H.; Lamb, A.L.; Neufeld, K.L. The 15-Amino Acid Repeat Region of Adenomatous Polyposis Coli Is Intrinsically Disordered and Retains Conformational Flexibility upon Binding β-Catenin. Biochemistry 2020, 59, 4039–4050. [Google Scholar] [CrossRef]
- Hemsath, L.; Dvorsky, R.; Fiegen, D.; Carlier, M.F.; Ahmadian, M.R. An electrostatic steering mechanism of Cdc42 recognition by Wiskott-Aldrich syndrome proteins. Mol. Cell 2005, 20, 313–324. [Google Scholar] [CrossRef]
- Japrung, D.; Dogan, J.; Freedman, K.J.; Nadzeyka, A.; Bauerdick, S.; Albrecht, T.; Kim, M.J.; Jemth, P.; Edel, J.B. Single-Molecule Studies of Intrinsically Disordered Proteins Using Solid-State Nanopores. Anal. Chem. 2013, 85, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M.; Steward, A.; Clarke, J. Folding and Binding of an Intrinsically Disordered Protein: Fast, but Not ‘Diffusion-limited’. J. Am. Chem. Soc. 2013, 135, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, D.U.; Cho, S.S.; Komives, E.A.; Wolynes, P.G. The energy landscape of modular repeat proteins: Topology determines folding mechanism in the ankyrin family. J. Mol. Biol. 2005, 354, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, D.U.; Walczak, A.M.; Komives, E.A.; Wolynes, P.G. The Energy Landscapes of Repeat-Containing Proteins: Topology, Cooperativity, and the Folding Funnels of One-Dimensional Architectures. PLoS Comput. Biol. 2008, 4, 1000070. [Google Scholar] [CrossRef]
- Hutton, R.D.; Wilkinson, J.; Faccin, M.; Sivertsson, E.M.; Pelizzola, A.; Lowe, A.R.; Bruscolini, P.; Itzhaki, L.S. Mapping the Topography of a Protein Energy Landscape. J. Am. Chem. Soc. 2015, 137, 14610–14625. [Google Scholar] [CrossRef] [Green Version]
- Lowe, A.R.; Itzhaki, L.S. Rational redesign of the folding pathway of a modular protein. Proc. Natl. Acad. Sci. USA 2007, 104, 2679–2684. [Google Scholar] [CrossRef] [Green Version]
- Tsytlonok, M.; Sormanni, P.; Rowling, P.J.E.; Vendruscolo, M.; Itzhaki, L.S. Subdomain architecture and stability of a giant repeat protein. J. Phys. Chem. B 2013, 117, 13029–13037. [Google Scholar] [CrossRef]
- Werbeck, N.D.; Rowling, P.J.E.; Chellamuthu, V.R.; Itzhaki, L.S. Shifting transition states in the unfolding of a large ankyrin repeat protein. Proc. Natl. Acad. Sci. USA 2008, 105, 9982–9987. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, M.O.; Oliveberg, M. Malleability of protein folding pathways: A simple reason for complex behaviour. Curr. Opin. Struct. Biol. 2007, 17, 21–29. [Google Scholar] [CrossRef]
- Oliveberg, M.; Wolynes, P.G. The experimental survey of protein-folding energy landscapes. Q. Rev. Biophys. 2005, 38, 245–288. [Google Scholar] [CrossRef]
- Otzen, D.E.; Fersht, A.R. Folding of circular and permuted chymotrypsin inhibitor 2: Retention of the folding nucleus. Biochemistry 1998, 37, 8139–8146. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCF7L2 Variant | Association | Dissociation | ||
|---|---|---|---|---|
| kon (×107 M−1·s−1) | koff,major (×10−4 s−1) | koff,minor (×10−3 s−1) | Relative Amplitude of Minor Phase% | |
| WT | 7.33 ± 0.14 | 5.73 ± 0.40 | 1.52 ± 0.28 | 23 ± 3 |
| L12A | 7.19 ± 0.13 | 9.77 ± 0.75 | 2.64 ± 0.20 | 19 ± 12 |
| N15A | 6.25 ± 0.23 | 4.42 ± 0.15 | 3.65 ± 0.83 | 18 ± 2 |
| E17A | 6.30 ± 0.42 | 89.1 ± 2.9 | 28.3 ± 1.9 | 28 ± 4 |
| I19A * | 7.04 ± 0.52 | 110 ± 0.5 | 207 ± 10 | 15 ± 0.1 |
| F21A * | 4.82 ± 0.17 | 258 ± 2 | 423 ± 15 | 20 ± 0.1 |
| D23A | 5.31 ± 0.25 | 3.00 ± 0.16 | 2.39 ± 0.70 | 15 ± 1 |
| E24A | 6.05 ± 0.12 | 5.03 ± 0.40 | 1.53 ± 0.21 | 26 ± 1 |
| E26A | 5.59 ± 0.22 | 4.74 ± 0.30 | 1.66 ± 0.16 | 24 ± 4 |
| E28A | 5.84 ± 0.35 | 4.96 ± 0.35 | 1.16 ± 0.28 | 23 ± 5 |
| E29A | 5.73 ± 0.43 | 8.97 ± 0.44 | 2.45 ± 0.21 | 18 ± 2 |
| E38A | 6.20 ± 0.19 | 5.39 ± 0.50 | 2.61 ± 0.28 | 30 ± 1 |
| L41A * | 5.95 ± 0.24 | 993 ± 17 | 616 ± 111 | 11 ± 0.4 |
| V44A * | 5.05 ± 0.46 | 742 ± 8 | 498 ± 70 | 11 ± 0.4 |
| K45A | 10.31 ± 0.19 | 21.9 ± 0.4 | 15.0 ± 1.3 | 19 ± 1 |
| L48A | 5.67 ± 0.60 | 13.2 ± 0.1 | 4.24 ± 0.13 | 17 ± 0.6 |
| E24A E29A | 6.13 ± 0.34 | 4.25 ± 0.29 | 1.73 ± 0.14 | 18 ± 4 |
| D23A E24A E26A E28A E29A (5X) | 4.26 ± 0.17 | 2.95 ± 0.23 | 2.34 ± 0.33 | 15 ± 2 |
| Instrument | k1 (×10−3 s−1) | A1 (%) | k2 (×10−3 s−1) | A2 (%) | k3 (×10−3 s−1) | A3 (%) | k4 (×10−3 s−1) | A4 (%) |
|---|---|---|---|---|---|---|---|---|
| Fluorimeter | - | - | 87.6 ± 18.8 | 10 ± 0.5 | 15.0 ± 0.8 | 75 ± 4.5 | 2.66 ± 0.06 | 15 ± 4 |
| Stopped flow | 535 ± 70 | 6 ± 0.5 | 65.2 ± 7.3 | 15 ± 0.8 | 9.1 ± 0.2 | 79 ± 0.3 | - | - |
| TCF7L2 | Salt Dependence | ||
|---|---|---|---|
| WT | −1.72 ± 0.08 | 1.50 ± 0.13 | 1.89 ± 0.20 |
| 5X | −1.33 ± 0.07 | 0.61 ± 0.06 | 0.70 ± 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, B.M.; Rowling, P.J.E.; Dobson, C.M.; Itzhaki, L.S. Parallel and Sequential Pathways of Molecular Recognition of a Tandem-Repeat Protein and Its Intrinsically Disordered Binding Partner. Biomolecules 2021, 11, 827. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060827
Smith BM, Rowling PJE, Dobson CM, Itzhaki LS. Parallel and Sequential Pathways of Molecular Recognition of a Tandem-Repeat Protein and Its Intrinsically Disordered Binding Partner. Biomolecules. 2021; 11(6):827. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060827
Chicago/Turabian StyleSmith, Ben M., Pamela J. E. Rowling, Christopher M. Dobson, and Laura S. Itzhaki. 2021. "Parallel and Sequential Pathways of Molecular Recognition of a Tandem-Repeat Protein and Its Intrinsically Disordered Binding Partner" Biomolecules 11, no. 6: 827. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060827