
HK2 Mediated Glycolytic Metabolism in Mouse Photoreceptors Is Not Required to Cause Late Stage Age-Related Macular Degeneration-Like Pathologies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Research Design and Methods
2.1. Animals
2.2. Funduscopy, Angiography, and Optical Coherence Tomography (OCT)
2.3. Identification of Pathologies by Funduscopy for Quantification Purposes
2.4. Electroretinography (ERG)
2.5. Histological Analyses
2.6. RPE Phagocytosis Activity Analysis
2.7. Lactate ASSAY
2.8. Quantitative Western Blot Analysis
2.9. Lipid Profiling
2.10. Statistical Analysis
3. Results
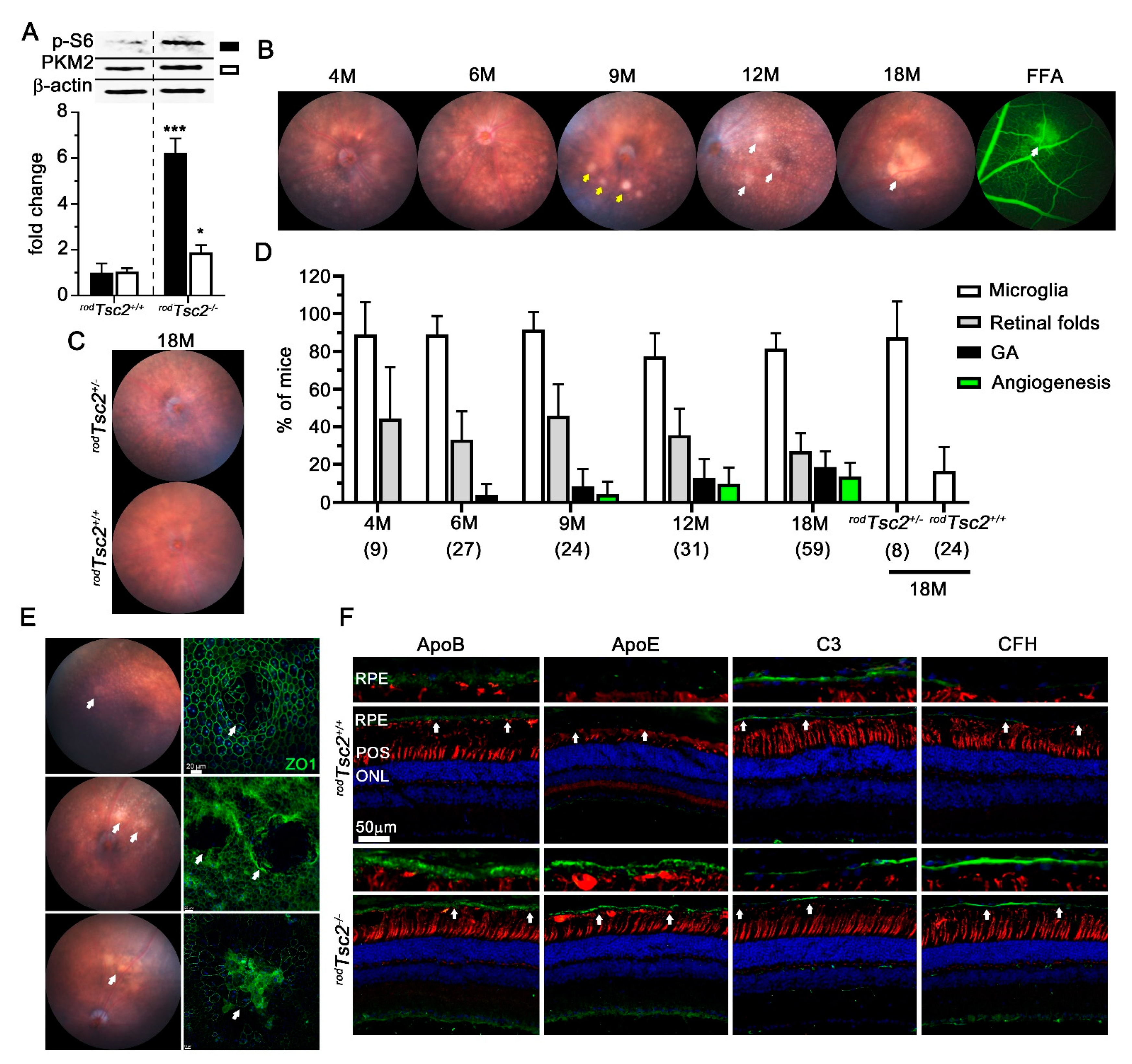
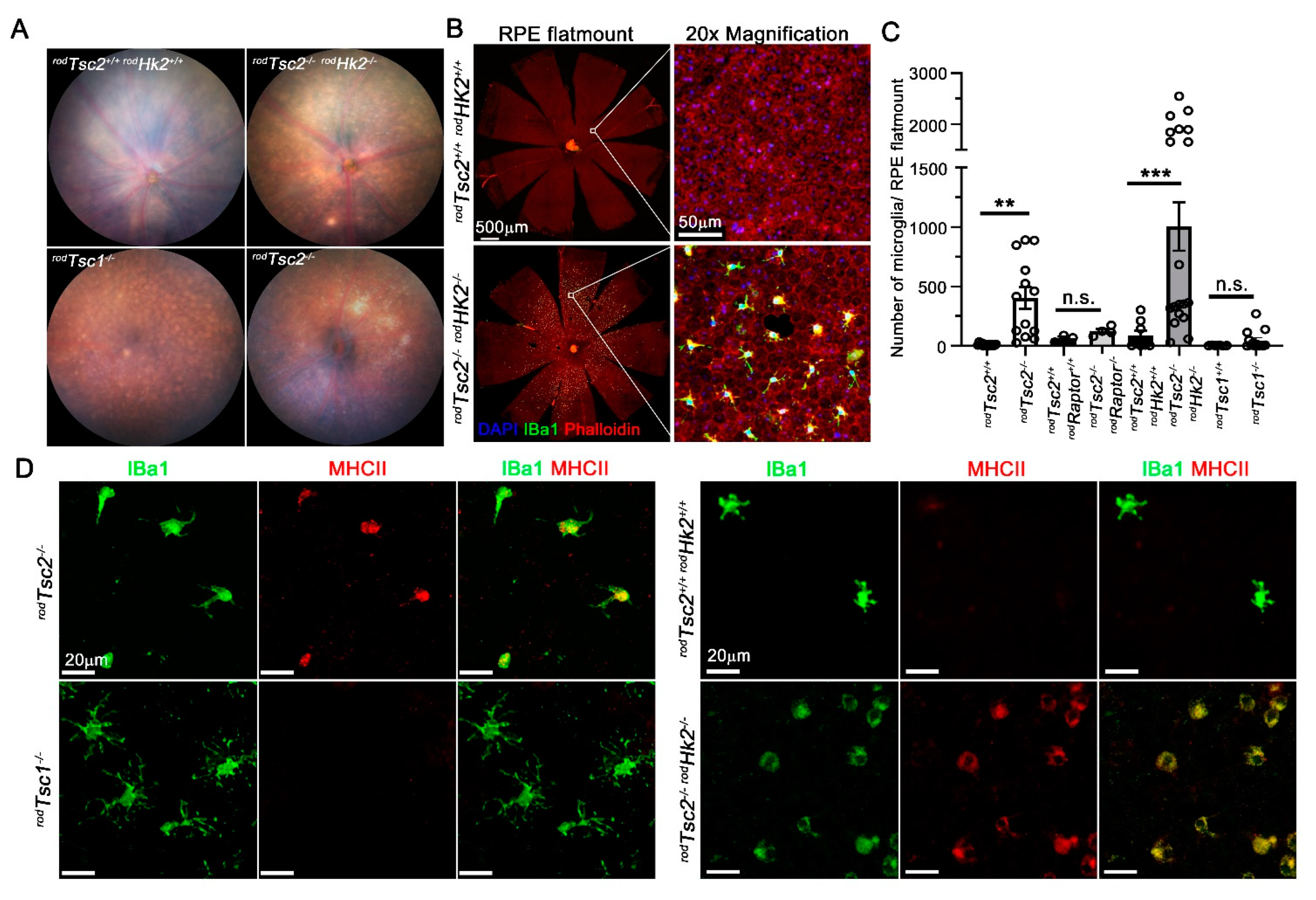
3.1. rodTsc2−/− Mice Develop AMD-Like Pathologies Similar to rodTsc1−/− Mice
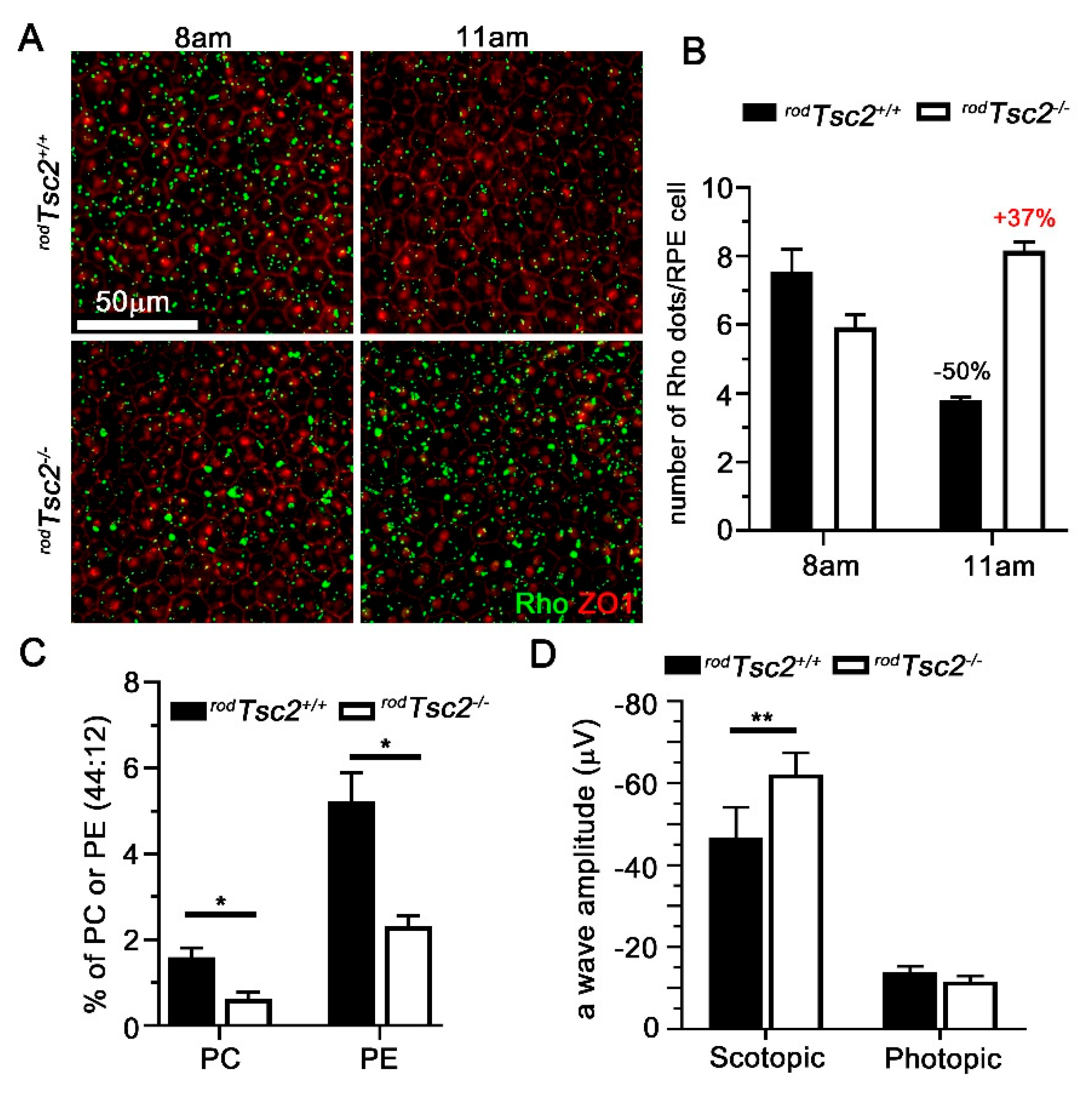
3.2. RPE Phagocytosis Activity Is Disrupted in rodTsc2−/− Mice
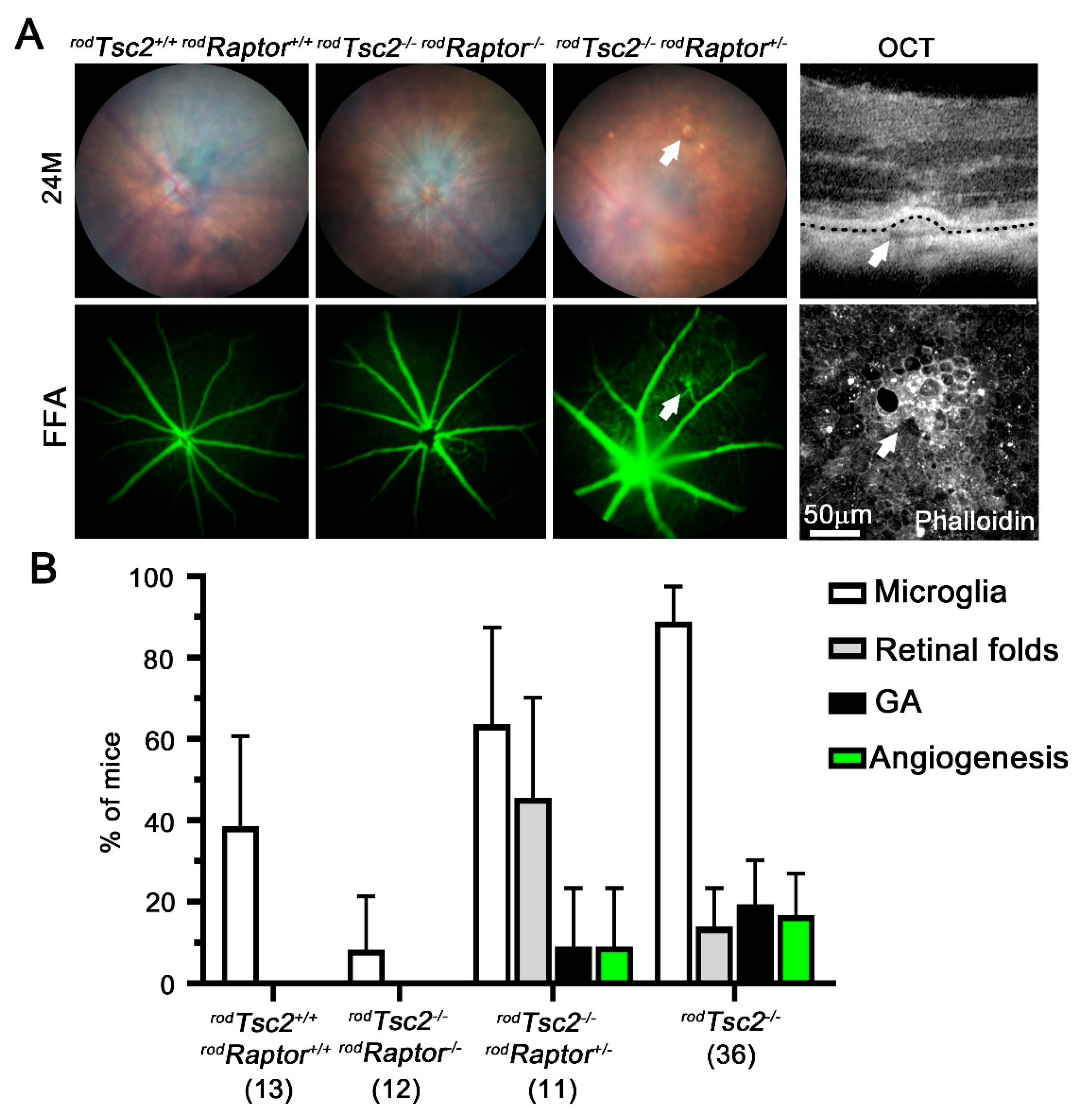
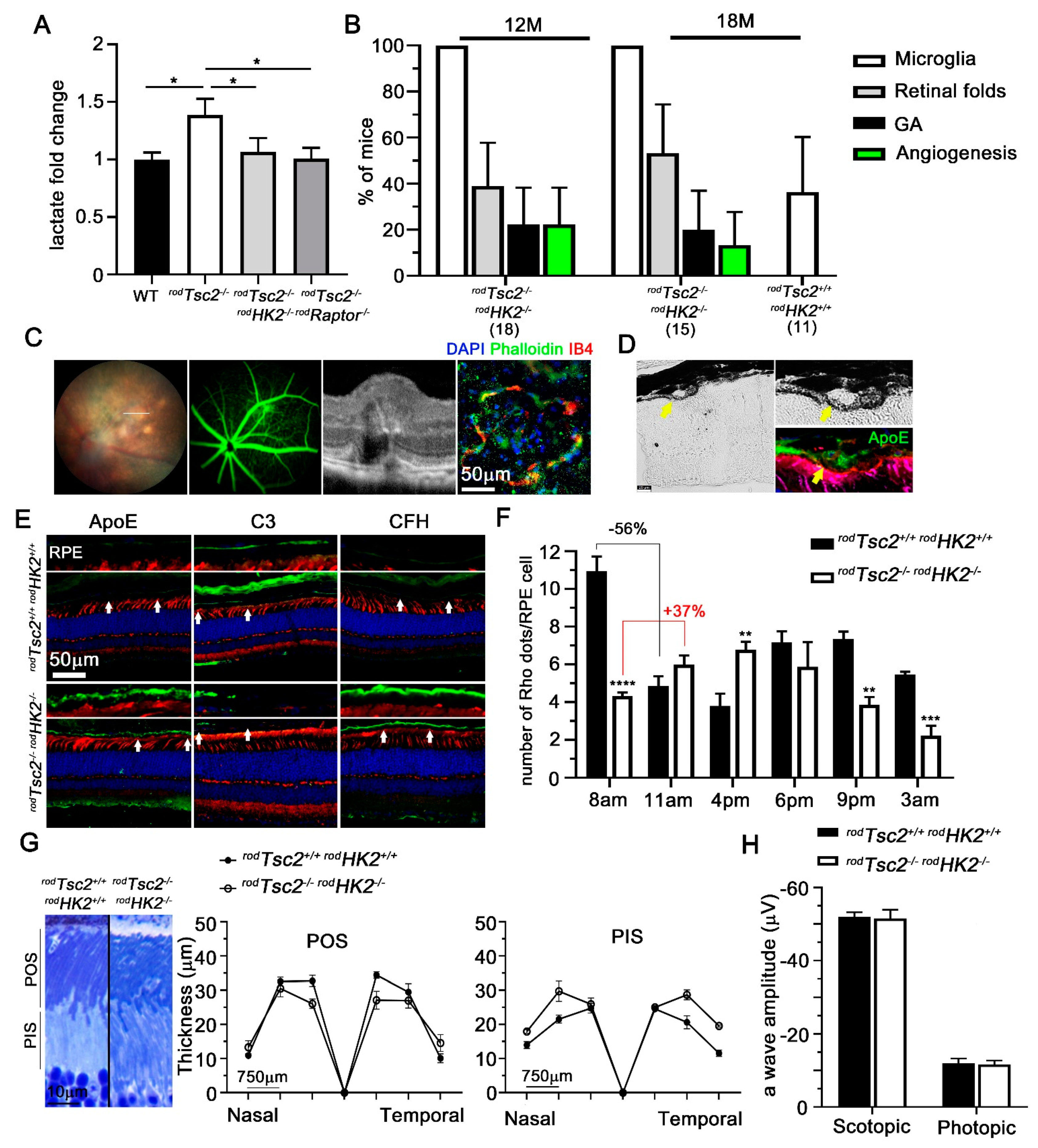
3.3. HK2-Mediated Aerobic Glycolysis Is Not Required for Severe AMD Pathologies to Develop
3.4. Microglia Migration and Reactivity Seen in the Subretinal Space
4. Discussion
Author Contributions
Funding
Institutional Animal Use Committee Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, D.S.; O’Colmain, B.J.; Munoz, B.; Tomany, S.C.; McCarty, C.; de Jong, P.T.; Nemesure, B.; Mitchell, P.; Kempen, J. Eye Diseases Prevalence Research, G. Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 2004, 122, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.W. Age-related macular degeneration revisited—Piecing the puzzle: The LXIX Edward Jackson memorial lecture. Am. J. Ophthalmol. 2013, 155, 1–35.e13. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Curcio, C.A. Antecedents of Soft Drusen, the Specific Deposits of Age-Related Macular Degeneration, in the Biology of Human Macula. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD182–AMD194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curcio, C.A. Soft Drusen in Age-Related Macular Degeneration: Biology and Targeting Via the Oil Spill Strategies. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD160–AMD181. [Google Scholar] [CrossRef] [Green Version]
- Geerlings, M.J.; de Jong, E.K.; den Hollander, A.I. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Grassmann, F.; Fleckenstein, M.; Chew, E.Y.; Strunz, T.; Schmitz-Valckenberg, S.; Gobel, A.P.; Klein, M.L.; Ratnapriya, R.; Swaroop, A.; Holz, F.G.; et al. Clinical and genetic factors associated with progression of geographic atrophy lesions in age-related macular degeneration. PLoS ONE 2015, 10, e0126636. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, D.; Seddon, J.M. Phenotypic Characterization of Complement Factor H R1210C Rare Genetic Variant in Age-Related Macular Degeneration. JAMA Ophthalmol. 2015, 133, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Malsy, J.; Alvarado, A.C.; Lamontagne, J.O.; Strittmatter, K.; Marneros, A.G. Distinct effects of complement and of NLRP3- and non-NLRP3 inflammasomes for choroidal neovascularization. Elife 2020, 9. [Google Scholar] [CrossRef]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [Green Version]
- Celkova, L.; Doyle, S.L.; Campbell, M. NLRP3 Inflammasome and Pathobiology in AMD. J. Clin. Med. 2015, 4, 172–192. [Google Scholar] [CrossRef] [Green Version]
- Soundara Pandi, S.P.; Ratnayaka, J.A.; Lotery, A.J.; Teeling, J.L. Progress in developing rodent models of age-related macular degeneration (AMD). Exp. Eye Res. 2021, 203, 108404. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Neuringer, M.; Courtney, R.J. Animal models of age related macular degeneration. Mol. Asp. Med. 2012, 33, 487–509. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.Y.; Cipi, J.; Ma, S.; Hafler, B.P.; Kanadia, R.N.; Brush, R.S.; Agbaga, M.P.; Punzo, C. Altered photoreceptor metabolism in mouse causes late stage age-related macular degeneration-like pathologies. Proc. Natl. Acad. Sci. USA 2020, 117, 13094–13104. [Google Scholar] [CrossRef]
- Petit, L.; Ma, S.; Cipi, J.; Cheng, S.Y.; Zieger, M.; Hay, N.; Punzo, C. Aerobic Glycolysis Is Essential for Normal Rod Function and Controls Secondary Cone Death in Retinitis Pigmentosa. Cell Rep. 2018, 23, 2629–2642. [Google Scholar] [CrossRef] [Green Version]
- Punzo, C.; Cepko, C. Cellular responses to photoreceptor death in the rd1 mouse model of retinal degeneration. Investig. Ophthalmol Vis. Sci. 2007, 48, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, A.; Ma, S.; Le, Y.Z.; Hall, M.N.; Ruegg, M.A.; Punzo, C. Activated mTORC1 promotes long-term cone survival in retinitis pigmentosa mice. J. Clin. Investig. 2015, 125, 1446–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, A.; Ma, S.; Punzo, C. TSC but not PTEN loss in starving cones of retinitis pigmentosa mice leads to an autophagy defect and mTORC1 dissociation from the lysosome. Cell Death Dis. 2016, 7, e2279. [Google Scholar] [CrossRef] [PubMed]
- Zieger, M.; Punzo, C. Improved cell metabolism prolongs photoreceptor survival upon retinal-pigmented epithelium loss in the sodium iodate induced model of geographic atrophy. Oncotarget 2016, 7, 9620–9633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Venkatesh, A.; Langellotto, F.; Le, Y.Z.; Hall, M.N.; Ruegg, M.A.; Punzo, C. Loss of mTOR signaling affects cone function, cone structure and expression of cone specific proteins without affecting cone survival. Exp. Eye Res. 2015, 135, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ames, A., 3rd. CNS energy metabolism as related to function. Brain Res. Brain Res. Rev. 2000, 34, 42–68. [Google Scholar] [CrossRef]
- Parker, R.O.; Crouch, R.K. Retinol dehydrogenases (RDHs) in the visual cycle. Exp. Eye Res. 2010, 91, 788–792. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; Kefalov, V.J. The cone-specific visual cycle. Prog. Retin. Eye Res. 2011, 30, 115–128. [Google Scholar] [CrossRef] [Green Version]
- LaVail, M.M. Circadian nature of rod outer segment disc shedding in the rat. Investig. Ophthalmol. Vis. Sci. 1980, 19, 407–411. [Google Scholar]
- Young, R.W. Shedding of discs from rod outer segments in the rhesus monkey. J. Ultrastruct. Res. 1971, 34, 190–203. [Google Scholar] [CrossRef]
- Young, R.W. The renewal of rod and cone outer segments in the rhesus monkey. J. Cell Biol. 1971, 49, 303–318. [Google Scholar] [CrossRef] [Green Version]
- Bownds, D.; Gordon-Walker, A.; Gaide-Huguenin, A.C.; Robinson, W. Characterization and analysis of frog photoreceptor membranes. J. Gen. Physiol. 1971, 58, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Lisman, J.E.; Bering, H. Electrophysiological measurement of the number of rhodopsin molecules in single Limulus photoreceptors. J. Gen. Physiol. 1977, 70, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Whikehart, D.R. Biochemistry of the Eye, 2nd ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2003; p. 512. [Google Scholar]
- Scott, B.L.; Racz, E.; Lolley, R.N.; Bazan, N.G. Developing rod photoreceptors from normal and mutant Rd mouse retinas: Altered fatty acid composition early in development of the mutant. J. Neurosci. Res. 1988, 20, 202–211. [Google Scholar] [CrossRef]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S.; Tsantilas, K.; Engel, A.L.; Du, J.; Linton, J.D.; Farnsworth, C.C.; Sloat, S.R.; Rountree, A.; et al. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. Elife 2017, 6. [Google Scholar] [CrossRef]
- Campbell, M.; Humphries, P. The blood-retina barrier: Tight junctions and barrier modulation. Adv. Exp. Med. Biol. 2012, 763, 70–84. [Google Scholar]
- Gardner, T.W.; Antonetti, D.A.; Barber, A.J.; Lieth, E.; Tarbell, J.A. The molecular structure and function of the inner blood-retinal barrier. Penn State Retina Research Group. Doc. Ophthalmol. 1999, 97, 229–237. [Google Scholar] [CrossRef]
- Tout, S.; Chan-Ling, T.; Hollander, H.; Stone, J. The role of Muller cells in the formation of the blood-retinal barrier. Neuroscience 1993, 55, 291–301. [Google Scholar] [CrossRef]
- Tyni, T.; Johnson, M.; Eaton, S.; Pourfarzam, M.; Andrews, R.; Turnbull, D.M. Mitochondrial fatty acid beta-oxidation in the retinal pigment epithelium. Pediatr. Res. 2002, 52, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reveles, J.; Dhingra, A.; Alexander, D.; Bragin, A.; Philp, N.J.; Boesze-Battaglia, K. Phagocytosis-dependent ketogenesis in retinal pigment epithelium. J. Biol. Chem. 2017, 292, 8038–8047. [Google Scholar] [CrossRef] [Green Version]
- Tyni, T.; Paetau, A.; Strauss, A.W.; Middleton, B.; Kivela, T. Mitochondrial fatty acid beta-oxidation in the human eye and brain: Implications for the retinopathy of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Pediatr. Res. 2004, 56, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Adijanto, J.; Du, J.; Moffat, C.; Seifert, E.L.; Hurle, J.B.; Philp, N.J. The retinal pigment epithelium utilizes fatty acids for ketogenesis. J. Biol. Chem. 2014, 289, 20570–20582. [Google Scholar] [CrossRef] [Green Version]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD Pathobiology: A Bioenergetic Crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.E.; DeWeerd, A.J.; Ildefonso, C.J.; Lewin, A.S.; Ash, J.D. Mitochondrial oxidative stress in the retinal pigment epithelium (RPE) led to metabolic dysfunction in both the RPE and retinal photoreceptors. Redox Biol. 2019, 24, 101201. [Google Scholar] [CrossRef] [PubMed]
- Grenell, A.; Wang, Y.; Yam, M.; Swarup, A.; Dilan, T.L.; Hauer, A.; Linton, J.D.; Philp, N.J.; Gregor, E.; Zhu, S.; et al. Loss of MPC1 reprograms retinal metabolism to impair visual function. Proc. Natl. Acad. Sci. USA 2019, 116, 3530–3535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, T.; Du, J.; Makia, M.S.; Hurley, J.B.; Naash, M.I.; Al-Ubaidi, M.R. Absence of retbindin blocks glycolytic flux, disrupts metabolic homeostasis, and leads to photoreceptor degeneration. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gu, S.; Chen, M.; Zhang, S.J.; Jiang, Z.; Chen, X.; Jiang, C.; Liu, G.; Radu, R.A.; Sun, X.; et al. Abnormal mTORC1 signaling leads to retinal pigment epithelium degeneration. Theranostics 2019, 9, 1170–1180. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Marth, J.D.; Orban, P.C.; Mossmann, H.; Rajewsky, K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science 1994, 265, 103–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, O.; Way, S.; McKenna, J., 3rd; Gambello, M.J. Generation of a conditional disruption of the Tsc2 gene. Genesis 2007, 45, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Romanino, K.; Cloetta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, K.C.; Wang, Q.; Bhaskar, P.T.; Miller, L.; Wang, Z.; Wheaton, W.; Chandel, N.; Laakso, M.; Muller, W.J.; Allen, E.L.; et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 2013, 24, 213–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Chen, D.; Sauve, Y.; McCandless, J.; Chen, Y.J.; Chen, C.K. Rhodopsin-iCre transgenic mouse line for Cre-mediated rod-specific gene targeting. Genesis 2005, 41, 73–80. [Google Scholar] [CrossRef]
- Mattapallil, M.J.; Wawrousek, E.F.; Chan, C.C.; Zhao, H.; Roychoudhury, J.; Ferguson, T.A.; Caspi, R.R. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2921–2927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, A.; Ma, S.; Langellotto, F.; Gao, G.; Punzo, C. Retinal gene delivery by rAAV and DNA electroporation. Curr. Protoc. Microbiol. 2013, 28, 14D.4.1–14D.4.32. [Google Scholar] [CrossRef] [Green Version]
- Law, A.L.; Ling, Q.; Hajjar, K.A.; Futter, C.E.; Greenwood, J.; Adamson, P.; Wavre-Shapton, S.T.; Moss, S.E.; Hayes, M.J. Annexin A2 regulates phagocytosis of photoreceptor outer segments in the mouse retina. Mol. Biol. Cell 2009, 20, 3896–3904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busik, J.V.; Reid, G.E.; Lydic, T.A. Global analysis of retina lipids by complementary precursor ion and neutral loss mode tandem mass spectrometry. Methods Mol. Biol. 2009, 579, 33–70. [Google Scholar] [CrossRef] [Green Version]
- Rajala, A.; Wang, Y.; Brush, R.S.; Tsantilas, K.; Jankowski, C.S.R.; Lindsay, K.J.; Linton, J.D.; Hurley, J.B.; Anderson, R.E.; Rajala, R.V.S. Pyruvate kinase M2 regulates photoreceptor structure, function, and viability. Cell Death Dis. 2018, 9, 240. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, E.L. Contribution of microglia and monocytes to the development and progression of age related macular degeneration. Ophthalmic. Physiol. Opt. 2020, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef]
- Jones, A.C.; Daniells, C.E.; Snell, R.G.; Tachataki, M.; Idziaszczyk, S.A.; Krawczak, M.; Sampson, J.R.; Cheadle, J.P. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum. Mol. Genet. 1997, 6, 2155–2161. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.H.; Rensing, N.R.; Zhang, B.; Gutmann, D.H.; Gambello, M.J.; Wong, M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 2011, 20, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Mietzsch, U.; McKenna, J., 3rd; Reith, R.M.; Way, S.W.; Gambello, M.J. Comparative analysis of Tsc1 and Tsc2 single and double radial glial cell mutants. J. Comp. Neurol. 2013, 521, 3817–3831. [Google Scholar] [CrossRef]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol. Cell Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Lambert, N.G.; ElShelmani, H.; Singh, M.K.; Mansergh, F.C.; Wride, M.A.; Padilla, M.; Keegan, D.; Hogg, R.E.; Ambati, B.K. Risk factors and biomarkers of age-related macular degeneration. Prog. Retin Eye Res. 2016, 54, 64–102. [Google Scholar] [CrossRef] [Green Version]
- Khurana, R.N.; Fujii, G.Y.; Walsh, A.C.; Humayun, M.S.; de Juan, E., Jr.; Sadda, S.R. Rapid recurrence of geographic atrophy after full macular translocation for nonexudative age-related macular degeneration. Ophthalmology 2005, 112, 1586–1591. [Google Scholar] [CrossRef]
- Cahill, M.T.; Mruthyunjaya, P.; Bowes Rickman, C.; Toth, C.A. Recurrence of retinal pigment epithelial changes after macular translocation with 360 degrees peripheral retinectomy for geographic atrophy. Arch. Ophthalmol. 2005, 123, 935–938. [Google Scholar] [CrossRef] [Green Version]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef]
- Ryu, J.K.; Cho, T.; Choi, H.B.; Wang, Y.T.; McLarnon, J.G. Microglial VEGF receptor response is an integral chemotactic component in Alzheimer’s disease pathology. J. Neurosci. 2009, 29, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Forstreuter, F.; Lucius, R.; Mentlein, R. Vascular endothelial growth factor induces chemotaxis and proliferation of microglial cells. J. Neuroimmunol. 2002, 132, 93–98. [Google Scholar] [CrossRef]
- Couturier, A.; Bousquet, E.; Zhao, M.; Naud, M.C.; Klein, C.; Jonet, L.; Tadayoni, R.; de Kozak, Y.; Behar-Cohen, F. Anti-vascular endothelial growth factor acts on retinal microglia/macrophage activation in a rat model of ocular inflammation. Mol. Vis. 2014, 20, 908–920. [Google Scholar]
- Uehara, H.; Mamalis, C.; McFadden, M.; Taggart, M.; Stagg, B.; Passi, S.; Earle, P.; Chakravarthy, U.; Hogg, R.E.; Ambati, B.K. The reduction of serum soluble Flt-1 in patients with neovascular age-related macular degeneration. Am. J. Ophthalmol. 2015, 159, 92–100.e2. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Uehara, H.; Zhang, X.; Das, S.K.; Olsen, T.; Holt, D.; Simonis, J.M.; Jackman, K.; Singh, N.; Miya, T.R.; et al. Photoreceptor avascular privilege is shielded by soluble VEGF receptor-1. Elife 2013, 2, e00324. [Google Scholar] [CrossRef]
- Goto, J.; Talos, D.M.; Klein, P.; Qin, W.; Chekaluk, Y.I.; Anderl, S.; Malinowska, I.A.; Di Nardo, A.; Bronson, R.T.; Chan, J.A.; et al. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc. Natl. Acad. Sci. USA 2011, 108, E1070–E1079. [Google Scholar] [CrossRef] [Green Version]
- Petrou, P.A.; Cunningham, D.; Shimel, K.; Harrington, M.; Hammel, K.; Cukras, C.A.; Ferris, F.L.; Chew, E.Y.; Wong, W.T. Intravitreal sirolimus for the treatment of geographic atrophy: Results of a phase I/II clinical trial. Investig. Ophthalmol. Vis. Sci. 2014, 56, 330–338. [Google Scholar] [CrossRef]
- Wong, W.T.; Dresner, S.; Forooghian, F.; Glaser, T.; Doss, L.; Zhou, M.; Cunningham, D.; Shimel, K.; Harrington, M.; Hammel, K.; et al. Treatment of geographic atrophy with subconjunctival sirolimus: Results of a phase I/II clinical trial. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2941–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.-Y.; Malachi, A.; Cipi, J.; Ma, S.; Brush, R.S.; Agbaga, M.-P.; Punzo, C. HK2 Mediated Glycolytic Metabolism in Mouse Photoreceptors Is Not Required to Cause Late Stage Age-Related Macular Degeneration-Like Pathologies. Biomolecules 2021, 11, 871. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060871
Cheng S-Y, Malachi A, Cipi J, Ma S, Brush RS, Agbaga M-P, Punzo C. HK2 Mediated Glycolytic Metabolism in Mouse Photoreceptors Is Not Required to Cause Late Stage Age-Related Macular Degeneration-Like Pathologies. Biomolecules. 2021; 11(6):871. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060871
Chicago/Turabian StyleCheng, Shun-Yun, Anneliese Malachi, Joris Cipi, Shan Ma, Richard S. Brush, Martin-Paul Agbaga, and Claudio Punzo. 2021. "HK2 Mediated Glycolytic Metabolism in Mouse Photoreceptors Is Not Required to Cause Late Stage Age-Related Macular Degeneration-Like Pathologies" Biomolecules 11, no. 6: 871. https://0-doi-org.brum.beds.ac.uk/10.3390/biom11060871